

Lattice Thermal Conductivity in XMg2Sb2(X = Ca or Mg) Compounds: Temperature and High-Order Anharmonicity Effect

Abstract
:1. Introduction
2. Computational Methods
3. Results and Discussion
3.1. Crystal-Structure and Elastic Properties
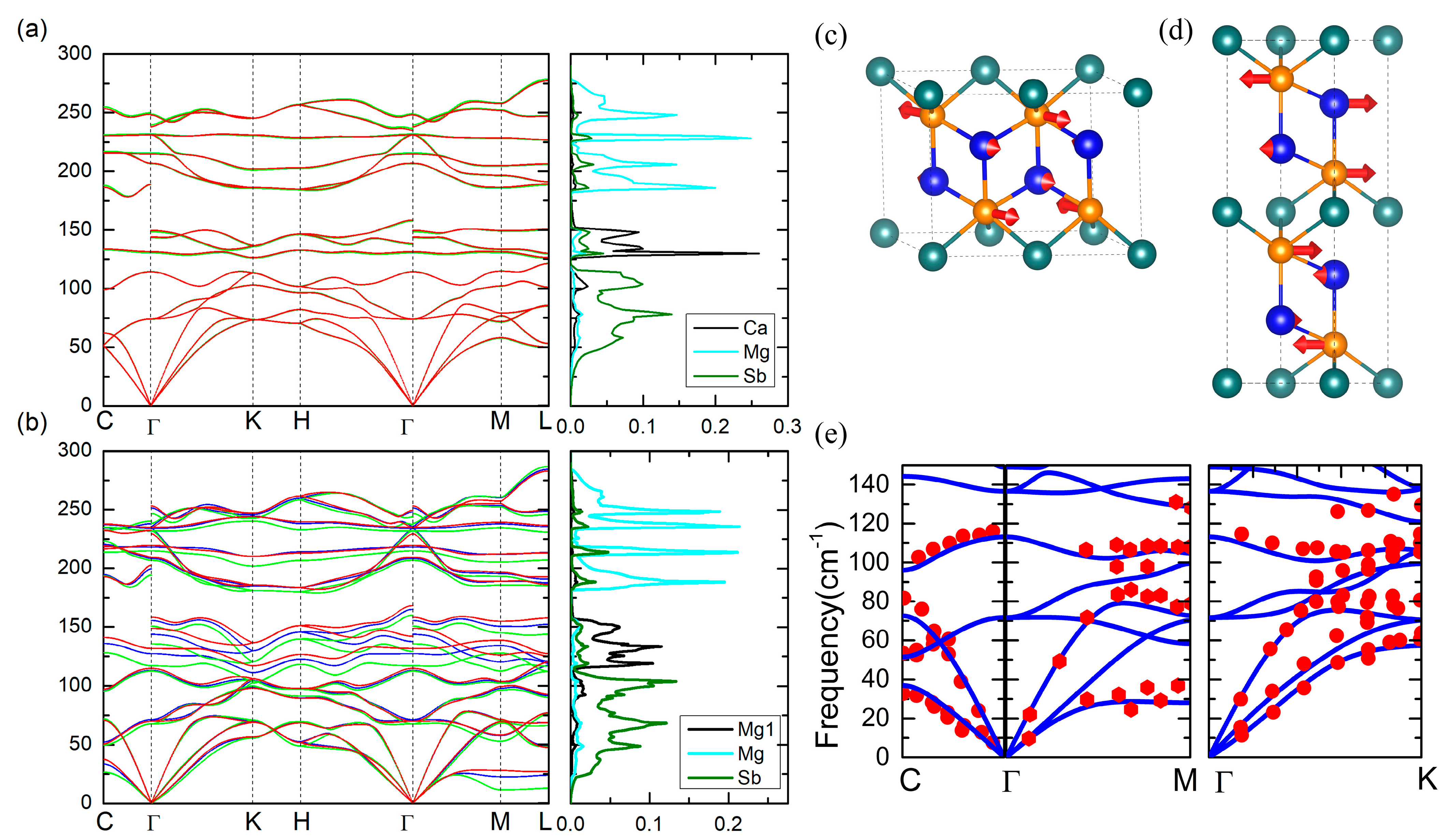
3.2. Phonon Dispersion
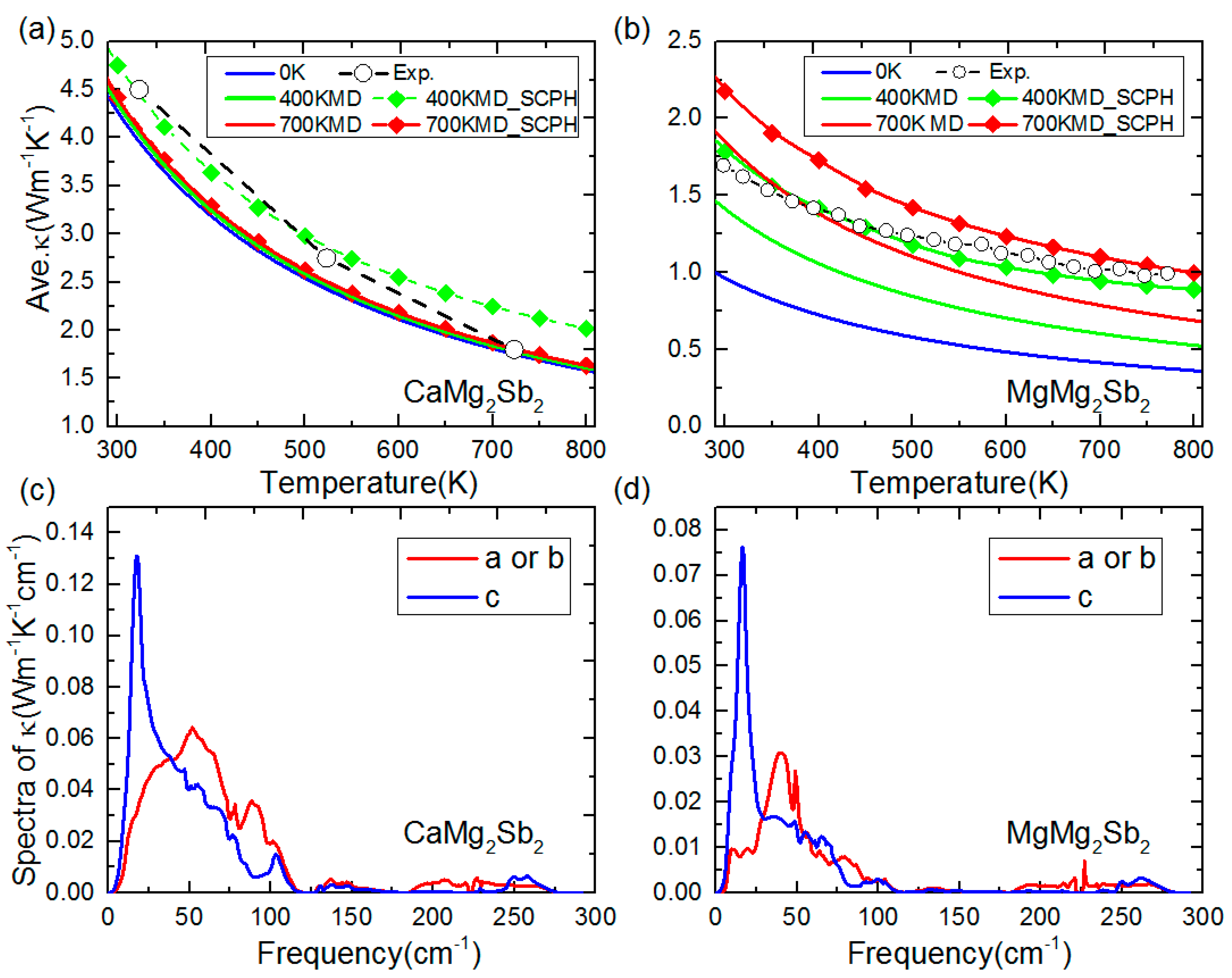
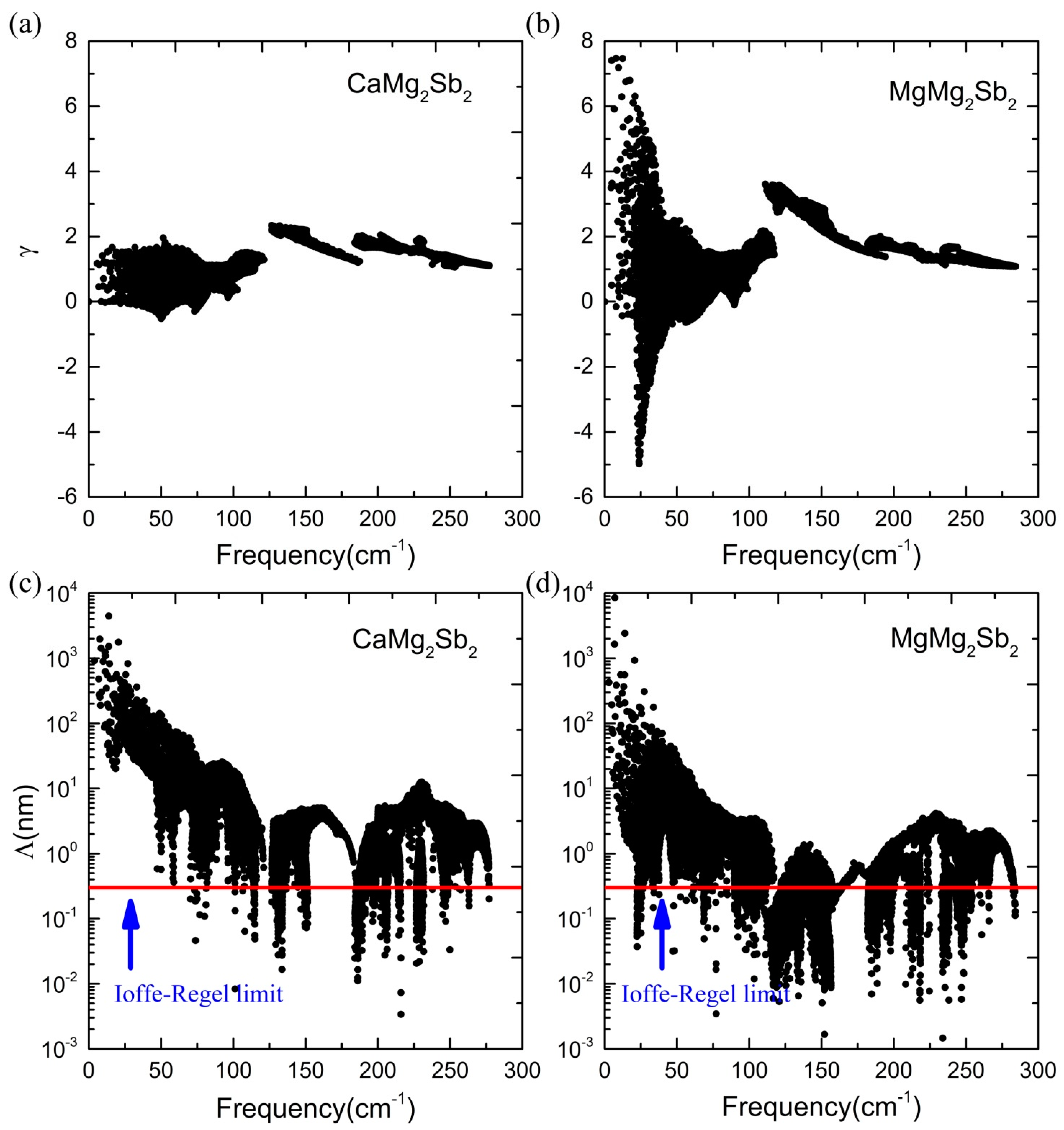
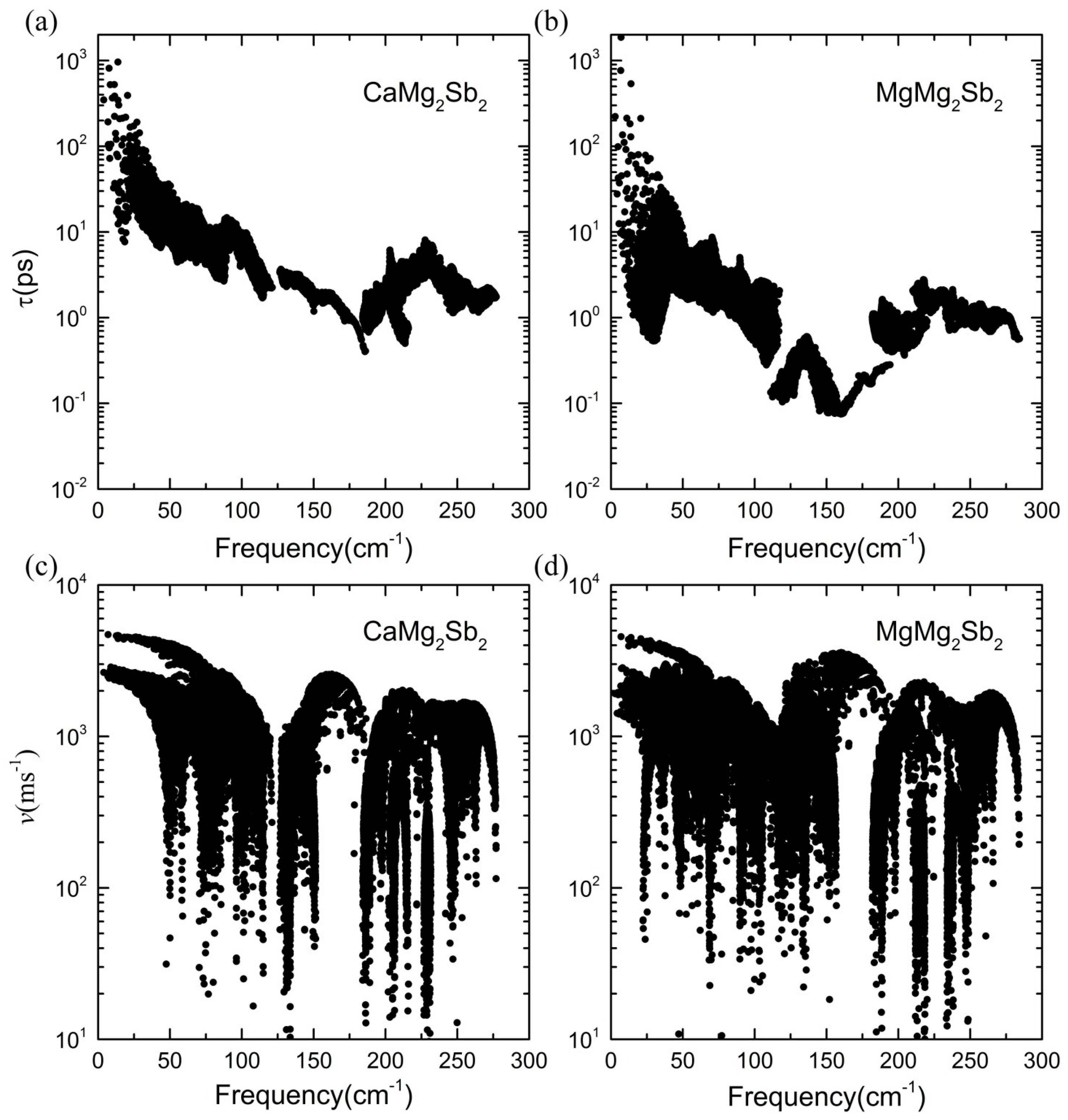
3.3. Lattice Thermal Conductivity
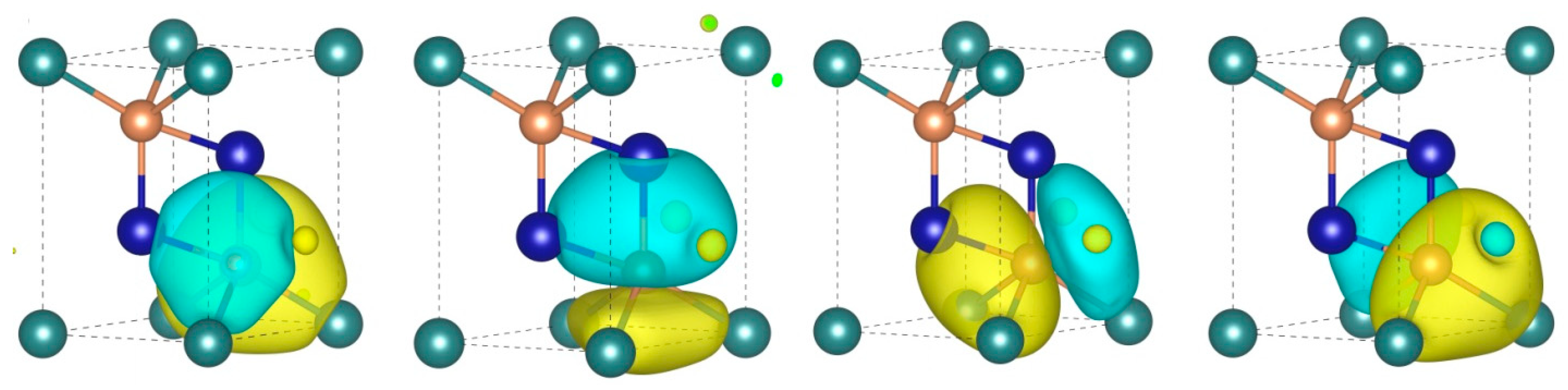
3.4. Chemical Bonding
4. Summary and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cahill, D.G.; Ford, W.K.; Goodson, K.E.; Mahan, G.D.; Majumdar, A.; Maris, H.J.; Merlin, R.; Phillpot, S.R. Nanoscale thermal transport. J. Appl. Phys. 2002, 93, 793–818. [Google Scholar] [CrossRef]
- Snyder, G.J.; Toberer, E.S. Complex thermoelectric materials. Nat. Mater. 2008, 7, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Bell, L.E. Cooling, Heating, Generating Power, and Recovering Waste Heat with Thermoelectric Systems. Science 2008, 321, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Ziman, J.M. Electrons and Phonons: The Theory of Transport Phenomena in Solids; Oxford University Press: Oxford, UK, 1960. [Google Scholar]
- Qian, X.; Zhou, J.; Chen, G. Phonon-engineered extreme thermal conductivity materials. Nat. Mater. 2021, 20, 1188–1202. [Google Scholar] [CrossRef]
- Poudel, B.; Hao, Q.; Ma, Y.; Lan, Y.; Minnich, A.; Yu, B.; Yan, X.; Wang, D.; Muto, A.; Vashaee, D. High-thermoelectric perfor-mance of nanostructured bismuth antimony telluride bulk alloys. Science 2008, 320, 634–638. [Google Scholar] [CrossRef]
- Heremans, J.P.; Jovovic, V.; Toberer, E.S.; Saramat, A.; Kurosaki, K.; Charoenphakdee, A.; Yamanaka, S.; Snyder, G.J. Enhancement of thermoelectric efficiency in PbTe by distortion of the electronic density of states. Science 2008, 321, 554–557. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Parker, D.S.; Sales, B.C.; Puretzky, A.A.; McGuire, M.A.; Lindsay, L. Two-channel model for ultralow thermal conductivity of crystalline Tl3VSe4. Science 2018, 360, 1455. [Google Scholar] [CrossRef]
- Wu, M.; Huang, L.E. Unusual lattice thermal conductivity in the simple crystalline compounds TlXTe2 (X=Ga,In). Phys. Rev. B 2019, 100, 075207. [Google Scholar] [CrossRef]
- Pei, Y.; Chang, C.; Wang, Z.; Yin, M.; Wu, M.; Tan, G.; Wu, H.; Chen, Y.; Zheng, L.; Gong, S. Multiple converged conduction bands in K2Bi8Se13: A promising thermoelectric material with extremely low thermal conductivity. J. Am. Chem. Soc. 2016, 138, 16364–16371. [Google Scholar] [CrossRef]
- Brown, S.R.; Kauzlarich, S.M.; Gascoin, F.; Snyder, G.J. Yb14MnSb11: New high efficiency thermoelectric material for power generation. Chem. Mater. 2006, 18, 1873–1877. [Google Scholar] [CrossRef]
- Christensen, M.; Abrahamsen, A.B.; Christensen, N.B.; Juranyi, F.; Andersen, N.H.; Lefmann, K.; Andreasson, J.; Bahl, C.R.H.; Iversen, B.B. Avoided crossing of rattler modes in thermoelectric materials. Nat. Mater. 2008, 7, 811–815. [Google Scholar] [CrossRef] [PubMed]
- Sales, B.C.; Mandrus, D.; Williams, R.K. Filled Skutterudite Antimonides: A New Class of Thermoelectric Materials. Science 1996, 272, 1325–1328. [Google Scholar] [CrossRef] [PubMed]
- Condron, C.L.; Kauzlarich, S.M.; Gascoin, F.; Snyder, G.J. Thermoelectric properties and microstructure of Mg3Sb2. J. Solid State Chem. 2006, 179, 2252–2257. [Google Scholar] [CrossRef]
- Mao, J.; Shuai, J.; Song, S.; Wu, Y.; Dally, R.; Zhou, J.; Liu, Z.; Sun, J.; Zhang, Q.; Cruz, C.D.; et al. Manipulation of ionized impurity scattering for achieving high thermoelectric performance in n-type Mg3Sb2-based materials. Proc. Natl. Acad. Sci. USA 2017, 114, 10548–10553. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Song, L.; Iversen, B.B. Insights into the design of thermoelectric Mg3Sb2 and its analogs by combining theory and experiment. Npj Comput. Mater. 2019, 5, 76. [Google Scholar] [CrossRef]
- Toberer, E.S.; May, A.F.; Snyder, G.J. Zintl Chemistry for Designing High Efficiency Thermoelectric Materials. Chem. Mater. 2010, 22, 624–634. [Google Scholar] [CrossRef]
- Bhardwaj, A.; Rajput, A.; Shukla, A.K.; Pulikkotil, J.J.; Srivastava, A.K.; Dhar, A.; Gupta, G.; Auluck, S.; Misra, D.K.; Budhani, R.C. Mg3Sb2-based Zintl compound: A non-toxic, inexpensive and abundant thermoelectric material for power generation. RSC Adv. 2013, 3, 8504–8516. [Google Scholar] [CrossRef]
- Peng, W.; Petretto, G.; Rignanese, G.-M.; Hautier, G.; Zevalkink, A. An Unlikely Route to Low Lattice Thermal Conductivity: Small Atoms in a Simple Layered Structure. Joule 2018, 2, 1879–1893. [Google Scholar] [CrossRef]
- Zhu, Y.; Xia, Y.; Wang, Y.; Sheng, Y.; Yang, J.; Fu, C.; Li, A.; Zhu, T.; Luo, J.; Wolverton, C. Violation of the T−1 Relationship in the Lattice Thermal Conductivity of Mg3Sb2 with Locally Asymmetric Vibrations. Research 2020, 2020, 4589786. [Google Scholar] [CrossRef]
- Hafner, J. Ab-initio simulations of materials using VASP: Density-functional theory and beyond. J. Comput. Chem. 2008, 29, 2044. [Google Scholar] [CrossRef]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244–13249. [Google Scholar] [CrossRef] [PubMed]
- Marzari, N.; Mostofi, A.A.; Yates, J.R.; Souza, I.; Vanderbilt, D. Maximally localized Wannier functions: Theory and applications. Rev. Mod. Phys. 2012, 84, 1419. [Google Scholar] [CrossRef]
- Mostofi, A.A.; Yates, J.R.; Lee, Y.-S.; Souza, I.; Vanderbilt, D.; Marzari, N. wannier90: A tool for obtaining maximally-localised Wannier functions. Comput. Phys. Commun. 2008, 178, 685–699. [Google Scholar] [CrossRef]
- Zhou, F.; Nielson, W.; Xia, Y.; Ozoliņš, V. Lattice Anharmonicity and Thermal Conductivity from Compressive Sensing of First-Principles Calculations. Phys. Rev. Lett. 2014, 113, 185501. [Google Scholar] [CrossRef] [PubMed]
- Tadano, T.; Gohda, Y.; Tsuneyuki, S. Anharmonic force constants extracted from first-principles molecular dynamics: Ap-plications to heat transfer simulations. J. Phys. Condens. Mattter. 2014, 26, 225402. [Google Scholar] [CrossRef]
- Tadano, T.; Tsuneyuki, S. Self-consistent phonon calculations of lattice dynamical properties in cubic SrTiO3 with first-principles anharmonic force constants. Phys. Rev. B 2015, 92, 054301. [Google Scholar] [CrossRef]
- Wood, M.; Aydemir, U.; Ohno, S.; Snyder, G.J. Observation of valence band crossing: The thermoelectric properties of CaZn2Sb2–CaMg2Sb2 solid solution. J. Mater. Chem. A 2018, 6, 9437–9444. [Google Scholar] [CrossRef]
- Martinez-Ripoll, M.; Haase, A.; Brauer, G. The crystal structure of α-Mg3Sb2. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1974, 30, 2006–2009. [Google Scholar] [CrossRef]
- Jia, T.; Chen, G.; Zhang, Y. Lattice thermal conductivity evaluated using elastic properties. Phys. Rev. B 2017, 95, 155206. [Google Scholar] [CrossRef]
- Golesorkhtabar, R.; Pavone, P.; Spitaler, J.; Puschnig, P.; Draxl, C. ElaStic: A tool for calculating second-order elastic constants from first principles. Comput. Phys. Commun. 2013, 184, 1861–1873. [Google Scholar] [CrossRef]
- Tian, Z.; Garg, J.; Esfarjani, K.; Shiga, T.; Shiomi, J.; Chen, G. Phonon conduction in PbSe, PbTe, and PbTe1-xSex from first-principles calculations. Phys. Rev. B 2012, 85, 184303. [Google Scholar] [CrossRef]
- Ji, H.S.; Togo, A.; Kaviany, M.; Tanaka, I.; Shim, J.H. Low phonon conductivity of layered BiCuOS, BiCuOSe, and BiCuOTe from first principles. Phys. Rev. B 2016, 94, 115203. [Google Scholar] [CrossRef]
- Xiao, Y.; Chang, C.; Pei, Y.; Wu, D.; Peng, K.; Zhou, X.; Gong, S.; He, J.; Zhang, Y.; Zeng, Z.; et al. Origin of low thermal conductivity in SnSe. Phys. Rev. B 2016, 94, 125203. [Google Scholar] [CrossRef]
- Chi, H.; Kim, H.; Thomas, J.C.; Shi, G.; Sun, K.; Abeykoon, M.; Bozin, E.S.; Shi, X.; Li, Q.; Shi, X.; et al. Low-temperature structural and transport anomalies in Cu2Se. Phys. Rev. B 2014, 89, 195209. [Google Scholar] [CrossRef]
- Kanno, T.; Tamaki, H.; Yoshiya, M.; Uchiyama, H.; Maki, S.; Takata, M.; Miyazaki, Y. High-Density Frenkel Defects as Origin of N-Type Thermoelectric Performance and Low Thermal Conductivity in Mg3Sb2-Based Materials. Adv. Funct. Mater. 2021, 31, 2008469. [Google Scholar] [CrossRef]
- Ding, J.; Lanigan-Atkins, T.; Calderón-Cueva, M.; Banerjee, A.; Abernathy, D.L.; Said, A.; Zevalkink, A.; Delaire, O. Soft an-harmonic phonons and ultralow thermal conductivity in Mg3(Sb, Bi)2 thermoelectrics. Sci. Adv. 2021, 7, eabg1449. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Song, L.; Sist, M.; Tolborg, K.; Iversen, B.B. Chemical bonding origin of the unexpected isotropic physical properties in thermoelectric Mg3Sb2 and related materials. Nat. Commun. 2018, 9, 4716. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Lattice Constant | Direct Chemical Bond | MLWF | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A (Å) | c/a | dWFC-Sb | dWFC-Mg | dWFC-X | ΩMg | ΩX | ΩSb | |||||||||
| CaMg2Sb2 | 4.69 4.65 a | 1.62 1.63 a | 3.28 | 2.94 | 2.87 | 0.75, 0.82 * | 0.18 | 0.32 | 0.14 | 0.43 | 2.91, 3.34 * | |||||
| MgMg2Sb2 | 4.60 4.58 b | 1.58 1.58 b | 3.11 | 2.93 | 2.82 | 0.72, 0.88 * | 0.18 | 0.18 | 0.14 | 0.14 | 3.15, 3.50 * | |||||
| Elastic properties | ||||||||||||||||
| C11 | C12 | C13 | C33 | C44 | C23 | C55 | B | G | E | ΘD | ||||||
| CaMg2Sb2 | 77.9 | 27.0 | 20.9 | 66.1 | 25.4 | 20.9 | 26.7 | 40.0 | 26.0 | 64.0 | 4423 4330 a | 2610 2716 a | 3536 | 34.1 | ||
| MgMg2Sb2 | 71.0 | 37.0 | 21.7 | 76.0 | 17.0 | 21.7 | 14.0 | 42.1 | 18.2 | 47.7 | 4099 | 2147 | 2960 | 28.8 | ||
| Mode | T(K) | CaMg2Sb2 | MgMg2Sb2 | ||||
|---|---|---|---|---|---|---|---|
| ϖ (cm−1) | Involved Atoms: Direction | ϖ (cm−1) | Involved Atoms: Direction | ||||
| DFT | Δω | DFT | Δω | ||||
| Eg (R, 1) | 0 | 74.0 | - | Mg, mainly Sb: One Mg, Sb pair a (b), One Mg, Sb pair minus a (b) | 67.6 | - | Mg, mainly Sb: One Mg, Sb pair a (b), One Mg, Sb pair minus a (b) |
| 300 | 74.1 | 0.4 | 71.2 | 0.2 | |||
| 700 | 74.6 | 0.2 | 69.8 | 1.9 | |||
| A1g (R) | 0 | 114.3 | - | Sb: One Sb c, One Sb minus c | 112.3 | - | Sb: One Sb c, One Sb minus c |
| 300 | 114.2 | 1.1 | 112.9 | 1.1 | |||
| 700 | 114.1 | 2.2 | 114.6 | 1.3 | |||
| Eu (IR, TO) | 0 | 130.6 | - | mainly Ca, Sb: Ca a, Sb minus a | 116.9 | - | mainly Mg1, Sb:Mg1a, Sb minus a |
| 300 | 131.5 | 4.1 | 127.1 | 9.4 | |||
| 700 | 132.4 | 7.8 | 131.6 | 14.6 | |||
| Eu (IR, LO) | 0 | 143.4 | - | mainly Ca, Sb: Ca b, Sb minus b | 150.6 | - | mainly Mg1, Sb:Mg1 b, Sb minus b |
| 300 | 143.9 | 3.9 | 155.5 | 6.9 | |||
| 700 | 144.5 | 7.0 | 158.2 | 11.1 | |||
| A2u (IR, 1) | 0 | 148.3 | - | All, Ca(most): Ca c, Sb minus c, Mg c | 135.5 | - | All, Mg1 (most): Mg1 c, Sb minus c, Mg c |
| 300 | 149.5 | 3.8 | 143.6 | 6.7 | |||
| 700 | 151.4 | 6.5 | 148.7 | 9.1 | |||
| A1g (R) | 0 | 206.6 | - | Mg: One Mg c, One Mg minus c | 206.7 | - | Mg: One Mg c, One Mg minus c |
| 300 | 206.8 | 3.5 | 208.3 | 2.2 | |||
| 700 | 206.5 | 6.4 | 209.3 | 5.3 | |||
| Eu (IR, TO) | 0 | 215.6 | - | Sb, Mg(most): Mg a, Sb minus a | 214.8 | - | Sb, Mg(most): Mg a, Sb minus a |
| 300 | 214.5 | 2.2 | 218.1 | −0.8 | |||
| 700 | 217.3 | 3.0 | 219.4 | −0.2 | |||
| Eu (IR, LO) | 0 | 231.6 | - | Sb, Mg(most): Mg b, Sb minus b | 231.8 | - | Sb, Mg(most): Mg b, Sb minus b |
| 300 | 230.9 | 3.0 | 233.9 | 1.5 | |||
| 700 | 234.3 | 4.3 | 234.8 | 2.4 | |||
| Eg (R, TO) | 0 | 231.6 | - | Sb, Mg(most): one Mg and Sb pair a, One Mg and Sb pair minus a, | 231.8 | - | Sb, Mg(most): one Mg and Sb pair a, One Mg and Sb pair minus a |
| 300 | 230.9 | 3.0 | 233.9 | 1.5 | |||
| 700 | 234.3 | 4.3 | 234.8 | 2.4 | |||
| Eg (R, LO) | 0 | 248.6 | - | Sb, Mg(most): one Mg and Sb pair b, One Mg and Sb pair minus b | 246.5 | - | Sb, Mg(most): one Mg and Sb pair b, One Mg and Sb pair minus b |
| 300 | 247.8 | 1.9 | 251.3 | −0.1 | |||
| 700 | 250.0 | 3.1 | 253.0 | 0.7 | |||
| A2u (IR) | 0 | 239.5 | - | Ca, Mg: Ca c, Mg minus c | 239.2 | - | Mg1, Mg: Mg1 c, Mg minus c |
| 300 | 237.8 | 2.7 | 236.7 | 1.2 | |||
| 700 | 238.5 | 4.5 | 230.8 | 3.4 | |||
| Compounds | ) | (m/s) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| X | Mg | Sb | X | Mg | Sb | a/b | c | Ave. | |||
| CaMg2Sb2 | (2.52, 2.75) | (1.87, 1.74) | (−3.14, −3.11) | 1.38 | 2.00 | −2.69 | (6.56, 8.03) | (11.0, 11.0) | (2575, 4657) | (2644, 4749) | (2598, 4687) |
| MgMg2Sb2 | (3.34, 3.20) | (1.91, 1.84) | (−3.58, −3.44) | 2.00 | 2.00 | −3.00 | (4.97, 6.41) | (15.2, 16.3) | (1756, 4434) | (1782, 4636) | (1765, 4501) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, M.; Yang, H.; Xie, F.; Huang, L. Lattice Thermal Conductivity in XMg2Sb2(X = Ca or Mg) Compounds: Temperature and High-Order Anharmonicity Effect. Materials 2023, 16, 7349. https://doi.org/10.3390/ma16237349
Wu M, Yang H, Xie F, Huang L. Lattice Thermal Conductivity in XMg2Sb2(X = Ca or Mg) Compounds: Temperature and High-Order Anharmonicity Effect. Materials. 2023; 16(23):7349. https://doi.org/10.3390/ma16237349
Chicago/Turabian StyleWu, Minghui, Hongping Yang, Fengyan Xie, and Li Huang. 2023. "Lattice Thermal Conductivity in XMg2Sb2(X = Ca or Mg) Compounds: Temperature and High-Order Anharmonicity Effect" Materials 16, no. 23: 7349. https://doi.org/10.3390/ma16237349