
Electrochemical Methods for the Analysis of Trace Tin Concentrations—Review


Abstract
:1. Introduction
2. Types of Techniques Used
2.1. Polarography
2.2. Voltammetry
2.3. Potentiometry
3. Measurement Conditions
3.1. Working Electrodes
3.2. Supporting Electrolyte and Complexing Agent
3.3. Detection Limits
4. Influence of Foreign Ions and Organic Matrix
5. Simultaneous Determination of Tin with Other Ions
6. Application
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Habashi, F. Tin, Physical and Chemical Properties. In Encyclopedia of Metalloproteins; Springer: New York, NY, USA, 2013; pp. 2233–2234. [Google Scholar]
- Smith, P.J. (Ed.) Chemistry of Tin; Springer Science & Business Media: London, UK, 2012. [Google Scholar]
- Gielen, M. Tin Chemistry: Fundamentals, Frontiers, and Applications; John Wiley & Sons: Chichester, UK, 2008. [Google Scholar]
- Schäfer, S.G.; Femfert, U. Tin—A toxic heavy metal? A review of the literature. Regul. Toxicol. Pharmacol. 1984, 4, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Schulz, K.J.; DeYoung, J.H., Jr.; Seal, R.R., II; Bradley, D.C. (Eds.) Critical Mineral Resources of the United States—Economic and Environmental Geology and Prospects for future Supply; Professional Paper 1802; U.S. Geological Survey: Reston, VA, USA, 2017; Chapter A; pp. 1–53.
- Rüdel, H. Case study: Bioavailability of tin and tin compounds. Ecotoxicol. Environ. Saf. 2003, 56, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Blunden, S.; Wallace, T. Tin in canned food: A review and understanding of occurrence and effect. Food Chem. Toxicol. 2003, 41, 1651–1662. [Google Scholar] [CrossRef] [PubMed]
- Ostrakhovitch, E.A.; Cherian, M.G. Tin. In Handbook on the Toxicology of Metals, 3rd ed.; Elsevier: Amsterdam, The Netherlands, 2007; Volume 2, pp. 839–860. [Google Scholar]
- Tuzen, M.; Uluozlu, O.D.; Mendil, D.; Soylak, M.; Machado, L.O.R.; dos Santos, W.N.L.; Ferreira, S.L.C. A simple, rapid and green ultrasound assisted and ionic liquid dispersive microextraction procedure for the determination of tin in foods employing ETAAS. Food Chem. 2018, 245, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Jang, G.; Roper, D.K. Spectrophotometric determination of tin(II) by redox reaction using 3,3′,5,5′–tetramethylbenzidine dihydrochloride and N–bromosuccinimide. J. Anal. Chem. 2015, 70, 566–572. [Google Scholar] [CrossRef]
- Olmedo, P.; Pla, A.; Hernández, A.F.; Barbier, F.; Ayouni, L.; Gil, F. Determination of toxic elements (mercury, cadmium, lead, tin and arsenic) in fish and shellfish samples. Risk assessment for the consumers. Environ. Int. 2013, 59, 63–72. [Google Scholar] [CrossRef]
- Khodadoust, S.; Cham Kouri, N. Preconcentration of Sn (II) using the methylene blue on the activated carbon and its determination by spectrophotometry method. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2014, 123, 85–88. [Google Scholar] [CrossRef]
- Huang, C.; Li, Q.; Mo, J.; Wang, Z. Ultratrace determination of tin, germanium, and selenium by hydride generation coupled with a novel solution-cathode glow discharge-atomic emission spectrometry method. Anal. Chem. 2016, 88, 11559–11567. [Google Scholar] [CrossRef]
- Muniz, L.P.; dos Santos, L.M.G.; do Couto, K.L.M.; Jacob, S. Evaluation of metals in tomato sauces stored in different types of packaging. Food Sci. Technol. 2018, 38, 383–389. [Google Scholar] [CrossRef]
- Morte, E.S.d.B.; Barbosa, I.d.S.; Santos, E.C.; Nobrega, J.A.; Korn, M.d.G.A. Axial view inductively coupled plasma optical emission spectrometry for monitoring tin concentration in canned tomato sauce samples. Food Chem. 2012, 131, 348–352. [Google Scholar] [CrossRef]
- Mino, Y. Determination of tin in canned foods by X-ray fluorescence spectrometry. J. Health Sci. 2006, 52, 67–72. [Google Scholar] [CrossRef]
- Fritz, J.S.; Goodkin, L. Separation and determination of tin by liquid-solid chromatography. Anal. Chem. 1974, 46, 959–962. [Google Scholar] [CrossRef]
- Ye, Y.; Sang, J.; Ma, H.; Tao, G. Gas-phase chemiluminescence with ozone oxidation for the determination of total tin in environmental samples using flow injection hydride generation and cryotrapping. Anal. Chim. Acta 2010, 677, 149–155. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R.; White, H.S. Electrochemical Methods: Fundamentals and Applications; John Wiley & Sons: Hoboken, NJ, USA, 2022. [Google Scholar]
- Phillips, S.L.; Shain, I. Application of stripping analysis to the trace determination of tin. Anal. Chem. 1962, 34, 262–265. [Google Scholar] [CrossRef]
- Méndez, J.H.; Martínez, R.C.; López, M.E.G. Simultaneous determination of tin and lead by a.c. anodic stripping voltammetry at a hanging mercury drop electrode sensitized by cetyltrimethylammoniu. Anal. Chim. Acta 1982, 138, 47–54. [Google Scholar] [CrossRef]
- Fano, V.; Zanotti, L. Trace determination of tin(II) and tin(IV) in different solvents (H2O, CH3OH) by anodic stripping voltammetry. Microchem. J. 1973, 18, 345–349. [Google Scholar] [CrossRef]
- Dirilgen, N.; Dogan, F. Anodic stripping voltammetry: Sn and Pb analysis in archaeometallurgical samples. Int. J. Environ. Anal. Chem. 2004, 84, 855–864. [Google Scholar] [CrossRef]
- Li, Y.-H.; Xie, H.-Q.; Zhou, F.-Q.; Guo, H.-S. Determination of trace tin by anodic stripping voltammetry at a carbon paste electrode. Electroanalysis 2006, 18, 976–980. [Google Scholar] [CrossRef]
- Prior, C.; Walker, G.S. The use of the bismuth film electrode for the anodic stripping voltammetric determination of tin. Electroanalysis 2006, 18, 823–829. [Google Scholar] [CrossRef]
- Hutton, E.A.; Hočevar, S.B.; Mauko, L.; Ogorevc, B. Bismuth film electrode for anodic stripping voltammetric determination of tin. Anal. Chim. Acta 2006, 580, 244–250. [Google Scholar] [CrossRef]
- Prior, C. Anodic stripping voltammetry of tin at the bismuth film electrode using cetyltrimethylammonium bromide. Electroanalysis 2010, 22, 1446–1454. [Google Scholar] [CrossRef]
- Frena, M.; Campestrini, I.; de Braga, O.C.; Spinelli, A. In situ bismuth-film electrode for square-wave anodic stripping voltammetric determination of tin in biodiesel. Electrochim. Acta 2011, 56, 4678–4684. [Google Scholar] [CrossRef]
- Tyszczuk-Rotko, K.; Metelka, R.; Vytřas, K.; Barczak, M.; Sadok, I.; Mirosław, B. A simple and easy way to enhance sensitivity of Sn(IV) on bismuth film electrodes with the use of a mediator. Monatsh. Für Chem. Chem. Mon. 2016, 147, 61–68. [Google Scholar] [CrossRef]
- Yang, G.; Wang, Y.; Qi, F. Differential pulse anodic stripping voltammetric determination of traces of tin using a glassy carbon electrode modified with bismuth and a film of poly(bromophenol blue). Microchim. Acta 2012, 177, 365–372. [Google Scholar] [CrossRef]
- Sobhanardakani, S.; Farmany, A.; Abbasi, S. A new modified multiwalled carbon nanotube paste electrode for quantification of tin in fruit juice and bottled water samples. J. Ind. Eng. Chem. 2014, 20, 3214–3216. [Google Scholar] [CrossRef]
- Faller, C.; Henze, G.; Stojko, N.; Saraeva, S.; Brainina, K. Modified solid electrodes for stripping voltammetric determination of tin. Fresenius J. Anal. Chem. 1997, 358, 670–676. [Google Scholar] [CrossRef]
- Yang, S.; Tian, H.; Wang, D.; Tang, Y. The determination of trace tin by cathodic stripping voltammetry with a Nafion®-modified electrode. J. Electroanal. Chem. 1995, 383, 31–35. [Google Scholar] [CrossRef]
- Wang, J.; Zadeii, J. Ultrasensitive and selective measurements of tin by adsorptive stripping voltammetry of the tin—Tropolone complex. Talanta 1987, 34, 909–914. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, C.M.G.; Khan, S.H.; Riley, J.P. Determination of tin in sea water by adsorptive cathodic stripping voltammetry. Anal. Chim. Acta 1989, 222, 43–54. [Google Scholar] [CrossRef]
- Adeloju, S.B. Adsorptive voltammetric stripping analysis of ultra-trace amounts of tin in natural waters and sediments. Anal. Sci. 1991, 7, 1099–1103. [Google Scholar] [CrossRef]
- Li, Y.-H.; Long, H.; Zhou, F.-Q. Determination of trace tin by catalytic adsorptive cathodic stripping voltammetry. Anal. Chim. Acta 2005, 554, 86–91. [Google Scholar] [CrossRef]
- Abbasi, S.; Hamdeghadareh, S.; Rezaei-Soufi, L.; Farmany, A. 2,2-Hydroxyphenylbenzoxazole as a selective chelating agent for complexation with Tin and Zinc: A voltammetry study. Eurasian Chem. Commun. 2020, 2, 128–137. [Google Scholar]
- Gao, Z.; Siow, K.S. Adsorptive stripping differential pulse voltammetric determination of trace amounts of tin in biological samples. Anal. Sci. 1996, 12, 267–271. [Google Scholar] [CrossRef]
- Adeloju, S.B.O.; Pablo, F. Determination of ultra-trace concentrations of tin by adsorptive cathodic stripping voltammetry on a glassy carbon mercury film electrode. Anal. Chim. Acta 1992, 270, 143–152. [Google Scholar] [CrossRef]
- Heppeler, F.; Sander, S.; Henze, G. Determination of tin traces in water samples by adsorptive stripping voltammetry. Anal. Chim. Acta 1996, 319, 19–24. [Google Scholar] [CrossRef]
- Adamczyk, M.; Grabarczyk, M. Application of a solid bismuth microelectrode in an adsorptive stripping voltammetric procedure of trace tin quantification. J. Electrochem. Soc. 2022, 169, 016515. [Google Scholar] [CrossRef]
- Grabarczyk, M.; Wlazłowska, E.; Wawruch, A. Stripping voltammetry with nanomaterials-based electrode in the environmental analysis of trace concentrations of tin. ChemPhysChem 2023, e202300633. [Google Scholar] [CrossRef]
- Glodowski, S.; Kublik, Z. Cyclic and stripping voltammetry of tin at mercury hanging drop and film electrodes in acidic o-diphenol media in the presence of lead and cadmium. Anal. Chim. Acta 1980, 115, 51–60. [Google Scholar] [CrossRef]
- Dadda, A.S.; Teixeira, A.C.; Feltes, P.K.; Campos, M.M.; Leite, C.E.; Moriguchi-Jeckel, C.M. Determination of Sn2+ in lyophilized radiopharmaceuticals by voltammetry, using hydrochloric acid as electrolyte. J. Braz. Chem. Soc. 2014, 25, 1621–1629. [Google Scholar] [CrossRef]
- Adeloju, S.B.O.; Pablo, F. Simultaneous determination of lead and tin in biological and environmental materials by differential pulse cathodic voltammetry on a glassy carbon mercury film electrode. Electroanalysis 1995, 7, 750–755. [Google Scholar] [CrossRef]
- McCrory-Joy, C.; Rosamilia, J.M. Differential pulse polarography of germanium(IV), tin(IV), arsenic(V), antimony(V), selenium(IV) and tellurium(VI) at the static mercury drop electrode in catechol—Perchlorate media. Anal. Chim. Acta 1982, 142, 231–238. [Google Scholar] [CrossRef]
- Ni, Y.; Wang, L. Simultaneous polarographic determination of lead(II) and tin(II) by multivariate calibration. Anal. Lett. 1999, 32, 2081–2089. [Google Scholar] [CrossRef]
- Hubert, C.; Ziémons, E.; Rozet, E.; Breuer, A.; Lambert, A.; Jasselette, C.; de Bleye, C.; Lejeune, R.; Hubert, P. Development and validation of a quantitative method for the selective determination of tin species in tin octoate by differential pulse polarography. Talanta 2010, 80, 1413–1420. [Google Scholar] [CrossRef]
- Weber, G. Determination of tin in the ng g−1 range differential pulse polarography. Anal. Chim. Acta 1986, 186, 49–56. [Google Scholar] [CrossRef]
- Qiong, L.; Guanghan, L.; Heng, W.; Xiaogang, W. Determination of trace tin in foods by single-sweep polarography. Food Chem. 1999, 64, 129–132. [Google Scholar] [CrossRef]
- Somer, G.; Ünal, Ü. Simultaneous determination of trace Sn(II) and Sn(IV) using differential pulse polarography and application. Turk. J. Chem. 2011, 35, 73–85. [Google Scholar] [CrossRef]
- Somer, G.; Arslantas, A. Simultaneous determination of tin, lead and molybdenum by differential-pulse polarography. Analyst 1994, 119, 1257. [Google Scholar] [CrossRef]
- Decristoforo, C.; Obendorf, D.; Reichart, E.; Stubauer, G.; Riccabona, G. Determination of Sn(II) in technetium cold kits by voltammetry at the hanging mercury drop electrode (HMDE) and relevant radiopharmaceutical applications. Nucl. Med. Biol. 1998, 25, 675–683. [Google Scholar] [CrossRef]
- Pérez-Herranz, V.; García-Gabaldón, M.; Guiñón, J.; García-Antón, J. Effect of citric acid and hydrochloric acid on the polarographic behaviour of tin. Anal. Chim. Acta 2003, 484, 243–251. [Google Scholar] [CrossRef]
- Taher, M.A.; Puri, B.K. Differential pulse polarographic determination of tin in alloys and environmental samples after preconcentration with the ion pair of 2-nitroso-1-naphthol-4-sulfonic acid and tetradecyldimethylbenzylammonium chloride onto microcrystalline naphthalene or by column method. Talanta 1999, 48, 355–362. [Google Scholar]
- Monticelli, D.; Psaro, R.; Pozzi, A.; Dossi, C.; Recchia, S. Differential pulse voltammetric determination of tin in the presence of noble metals. Anal. Bioanal. Chem. 2005, 383, 115–121. [Google Scholar] [CrossRef]
- Hosseini, M.; Bagheri Sadeghi, H.; Rahimi, M.; Salavati-Niasari, M.; Dehghan Abkenar, S.; Alizadeh, K.; Reza Ganjali, M. Highly selective and sensitive tin(II) membrane electrode based on a new synthesized schiff’s base. Electroanalysis 2009, 21, 859–866. [Google Scholar] [CrossRef]
- Aghaie, H.; Giahi, M.; Monajjemi, M.; Arvand, M.; Nafissi, G.H.; Aghaie, M. Tin(II)-selective membrane potentiometric sensor using a crown ether as neutral carrier. Sens. Actuators B Chem. 2005, 107, 756–761. [Google Scholar] [CrossRef]
- Arvand, M.; Moghimi, A.M.; Afshari, A.; Mahmoodi, N. Potentiometric membrane sensor based on 6-(4-nitrophenyl)-2,4-diphenyl-3,5-diaza-bicyclo[3.1.0]hex-2-ene for detection of Sn(II) in real samples. Anal. Chim. Acta 2006, 579, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Demkin, A.M. Determination of trace tin by stripping potentiostatic coulometry. J. Anal. Chem. 2006, 61, 153–157. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R. Fundamentals and applications. Electrochem. Methods 2001, 2, 580–632. [Google Scholar]
- Borrill, A.J.; Reily, N.E.; Macpherson, J.V. Addressing the practicalities of anodic stripping voltammetry for heavy metal detection: A tutorial review. Analyst 2019, 144, 6834–6849. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Mehtab, S.; Zaidi, M.G. Voltammetry: An Electrochemical Analytical Method. Chem. Sci. 2020, 1, 129–140. [Google Scholar]
- Heyrovský, M. Polarography—Past, present, and future. J. Solid State Electrochem. 2011, 15, 1799–1803. [Google Scholar] [CrossRef]
- Gajdar, J.; Horakova, E.; Barek, J.; Fischer, J.; Vyskocil, V. Recent applications of mercury electrodes for monitoring of pesticides: A critical review. Electroanalysis 2016, 28, 2659–2671. [Google Scholar] [CrossRef]
- Bobacka, J.; Ivaska, A.; Lewenstam, A. Potentiometric ion sensors. Chem. Rev. 2008, 108, 329–351. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Qin, W. Recent advances in potentiometric biosensors. TrAC Trends Anal. Chem. 2020, 124, 115803. [Google Scholar] [CrossRef]
- Shao, Y.; Ying, Y.; Ping, J. Recent advances in solid–contact ion–selective electrodes: Functional materials, transduction mechanisms, and development trends. Chem. Soc. Rev. 2020, 49, 4405–4465. [Google Scholar] [CrossRef] [PubMed]
- Blair, E.O.; Corrigan, D.K. A review of microfabricated electrochemical biosensors for DNA detection. Biosens. Bioelectron. 2019, 134, 57–67. [Google Scholar] [CrossRef]
- Porada, R.; Jedlińska, K.; Lipińska, J.; Baś, B. Review–voltammetric sensors with laterally placed working electrodes: A review. J. Electrochem. Soc. 2020, 167, 037536. [Google Scholar] [CrossRef]
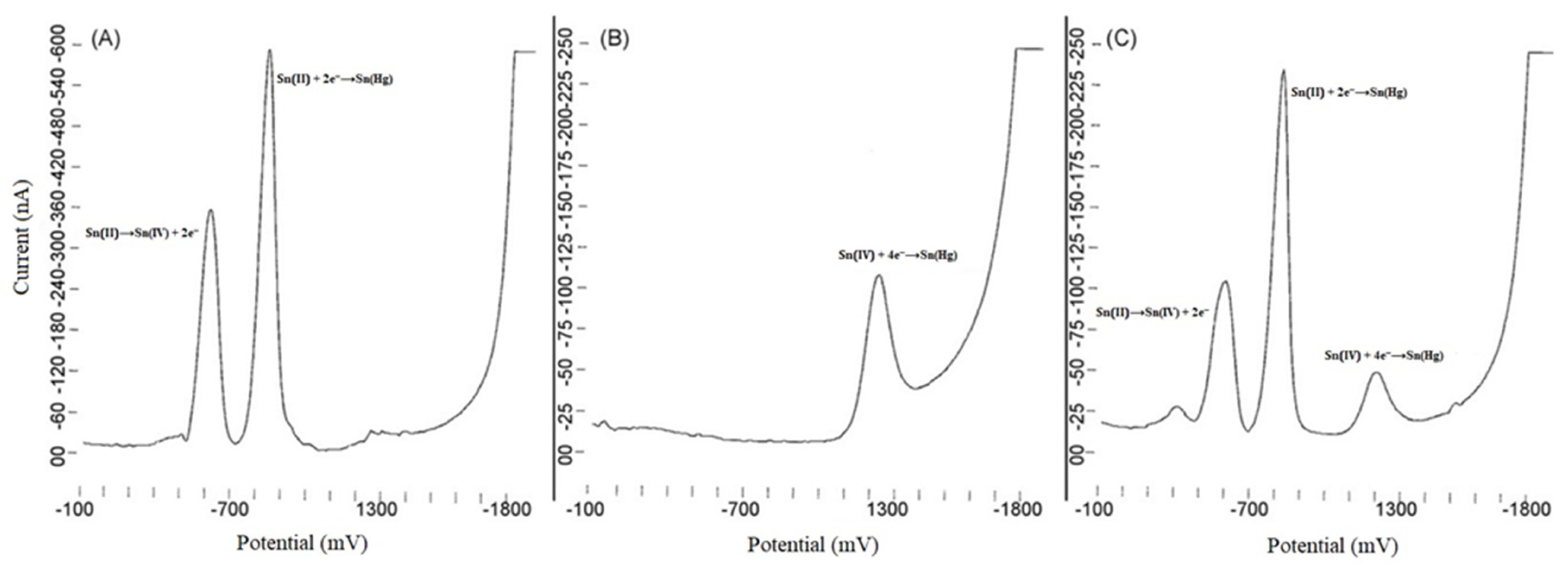
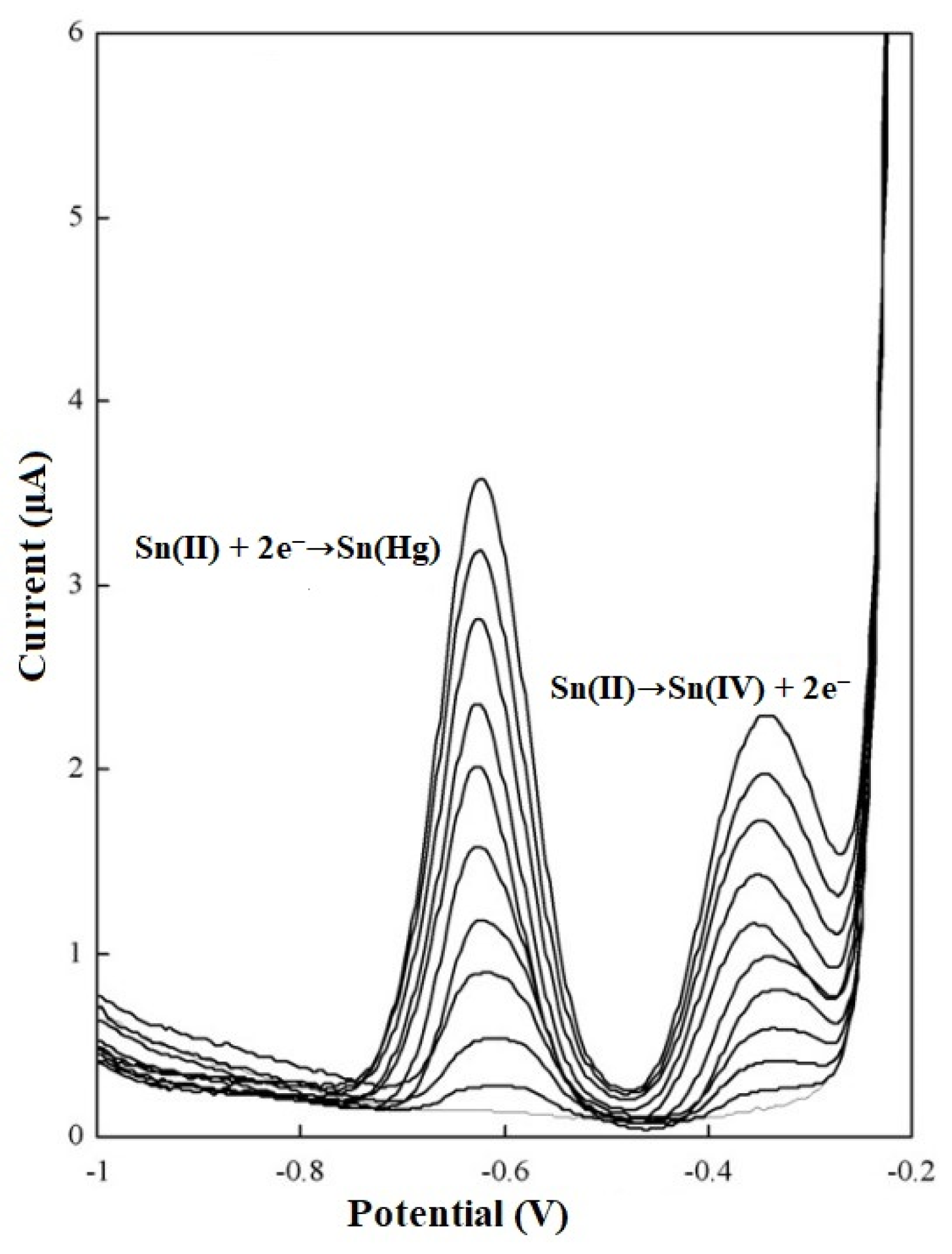
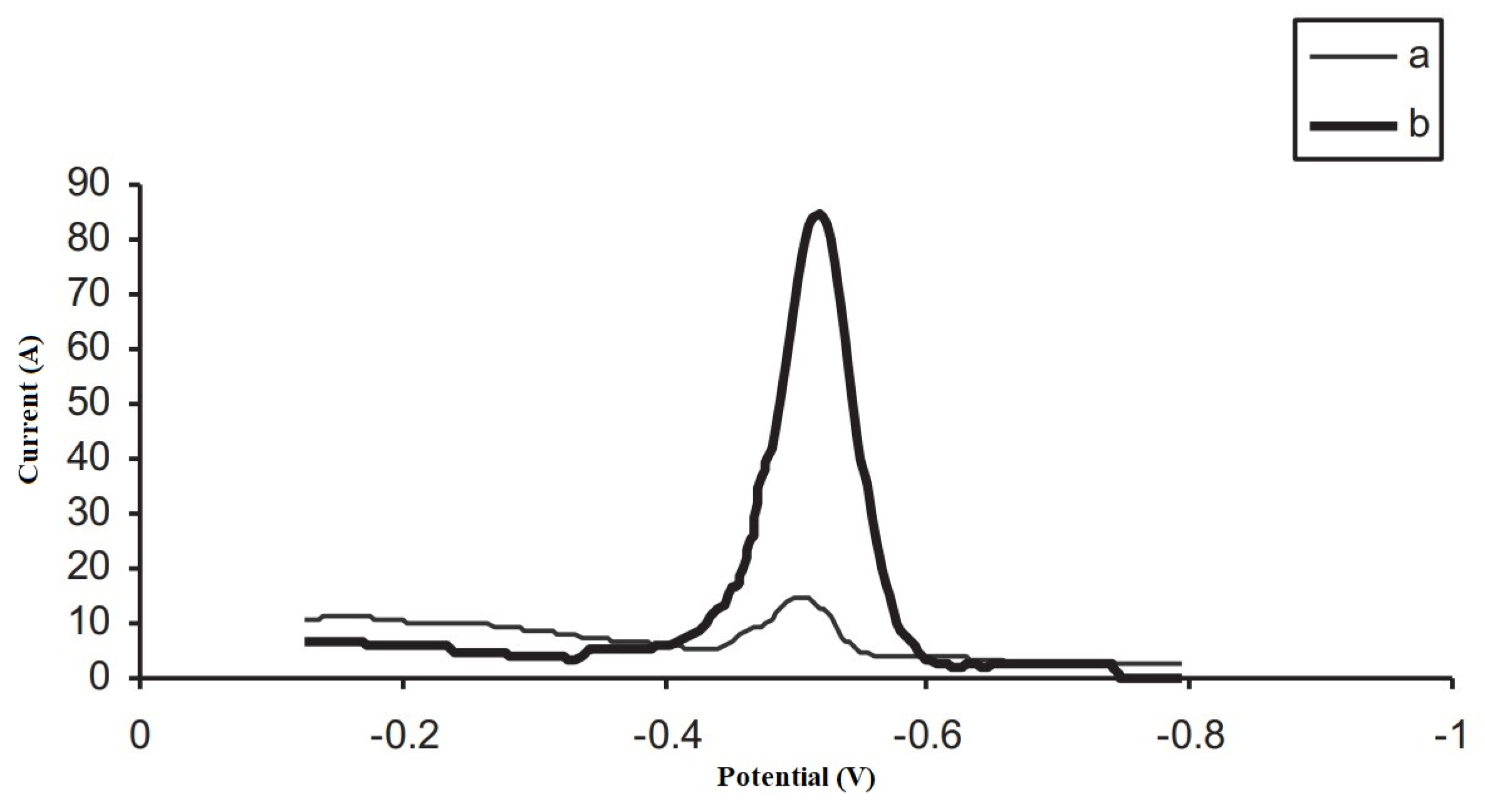
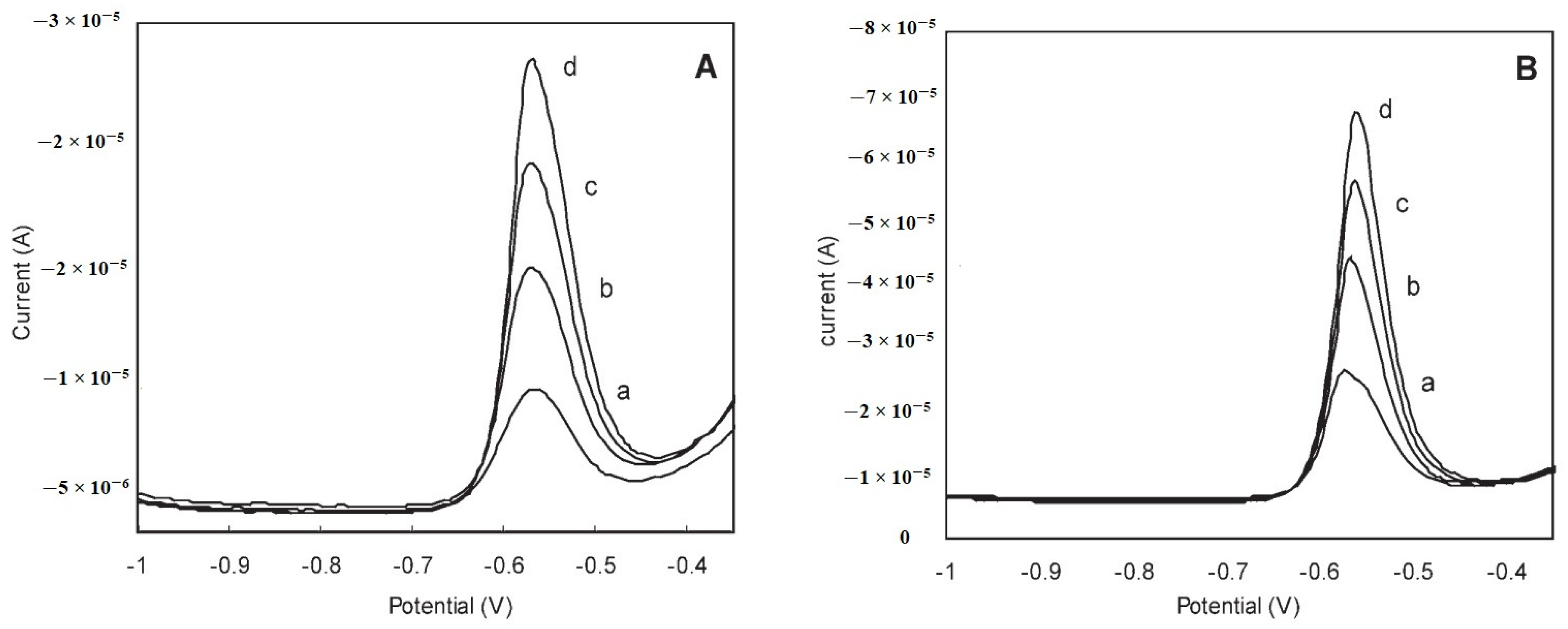

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Working Electrode | Supporting Electrolyte | Complexing Agent | Linear Range [mol L−1] | Detection Limit [mol L−1] | Accumulation Time [s] | Investigated Interferents | Application | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| AdSV | HMDE | (pH 2.1) | tropolone | no data–4.0 × 10−9 | 5.0 × 10−12 | 600 | In(III), Ni(II), Cr(VI), Al(III), As(III), V(V), Zn(II), Se(IV), Zr(II), W(VI), Bi(III), U(VI), Ba(II), Te(IV), Ti(IV), Mn(II), Fe(III), Mo(VI) Triton X–100 | sea water, estuarine water | [35] |
| AdSV | HMDE | 0.1 mol L−1 acetate buffer (pH 4.2) | catechol | 0–1.3 × 10−8 | 4.2 × 10−11 | 180 | Ni(II), Co(II), Fe(III), Al(III), Cu(II), Cd(II) EDTA, Triton X–100 | water, sediment samples | [36] |
| AdSV | HMDE | 0.1 mol L−1 formate buffer (pH 3.1) | DHBA | 8.4 × 10−11–3.4 × 10−7 | 4.2 × 10−11 | 60 | Ca(II), Mg(II), Al(III), Zn(II), Mn(II), Co(II), As(III), Be(II), Fe(III), Ni(II), Ga(III), Cu(II), Pb(II), Bi(III), Cd(II), Hg(II), Sb(III), Ti(III), In(III), W(VI), Cr(VI), Se(IV), Mo(VI), V(V), Ge(IV) Triton X–100 | canned food (fermented bean curd, grape juice, milk tea), human hair, waste water | [37] |
| AdSV | MFE | 0.01 mol L−1 acetate buffer (pH 4.3) | chloranilic acid | 4.2 × 10−9–1.7 × 10−7 | 1.9 × 10−11 | 400 | Sb(III), V(V), Fe(III), W(VI) Triton X–100, HA | drainage water | [41] |
| CSV | CME | 0.01 mol L−1 sodium chloride (pH 1.9) | – | 8.4 × 10−10–7.6 × 10−9 | 8.4 × 10−10 | 180 | K(I), Ca(II), Ba(II), Tl(I), Al(III), Bi (III), Cr(III), Ce(IV), Co(II), Fe(III), As(V), V(V), Ag(I), Mg(II), Cd(II), Hg(II), Pb(II), Ge(IV), Zn(II), Cu(II), Se(IV), Ni(II), Te(III), NO3−, CH3COO−, SCN−, H2PO4−, SO42−, Cr2O72− | human hair | [33] |
| AdSV | HMDE | 0.1 mol L−1 acetate buffer (pH 5.0) | HBO | 1.7 × 10−9–8.4 × 10−7 | 7.6 × 10−10 | 90 | Na(I), Cs(I), Ag(I), Al(III), Fe(III), Co(II), Mn(II), Cr(III), Cd(II),Pb(II) Sn(IV), Li(I), Ni(II), Cu(II), Mg(II), NH4+, Br−, Cl−, SCN−, H3PO4−, NO2−, CLO4−, SO32−, BrO3−, I− | blood serum, human hair | [38] |
| ASV | CPE | 0.1 mol L−1 acetate buffer (pH 4.5) | BPR | 8.4 × 10−10–4.2 × 10−7 | 5.0 × 10−10 | 120 | Ca(II), Mg(II), Al(III), Zn(II), Ba(II), Co(II), Hg(II), Mn(II), Cd(II), Ga(III), As(III), Cr(III), Ti(IV), Ge(IV), Fe(III), W(VI), Mo(VI), Se(IV), Pb(II), Ag(I), V(V), Sb(III), Cu(II) | canned food (fermented bean curd lychee juice orange juice milk tea), waste water | [24] |
| AdSV | HMDE | 0.1 mol L−1 acetate buffer (pH 4.3) | oxine | – | 3.5 × 10−10 7.0 × 10−11 | 60 300 | Zn(II), Mn(II), Co(II), Fe(II), Fe(III), Ni(I), Al(III), Ga(III), Hg(II); Cr(III), Cu(II), V(V), Ce(IV), Bi(III), Ag(I), Pb(II), Cd(II), In(III), W(VI), Mo(VI), Cd(II) | blood, serum, plants (orange, grass, corn) | [39] |
| AdSV | CNTs/SGC | 0.1 mol L−1 acetate buffer (pH 5.7) | cupferron | 1.0 × 10−9–1.0 × 10−7 | 3.1 × 10−10 | 95 | Au(III), Cd(II), Pt(IV), Se(IV), Zn(II), Fe(III), Pb(II), Hg(II), V(V), In(III), Ti(IV), Mo(VI), Pt(IV) SDS, CTAB, Triton X–100, Rhamnolipid, HA, FA | CRM (SPS–WW1 Waste Water), river water, tap water | [43] |
| AdSV | HMDE | 0.1 mol L−1 acetate buffer (pH 4.0) | tropolone | – | 2.3 × 10−10 | 560 | – | river water, orange juice | [34] |
| ASV | modified thick film electrodes | 1.0 mol L−1 hydrochloric acid | – | 8.4 × 10−9–8.4 × 10−7 | 7.6 × 10−9 | 60 | Cd(II), Pb(II), Cu(II), Zn(II), Tl(III) Triton X–100, HA | canned fruit juices (orange, pineapple, orange with pulp) | [32] |
| ASV | Bi/poly(BPB)GCE | 1.0 mol L−1 hydrochloric acid | – | 2.0 × 10−8–2.0 × 10−5 | 7.0 × 10−9 | 60 | Na(I), K(I), Ca(II), Mg(II), Mn(II), Ni(II), Co(II), Hg(II), Zn(II), Pb(II), Cu(II), Cd(II), Fe(III), SO42−, Cl− | canned fruit juice (pineapple, orange, grape, mango), | [30] |
| AdSV | GCMFE | 0.1 mol L−1 acetate buffer (pH 4.2–4.7) | catechol | 0–2.9 × 10−8 | 4.2 × 10−9 | 300 | Ca(II), Zn(II), Fe(III), Ni(II), Mg(II), Pb(II), Cu(II), Cd(II) | canned fruit juice (pineapple juice, apricot nectar, grapefruit juice, tomato juice, peach nectar) | [40] |
| ASV | BiFE | 0.1 mol L−1 acetate buffer (pH 4.5) | – | 8.4 × 10−9–8.4 × 10−7 | 2.2 × 10−9 | 60 | – | sea water | [26] |
| AdSV | BiFµE | 0.1 mol L−1 acetate buffer (pH 4.6) | cupferron | 8.0 × 10−9–8.0 × 10−7 | 2.1 × 10−9 | 40 | Au(III), Cd(II), Co(II), Cr(III), Cr(VI), Cu(II), Ge(IV), Mn(II), Ni(II), Pt(IV), Sb(III), Se(IV), Ti(IV), Tl(I), W(VI), Zn(II), Fe(III), Pb(II), Ga(III), Hg(II) SDS, CTAB, Triton X–100, Rhamnolipid, HA, FA | CRM (SPS–WW1 Waste Water, SPS –SW1 Surface Water), river water, rain water | [42] |
| ASV | CNT | borate buffer (pH 7.5) | – | 2.5 × 10−9–2.1 × 10−6 | 1.0 × 10−9 | 240 | K(I), Li(I), Ag(I), Mg(II), Ca(II), Ba(II), La(III), Cr(III), Fe(II), Fe(III), Na(I), Al(III), Co(II), Ti(III), Cd(II), Cu(II), Zn(II), HPO4−, CO32−, CN− | fruit juice, bottled water | [31] |
| ASV | BiFE | 0.1 mol L−1 acetate buffer (pH 4.5) | caffeic acid | 5.0 × 10−8–5.0 × 10−6 | 1.8 × 10−8 | 90 | – | – | [29] |
| ASV | BiFE | 75 mmol L−1 oxalic acid, 75 µmol L−1 CTAB | – | 4.2 × 10−8–2.1 × 10−6 | 1.6 × 10−8 | 120 | Cu(II), Se(IV), Mn(II), Fe(III), Mo(VI), V(V), Cr(III), Ni(II), Sb(III), Zn(II), Bi(III) | canned fruit juice (pineapple) | [27] |
| ASV | BiFE | 0.1 mol L−1 acetate buffer (pH 4.5) | caffeic acid | 1.7 × 10−7–7.83 × 10−6 | 1.4 × 10−7 | 90 | – | biodiesel | [28] |
| ASV | BiFE | 2.5 mol L−1 sodium bromide | – | 2.0 × 10−5–2.0 × 10−4 | – | 120 | – | canned fruit juice | [25] |
| Method | Working Electrode | Conditions (Supporting Electrolyte, pH, Temperature) | Linear Range [mol L−1] | Detection Limit [mol L−1] | Investigated Interferents | Application | Ref. |
|---|---|---|---|---|---|---|---|
| DPP | DME | 0.2 mol L−1 sodium acetate/acetic acid buffer of pH 4.7, 8.2 × 10−4 mol L−1 tropolone | 8.4 × 10−9–4.2 × 10−5 | 8.4 × 10−10 | Al(III), As(V), Be(II), Bi(III), Ca(II), Cd(II), Co(II), Cr(II), Cu(II), Fe(III), Ga(III), In(III), V(V), W(VI), Ti(IV), Mo(VI) | river rhine, fruit juice (pineaplle) | [50] |
| SSP | DME | 0.5 mol L−1 oxalic acid, 0.02% methylene blue | Sn(IV) 1.7 × 10−7–8.4 × 10−6 | 8.4 × 10−8 | Cd(II), Pb(II), Fe(II), Ag(I), Zn(II), Ca(II), Mg(II), HCO3−, Br−, CO32− | canned fruits (pineapple, orange, pear) | [51] |
| DPP | HMDE | methanol, perchloric acid | Sn(II) 8.4 × 10−6–4.2 × 10−4 | 4.2 × 10−8 | – | – | [54] |
| DPP | DME | 0.1 mol L−1 NaOH, 0.1 mol L−1 KNO3 | – | Sn(II) 5.5 × 10−7 Sn(IV) 8.2 × 10−7 | Fe(III), Cu(II), Pb(II), Cd(II), Zn(II), | canned tomato sauce | [52] |
| DPP | DME | 0.05 mol L−1 EDTA, 0.2 mol L−1 NaOAc, pH 3.5 | Sn(IV) 2.0 × 10−6–1.0 × 10−5 | Sn(IV) 3.0 × 10−7 | – | shipyard water, canned tomato sauce | [53] |
| DPP | SMDE | 0.1 mol L−1 catechol, 0.1 mol L−1 NaCIO4, 0.1 mol L−1 HCIO4 | Sn(IV) 8.4 × 10−6–8.4 × 10−5 | 2.4 × 10−7 | – | – | [47] |
| DPV | HMDE | 0.25 mol L−1 HCl, 5.1 × 10−7 mol L−1 Pt | 4.2 × 10−6–8.4 × 10−6 | 2.5 × 10−6 | Ir(III), Pd(II), Rh(II), Ru(I), Re(I), Pt(IV) | – | [57] |
| DPP | gold amalgamated electrode | 3.5 mol L−1 HCl | 4.2 × 10−6–1.9 × 10−3 | 1.3 × 10−6 | Zn(II), Mo(VI), Mn(II), Cr(VI), Se(VI), Ti(VI), Ga(II), Al(III), U(VI), V(V), Te(IV), Bi(III), Fe(III) Rh(III), Ru(III), Cd(II), Pd(II), Os(VIII), Co(II), Ni(II), Cu(II), Pb(II), Cr(VI), Sb(III), Tl(I), CH3COO−, NO3−, NH4+, F−, Br−, SCN−, I−, Cl−, CO32−, EDTA | fly ash, soil, sediment river, industrial effluents | [56] |
| DPP | SMDE | 0.1 mol L−1 NaOH, 0.5 mol L−1 sodium malonate, pH 9.8 | Sn(IV) 8.4 × 10−5–3.4 × 10−4 | 1.3 × 10−5 | – | – | [49] |
| DPP | SMDE | 0.1 mol L−1 HCl, 0.01 mol L−1 KSCN | Sn(II) 4.2 × 10−7–2.9 × 10−5 | – | – | zinc chamber of battery | [48] |
| DPP | HMDE | 0.2 mol L−1 HCl, 0.2 mol L−1 citric acid | 0–1.0 × 10−3 | – | – | – | [55] |
| Membrane Composition | pH | Concentration Range [mol L−1] | Detection Limit [mol L−1] | Response Time [s] | Life Time [Days] | Interference | Application | Ref. |
|---|---|---|---|---|---|---|---|---|
| PVC–NDDBH | 1.0 | 1.0 × 10−5–1.0 × 10−1 | 4.0 × 10−6 | 20 | 42 | Pb(II), Bi(III), Al(III), Ca(II), Co(II), Zn(II), Cd(II), Ba(II), Mg(II), Ni(II), Cu(II), Cr(III), Li(I), Na(I), Tl(I), Cs(I), NH4+ | synthetic solutions, standard copper–based alloy, Sn–Pb alloy, industrial wastewater, Canned meat | [60] |
| PVC–THPA | 3.0 | 5.0 × 10−7–1.0 × 10−1 | 2.0 × 10−7 | ˂15 | 56 | Ag(I), Tl(I), Pb(II), Mg(II), Na(I), Cu(II), K(I), Cs(I), Bi(III), Hg(II), Co(II), Cd(II), Al.(III), Ca(II), Cr(III), La(III), Ce(III), Ni(II) | synthetic solutions, industrial wastewater, orange juice, grapes juice, canned fish | [58] |
| PVC–DB18C6 | 1.0 | 1.0 × 10−6–1.0 × 10−2 | 8.0 × 10−7 | ˂15 | 90 | Al(III), Mn(II), Mg(II), Zn(II), Cu(II), Co(II), Fe(II), Cd(II), Ca(II), Pb(II), Hg(II), Sr(II), Bi(III), Fe(III) | alloy Sn–Pb | [59] |
| Method | Sample | Added Sn(II) [mg L−1] | Added Pb(II) [mg L−1] | Found Sn(II) [mg L−1] | Found Pb(II) [mg L−1] | Ref. |
|---|---|---|---|---|---|---|
| ASV | Orange juice | – | – | 0.372 | 0.118 | [32] |
| 0.5 | 0.1 | 0.875 | 0.231 | |||
| Pineapple juice | – | – | 0.283 | 0.099 | ||
| 0.25 | 0.1 | 0.535 | 0.189 | |||
| Orange with pulpe drink | – | – | 237.0 | 0.25 | ||
| 200.0 | 0.2 | 444.0 | 0.53 | |||
| Method | Sample | Add Sn(II) [mol L−1] | Found Sn(II) [mol L−1] | Ref. | ||
| ASV | Pineapple juice | 3 × 10−7 | 4.19 × 10−7 | [30] | ||
| 1 × 10−6 | 11.03 × 10−7 | |||||
| 3 × 10−6 | 32.41 × 10−7 | |||||
| 8 × 10−6 | 78.84 × 10−7 | |||||
| Orange juice | 3 × 10−7 | 7.43 × 10−7 | ||||
| 1 × 10−6 | 14.82 × 10−7 | |||||
| 3 × 10−6 | 33.53 × 10−7 | |||||
| 8 × 10−6 | 85.87 × 10−7 | |||||
| Grape juice | 3 × 10−7 | 5.93 × 10−7 | ||||
| 1 × 10−6 | 12.69 × 10−7 | |||||
| 3 × 10−6 | 31.85 × 10−7 | |||||
| 8 × 10−6 | 84.01 × 10−7 | |||||
| Mango Juice | 3 × 10−7 | 8.54 × 10−7 | ||||
| 1 × 10−6 | 17.79 × 10−7 | |||||
| 3 × 10−6 | 37.02 × 10−7 | |||||
| 8 × 10−6 | 86.13 × 10−7 | |||||
| Method | Sample | Added Sn(II) [µg L−1] | Added Zn(II) [µg L−1] | Found Sn(II) [µg L−1] | Found Zn(II) [µg L−1] | Ref. |
| AdSV | Blood Setum | 0 | 0 | 10.81 | 17.0 | [38] |
| 10 | 10 | 19.92 | 27.2 | |||
| 15 | 15 | 26.42 | 31.9 | |||
| Human Hair | 0 | 0 | 14.65 | 22.55 | ||
| 10 | 10 | 24.0 | 33.0 | |||
| 15 | 15 | 30.0 | 37.9 | |||
| Method | Sample | Added Sn(II) [ng mL−1] | Found Sn(II) [ng mL−1] | Ref. | ||
| ASV | Fruit juice | – | 0 | [31] | ||
| 99.5 | 10 | |||||
| 93.45 | 20 | |||||
| Bottled water | 0.63 | – | ||||
| 98.2 | 10 | |||||
| 100.4 | 20 | |||||
| Method | Sample | Added Sn [mg L−1] | Found Sn [mg L−1] | Ref. | ||
| AdSV | Pineapple juice | 10.00 | 10.83 | [40] | ||
| Apricot nectar | 5.00 | 3.18 | ||||
| Grapefruit juice | 10.00 | 9.46 | ||||
| Tomato juice | 5.00 | 3.23 | ||||
| Peach nectar | 10.00 | 10.19 | ||||
| Method | Sample | Added Sn(IV) [ng g−1] | Found Sn(IV) [ng g−1] | Ref. | ||
| CSV | Human hair | [33] | ||||
| I | 50 | 50.85 | ||||
| II | 50 | 49.10 | ||||
| II | 50 | 48.55 | ||||
| Method | Sample | Sn(II) added [nmol L−1] | Found Sn(II) [nmol L−1] | Ref. | ||
| AdSV | SPS–WW1 Waste Water | 30.0 | 29.0 | [42] | ||
| 80.0 | 83.0 | |||||
| 160.0 | 169.0 | |||||
| SPS–SW1 Surface Water | 30.0 | 28.7 | ||||
| 80.0 | 84.6 | |||||
| 160.0 | 153.0 | |||||
| Bystrzyca river water | 30.0 | 31.0 | ||||
| 80.0 | 76.0 | |||||
| 160.0 | 167.5 | |||||
| Rain water | 30.0 | 30.8 | ||||
| 80.0 | 87.0 | |||||
| 160.0 | 151.5 | |||||
| Method | Sample | Sn(II) Added [nmol L−1] | Found Sn(II) [nmol L−1] | Ref. | ||
| AdSV | SPS–WW1 Waste Water | – | – | [43] | ||
| 5.00 | 5.35 | |||||
| 10.00 | 10.34 | |||||
| Bystrzyca river water | 5.00 | 4.77 | ||||
| 10.00 | 9.55 | |||||
| Tap water | 5.00 | 5.15 | ||||
| 10.00 | 10.23 | |||||
| Sample | Found Sn(II) by ASV Method [µg g−1] | Found Sn(II) by GF–AAS Method [µg g−1] | Ref. | |
|---|---|---|---|---|
| Fermented bean curd | 48.3 | 46.7 | [24] | |
| Lychee juice | 41.9 | 43.7 | ||
| Orange juice | 23.1 | 22.3 | ||
| Milk tea | 8.8 | 9.3 | ||
| Sample | Found Sn(II) by ASV Method [×10−5 mol L−1] | Found Sn(II) by GF–AAS Method [×10−5 mol L−1] | Ref. | |
| Pineapple juice | 1.14 ± 0.05 | 1.17 ± 0.05 | [30] | |
| Orange juice | 4.51 ± 0.20 | 4.47 ± 0.18 | ||
| Grape juice | 2.82 ± 0.11 | 2.86 ± 0.10 | ||
| Mango Juice | 0.750 ± 0.03 | 7.33 ± 0.03 | ||
| Sample | Found Sn(IV) by AdSV Method [mg L−1] | Found Sn(IV) by AAS Method [mg L−1] | Ref. | |
| Pineapple juice | 100.3 ± 10.9 | 96.9 ± 3.4 | [40] | |
| Apricot nectar | 7.7 ± 2.5 | 7.16 ± 1.14 | ||
| Grapefruit juice | 54.2 ± 5.4 | 67.56 ± 1.53 | ||
| Tomato juice | 51.0 ± 5.1 | 58.17 ± 1.46 | ||
| Peach nectar | 173.5 ± 14.1 | 201.8 ± 6.6 | ||
| Sample | Found Sn by AdSV Method [µg L−1] | Found Sn by AAS Method [µg L−1] | Ref. | |
| Drainage water 1 | 38.5 ± 2.5 | 44 | [41] | |
| Drainage water 2 | 88 ± 4.8 | 90 | ||
| Sample | Found Sn(IV) by AdSV Method [µg g−1] | Found Sn(IV) by GF–AAS Method [µg g−1] | Ref. | |
| Fermented bean curd | 39.4 | 37.9 | [37] | |
| Grape juice | 25.4 | 26.5 | ||
| Milk tea | 10.8 | 11.5 | ||
| Human hair | [ng g−1] | [ng g−1] | ||
| A | 58.3 | 56.9 | ||
| B | 47.3 | 49.1 | ||
| C | 28.7 | 29.9 | ||
| Waste water | [µg L−1] | [µg L−1] | ||
| A | 5.1 | 5.5 | ||
| B | 3.3 | 2.9 | ||
| C | 1.2 | 0.9 | ||
| Sample | Found Sn(IV) by Single–sweep polarography Method [µg mL−1] | Found Sn(IV) by Spectrophotometric Method [µg mL−1] | Ref. | |
| Pineapple juice | 2.30 ± 0.23 | 2.40 ± 0.21 | [51] | |
| Orange juice | 8.50 ± 0.41 | 8.20 ± 0.52 | ||
| Pear juice | 0.50 ± 0.05 | 8.20 ± 0.52 | ||
| Sample | Found Sn(IV) by DPP Method [µg g−1] | Found Sn(IV) by AAS Method [µg g−1] | Ref. | |
| Fly ash near Indraprastha | 5.26 ± 0.03 | 5.25 ± 0.06 | [56] | |
| Soil near Wazirpur industrial area | 13.45 ± 0.06 | 13.48 ± 0.08 | ||
| Sediment of Yamuna river | 2.45 ± 0.04 | 2.44 ± 0.05 | ||
| Industrial effluents from electroplating and ceramic industry | 310 ± 4 µg mL−1 | 312 ± 3 µg mL−1 | ||
| Sample | Found Sn by DPP Method [ng mL−1] | Found Sn by AAS Method [ng mL−1] | Found Sn by ASV Method [ng mL−1] | Ref. |
| River Rhine | 12.3 ± 0.7 | 13.1 ± 0.8 | 12.0 ± 0.5 | [50] |
| Pineapple juice | 668 ± 13 | 680 ± 21 | 695 ± 15 | |
| Sample | Found Sn(II) by Potentiometry Method [µg g−1] | Found Sn(II) by ICP Method [µg g−1] | Ref. | |
| Synthetic solution | [mg mL−1] | [mg mL−1] | [58] | |
| (2.2% Co2+, 1.5% Cr, 0.6% Sn) | 5.63 ± 0.11 | 6.12 ± 0.04 | ||
| (2.0% Fe, 1.5% Cd, 0.5% Sn) | 4.83 ± 0.20 | 5.10 ± 0.03 | ||
| (1.0% Al, 2.0% Cu, 0.5% Sn) | 4.75 ± 0.15 | 5.08 ± 0.08 | ||
| Wastewater sample | 300.7 ± 2.2 | 299.5 ± 0.4 | ||
| Orange juice | 50.3 ± 0.1 | 49.5 ± 0.1 | ||
| Canned fish | 100.5 ±0.1 | 99.7 ± 0.1 | ||
| Grape juice | 58.3 ± 0.3 | 59.0 ± 0.1 | ||
| Sample | Found Sn(II) by Potentiometric Method | Found Sn(II) by AAS Method | Ref. | |
| Synthetic solutions | [60] | |||
| (1.2% Ni, 0.5% Sn) | 5.37 ± 0.06 mg mL−1 | 5.12 ± 0.03 mg mL−1 | ||
| (1.5% Fe, 0.5% Cr, 0.4% Sn) | 4.23 ± 0.12 mg mL−1 | 4.09 ± 0.08 mg mL−1 | ||
| Standard copper–based alloy | 3.7 ± 0.2 mg g−1 | 3.8 ± 0.1 mg g−1 | ||
| Canned meat | 281.7 ± 0.6 µg g−1 | 282.4 ± 0.2 µg g−1 | ||
| Sn–Pb alloy | 625.4 ± 0.7 mg g−1 | 632.2 ± 0.4 mg g−1 | ||
| Industrial wastewater | 310.5 ± 2.3 mg mL−1 | 311.8 ± 1.2 mg mL−1 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grabarczyk, M.; Wlazlowska, E.; Fialek, M. Electrochemical Methods for the Analysis of Trace Tin Concentrations—Review. Materials 2023, 16, 7545. https://doi.org/10.3390/ma16247545
Grabarczyk M, Wlazlowska E, Fialek M. Electrochemical Methods for the Analysis of Trace Tin Concentrations—Review. Materials. 2023; 16(24):7545. https://doi.org/10.3390/ma16247545
Chicago/Turabian StyleGrabarczyk, Malgorzata, Edyta Wlazlowska, and Marzena Fialek. 2023. "Electrochemical Methods for the Analysis of Trace Tin Concentrations—Review" Materials 16, no. 24: 7545. https://doi.org/10.3390/ma16247545
APA StyleGrabarczyk, M., Wlazlowska, E., & Fialek, M. (2023). Electrochemical Methods for the Analysis of Trace Tin Concentrations—Review. Materials, 16(24), 7545. https://doi.org/10.3390/ma16247545