
Thiol-Ene Photo-Click Hydrogels with Tunable Mechanical Properties Resulting from the Exposure of Different -Ene Moieties through a Green Chemistry

 , ,
, ,  , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Poly(ether urethane) Synthesis and -NH Group Exposure
2.3. Poly(ether urethane) Chemical Characterization and -NH Group Quantification
2.4. Poly(ether urethane) Functionalization with Photo-Sensitive Moieties
2.5. Chemical Characterization of Functionalized Poly(ether urethane)s
2.5.1. Attenuated Total Reflectance Fourier Transform Infrared Spectroscopy
2.5.2. Size Exclusion Chromatography
2.5.3. Proton Nuclear Magnetic Resonance Spectroscopy
2.5.4. Functional Group Quantification
2.6. Evaluation of Functionalized Poly(ether urethane) Thermo-Responsiveness at the Nano-Scale
2.6.1. Dynamic Light Scattering
2.6.2. Critical Micellar Temperature Evaluation
2.7. Thiol-Ene Hydrogel Preparation
2.8. Rheological Characterization of Thiol-Ene Hydrogels
2.9. Chemical Characterization of Photo-Crosslinked Thiol-Ene Hydrogels
2.9.1. Proton Nuclear Magnetic Resonance Spectroscopy
2.9.2. Carbon Nuclear Magnetic Resonance Spectroscopy
2.10. Statistical Analysis
3. Results
3.1. Chemical Characterization of the Synthesized Poly(ether urethane)
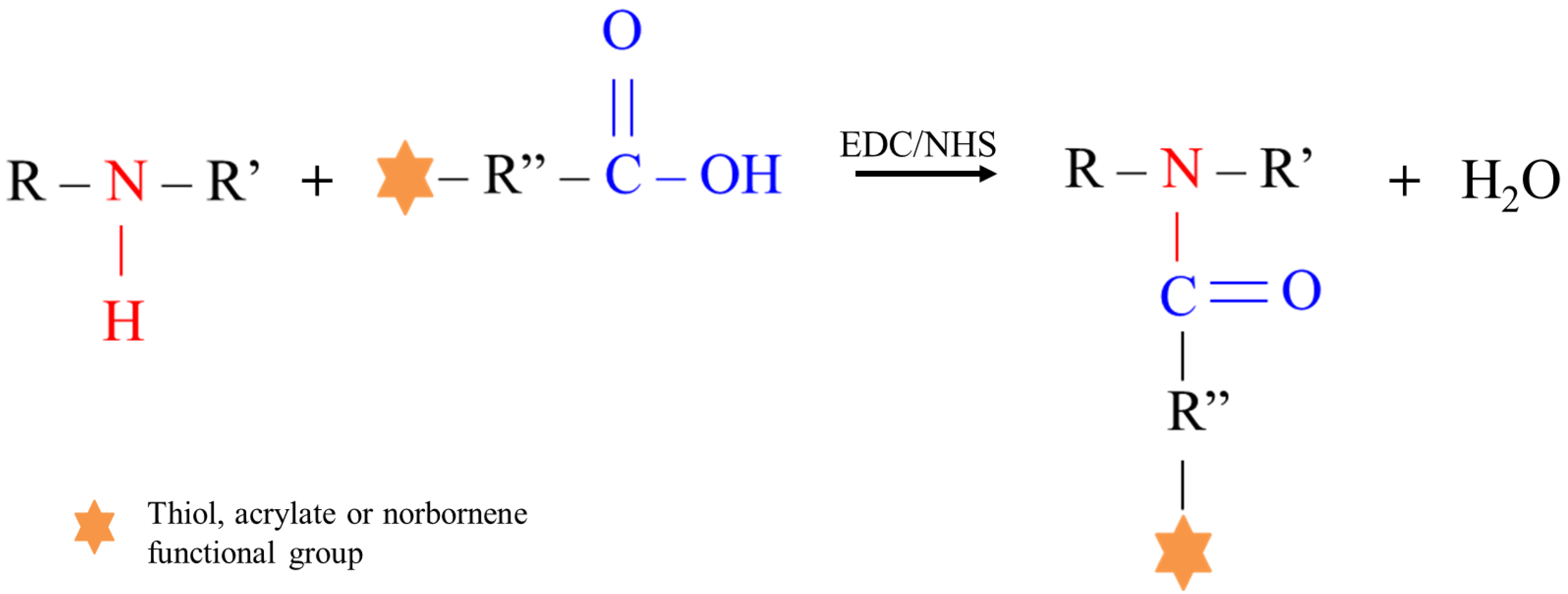
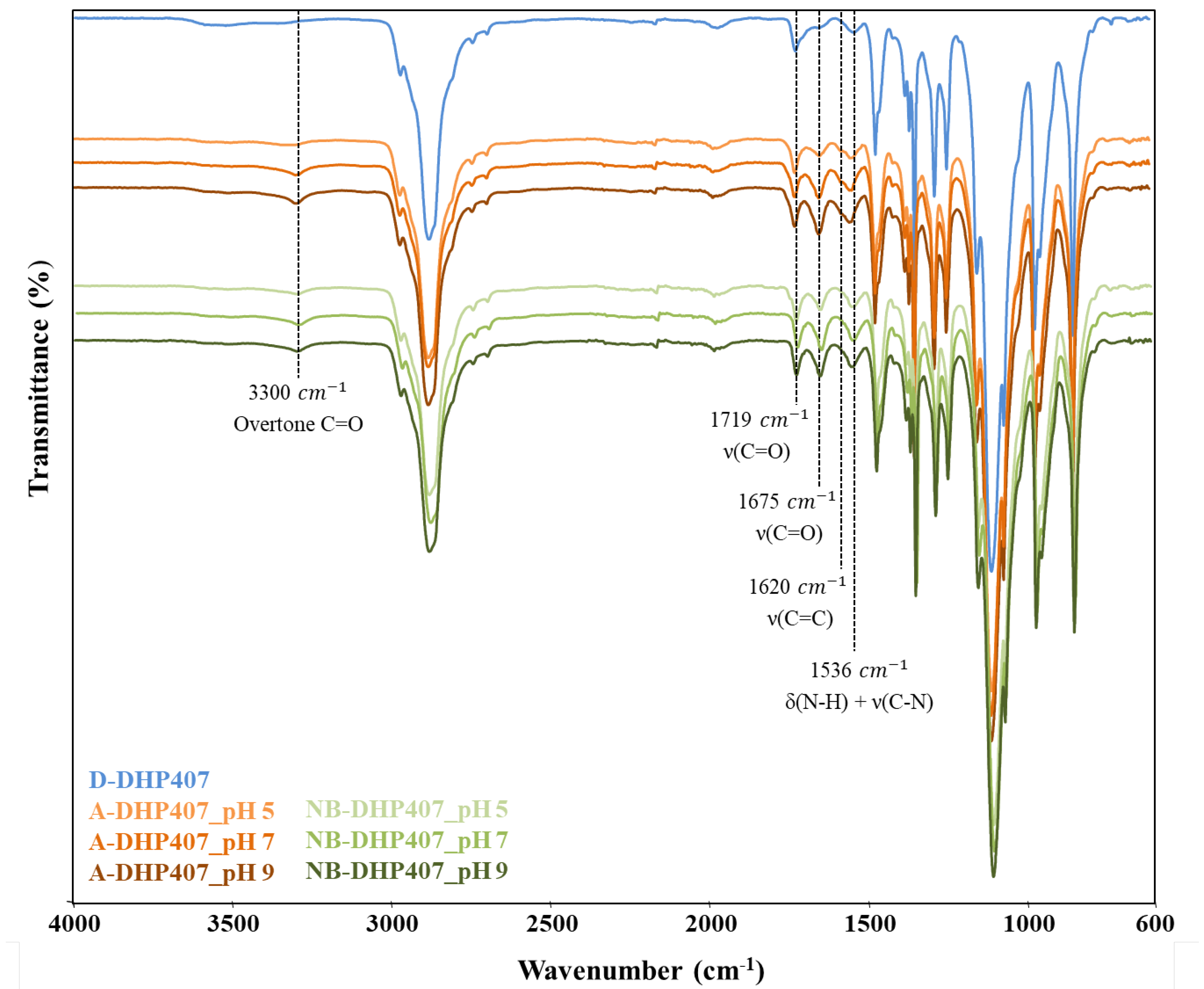
3.2. D-DHP407 Functionalization with Photo-Sensitive Groups through Water-Based Carbodiimide Chemistry
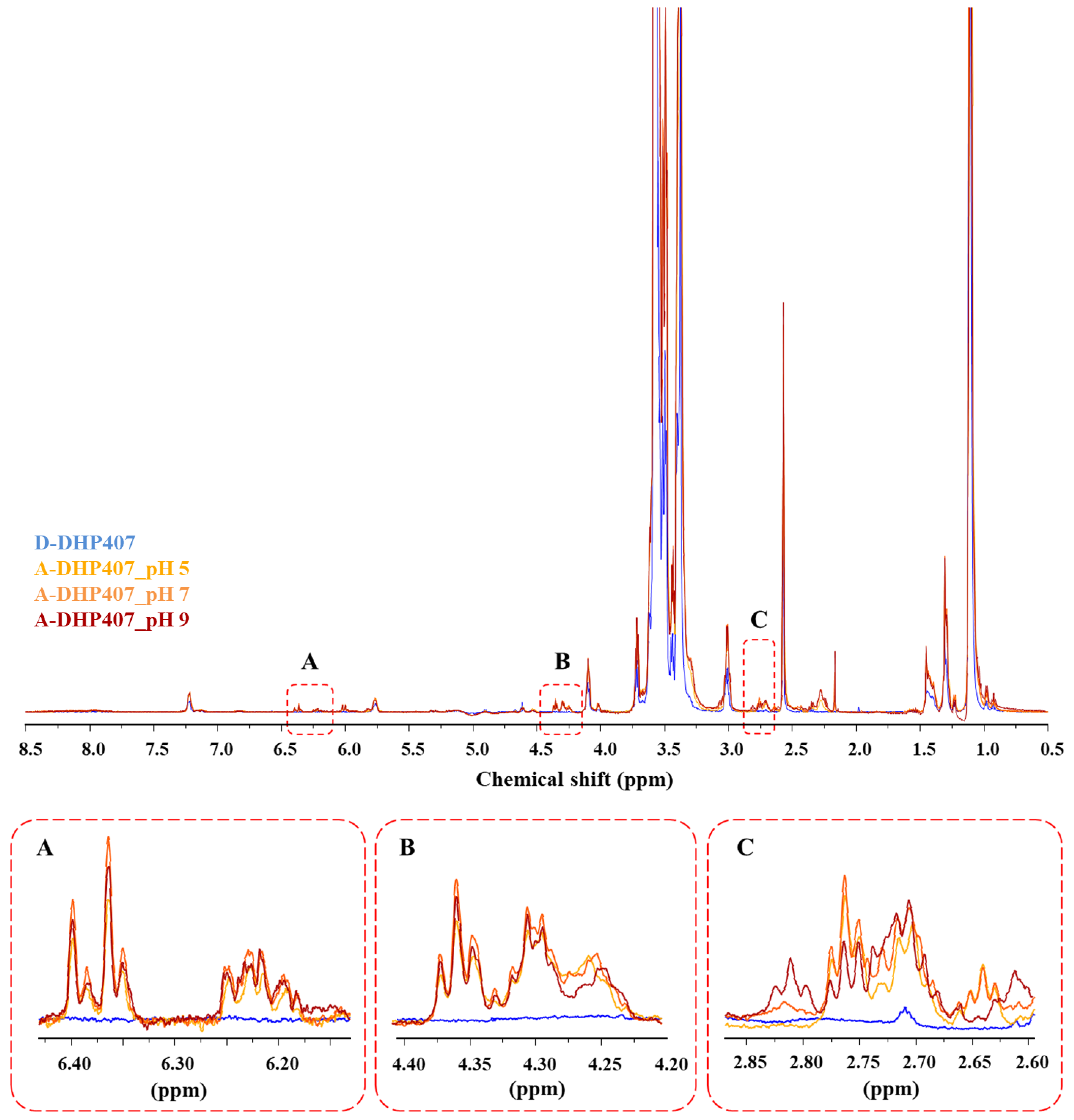
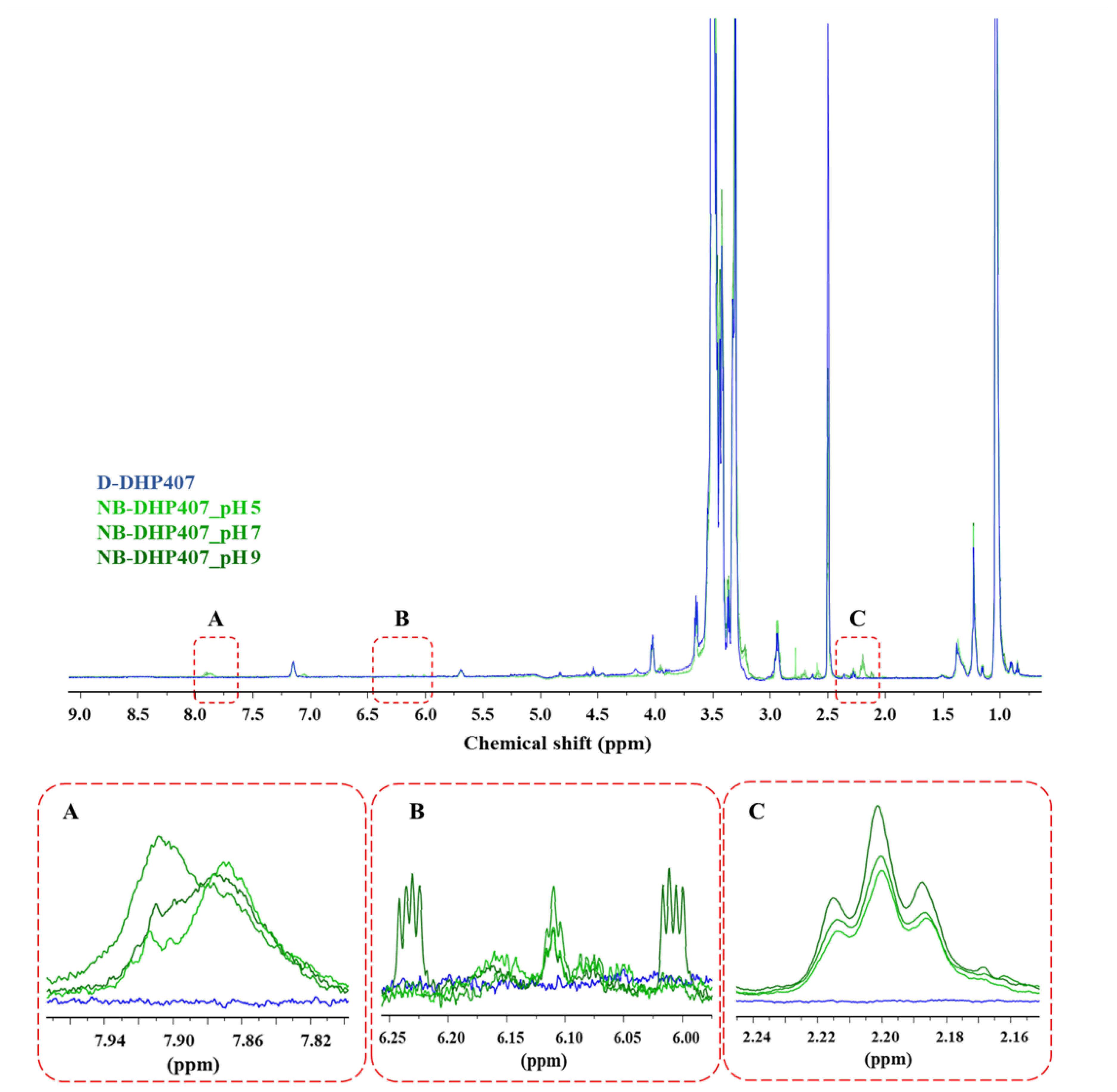
3.2.1. Influence of Grafting Reaction pH on the Degree of S-DHP407 Functionalization
3.2.2. Influence of Grafting Reaction pH on the Degree of the -Ene Moiety Functionalization
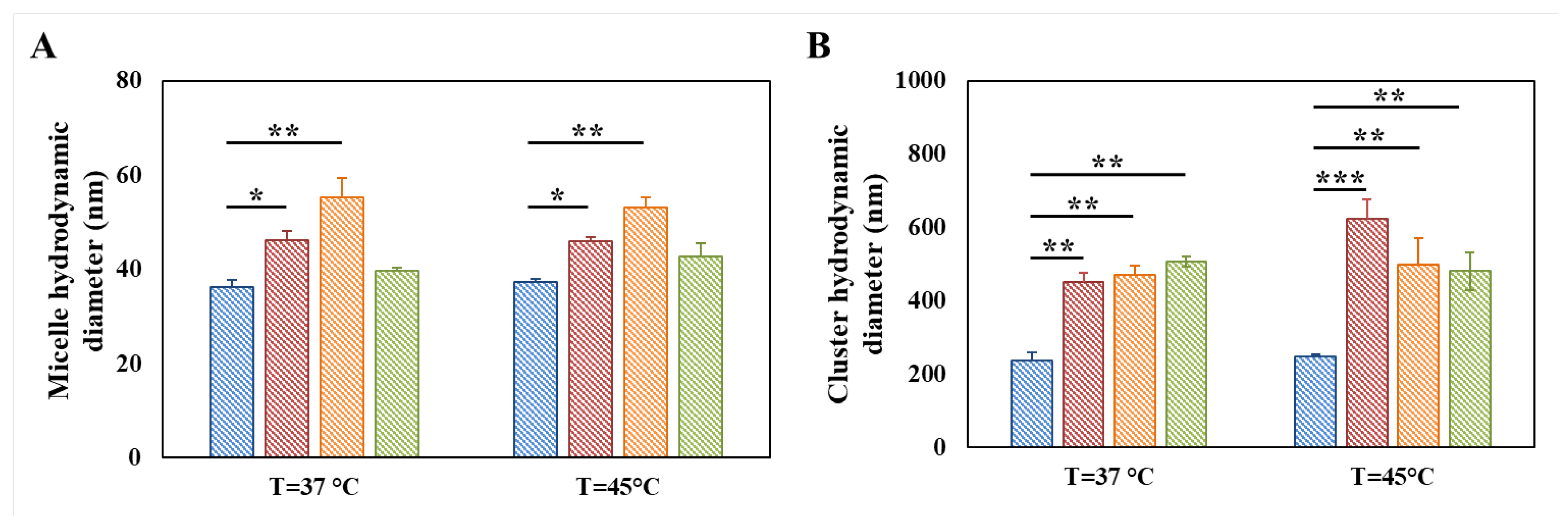
3.3. Evaluation of the Thermo-Responsiveness of Functionalized Poly(ether urethane)s
3.4. Design of Thermosensitive and Photo-Click Thiol-Ene Formulations
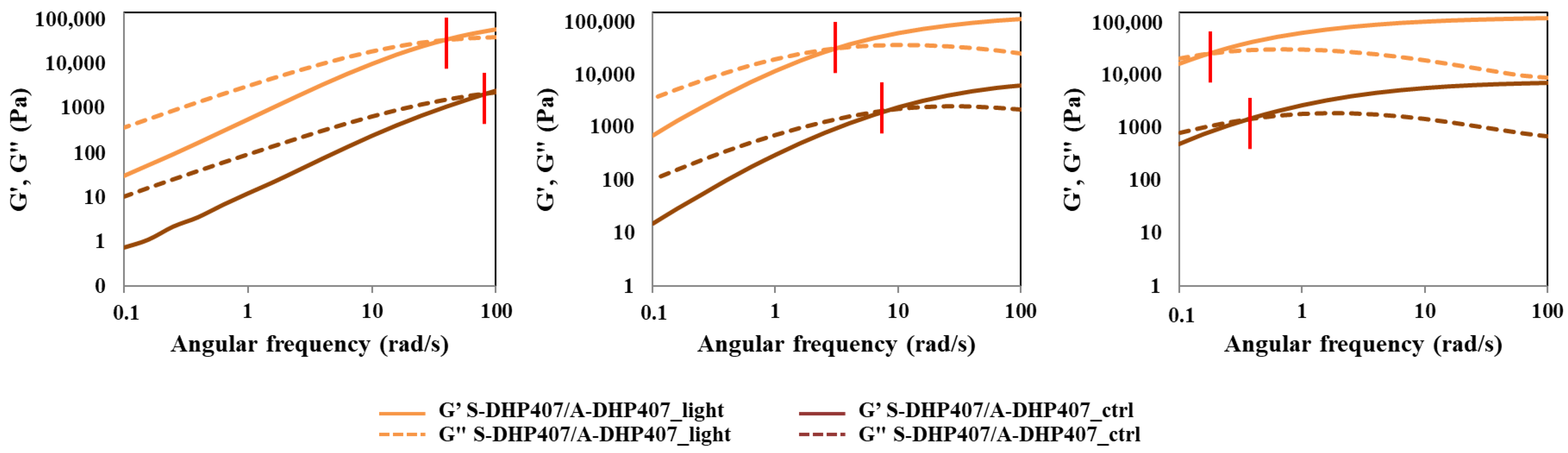
3.4.1. Thiol-Acrylate Photo-Click Hydrogels
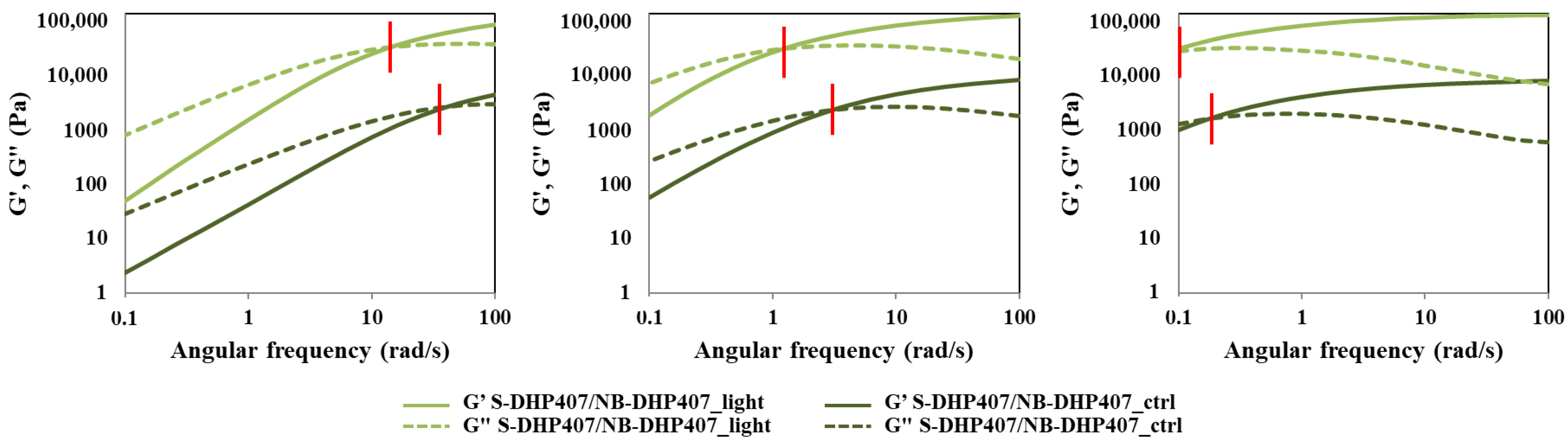
3.4.2. Thiol-Norbornene Photo-Click Hydrogels
3.4.3. The Role Exerted by the Co-Initiator in the Thiol-Ene Photo-Crosslinking Process
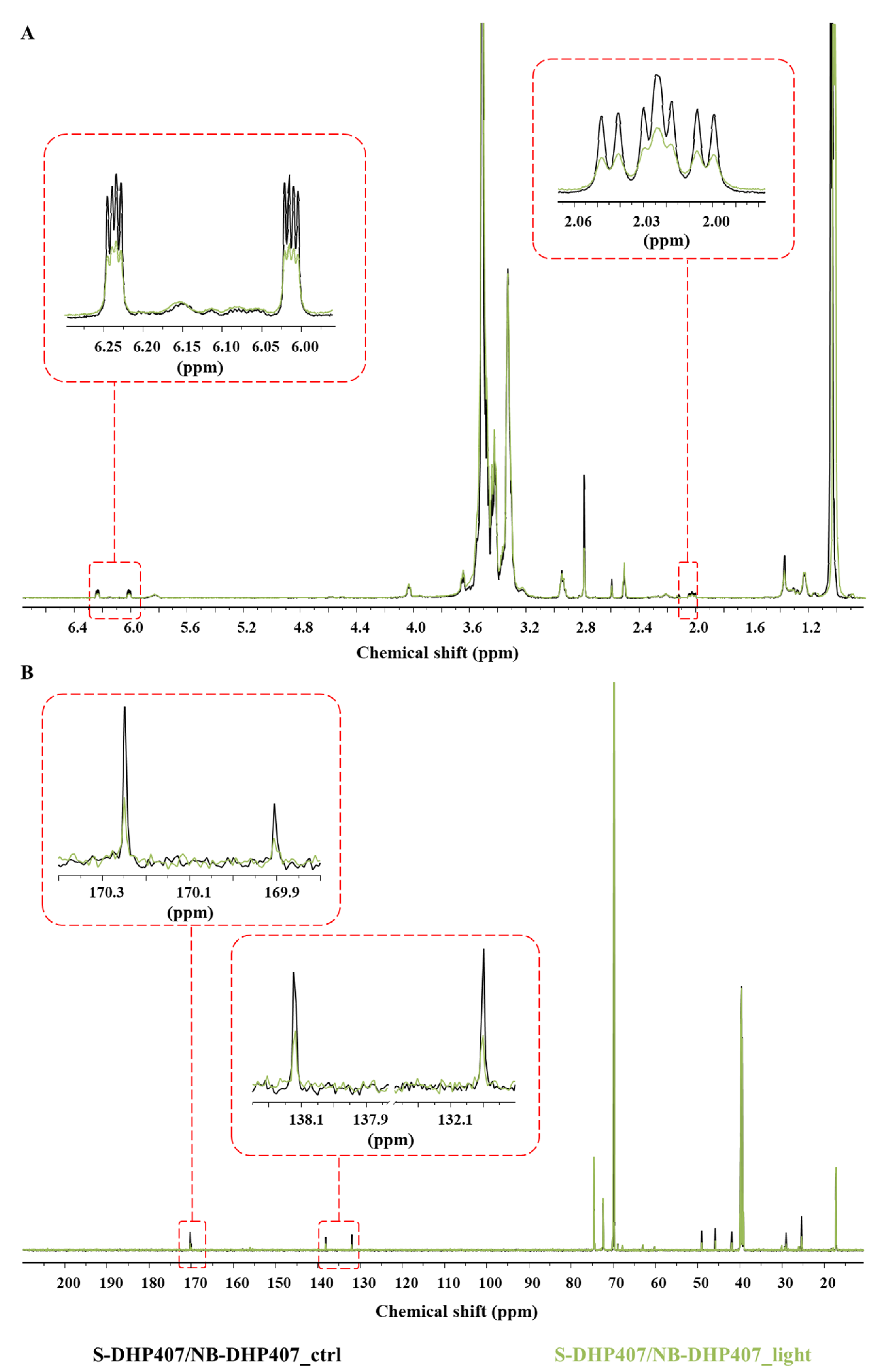
3.4.4. Chemical Characterization of Thiol-Ene Photo-Crosslinked Hydrogels
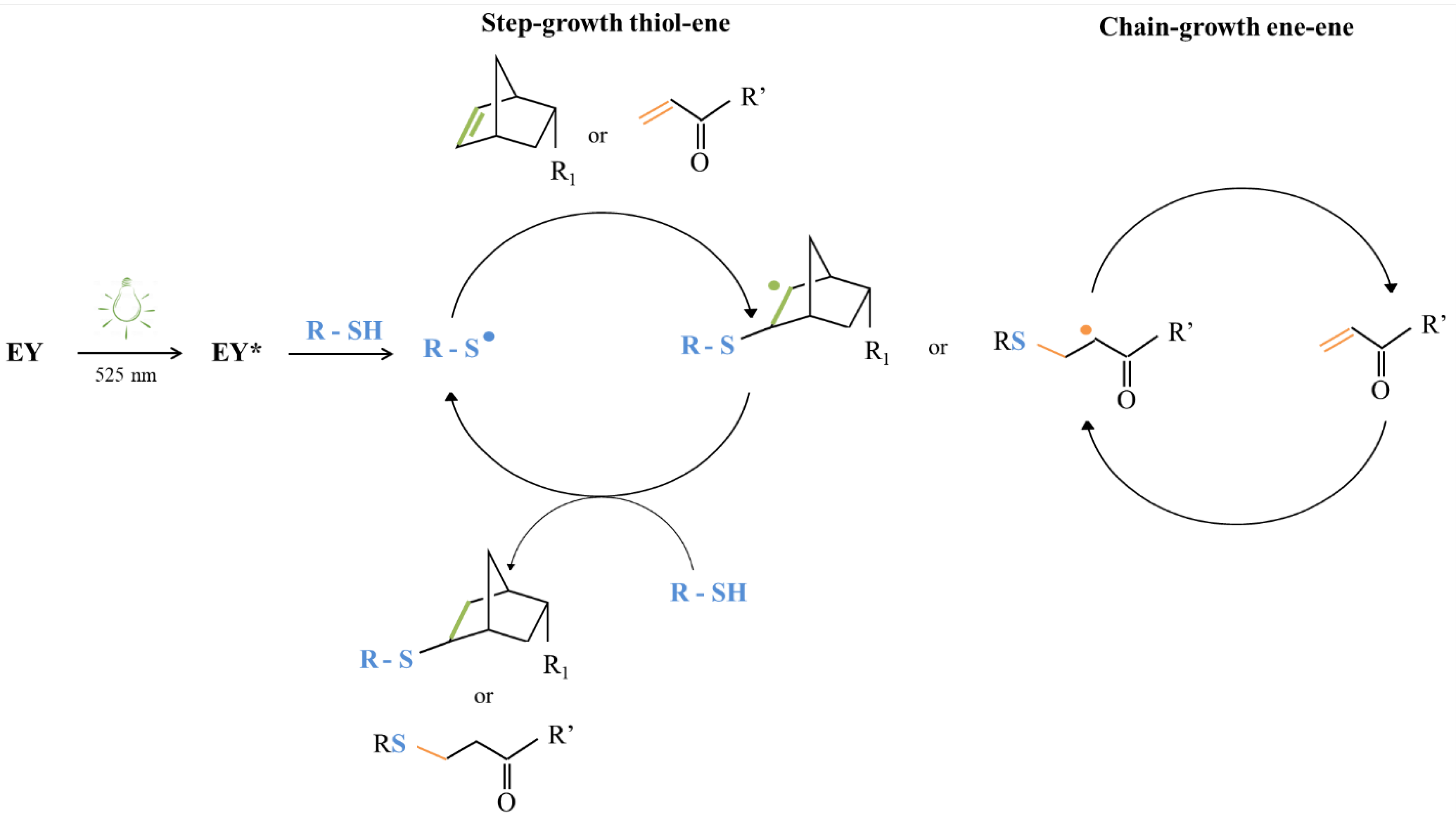
3.5. The Influence of Vis Light-Induced Photo-Crosslinking Mechanism Based on the -Ene Moiety
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- González-Díaz, E.C.; Varghese, S. Hydrogels as Extracellular Matrix Analogs. Gels 2016, 2, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ke, X.; Li, M.; Wang, X.; Liang, J.; Wang, X.; Wu, S.; Long, M.; Hu, C. An injectable chitosan/dextran/β-glycerophosphate hydrogel as cell delivery carrier for therapy of myocardial infarction. Carbohydr. Polym. 2020, 229, 115516. [Google Scholar] [CrossRef]
- Laomeephol, C.; Guedes, M.; Ferreira, H.; Reis, R.L.; Kanokpanont, S.; Damrongsakkul, S.; Neves, N.M. Phospholipid-induced silk fibroin hydrogels and their potential as cell carriers for tissue regeneration. J. Tissue Eng. Regen. Med. 2020, 14, 160–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, J.D.; Roloson, E.B.; Amelung, C.D.; Lampe, K.J. Rapidly Assembling Pentapeptides for Injectable Delivery (RAPID) Hydrogels as Cytoprotective Cell Carriers. ACS Biomater. Sci. Eng. 2019, 5, 2117–2121. [Google Scholar] [CrossRef] [PubMed]
- Fatimi, A.; Okoro, O.V.; Podstawczyk, D.; Siminska-Stanny, J.; Shavandi, A. Natural Hydrogel-Based Bio-Inks for 3D Bioprinting in Tissue Engineering: A Review. Gels 2022, 8, 179. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wu, C.; Chu, P.K.; Gelinsky, M. 3D printing of hydrogels: Rational design strategies and emerging biomedical applications. Mater. Scie. Eng. R Rep. 2020, 140, 100543. [Google Scholar] [CrossRef]
- Taneja, H.; Salodkar, S.M.; Parmar, A.S.; Chaudhary, S. Hydrogel based 3D printing: Bio ink for tissue engineering. J. Mol. Liq. 2022, 367, 120390. [Google Scholar] [CrossRef]
- Gong, Y.; He, L.; Li, J.; Zhou, Q.; Ma, Z.; Gao, C.; Shen, J. Hydrogel-filled polylactide porous scaffolds for cartilage tissue engineering. J. Biomed. Mater. Res. 2007, 82B, 192–204. [Google Scholar] [CrossRef] [PubMed]
- Colucci, F.; Mancini, V.; Mattu, C.; Boffito, M. Designing Multifunctional Devices for Regenerative Pharmacology Based on 3D Scaffolds, Drug-Loaded Nanoparticles, and Thermosensitive Hydrogels: A Proof-of-Concept Study. Pharmaceutics 2021, 13, 464. [Google Scholar] [CrossRef]
- Jacob, S.; Nair, A.B.; Shah, J.; Sreeharsha, N.; Gupta, S.; Shinu, P. Emerging Role of Hydrogels in Drug Delivery Systems, Tissue Engineering and Wound Management. Pharmaceutics 2021, 13, 357. [Google Scholar] [CrossRef]
- Alvarez-Lorenzo, C.; Grinberg, V.Y.; Burova, T.V.; Concheiro, A. Stimuli-sensitive cross-linked hydrogels as drug delivery systems: Impact of the drug on the responsiveness. Int. J. Pharma. 2020, 579, 119157. [Google Scholar] [CrossRef] [PubMed]
- Cascone, S.; Lamberti, G. Hydrogel-based commercial products for biomedical applications: A review. Int. J. Pharma. 2020, 573, 118803. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Tiwari, S. A review on biomacromolecular hydrogel classification and its applications. Int. J. Biol. Macromol. 2020, 162, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Manna, K.; Pal, S. Recent advances in various stimuli-responsive hydrogels: From synthetic designs to emerging healthcare applications. Mater. Chem. Front. 2022, 6, 2338–2385. [Google Scholar] [CrossRef]
- Bustamante-Torres, M.; Romero-Fierro, D.; Arcentales-Vera, B.; Palomino, K.; Magaña, H.; Bucio, E. Hydrogels Classification According to the Physical or Chemical Interactions and as Stimuli-Sensitive Materials. Gels 2021, 7, 182. [Google Scholar] [CrossRef] [PubMed]
- Chamkouri, H.; Chamkouri, M. A Review of Hydrogels, Their Properties and Applications in Medicine. Am. J. Biomed. Sci. Res. 2021, 11, 485–493. [Google Scholar] [CrossRef]
- Kang, J.; Yun, S.I. Double-Network Hydrogel Films Based on Cellulose Derivatives and κ-Carrageenan with Enhanced Mechanical Strength and Superabsorbent Properties. Gels 2023, 9, 20. [Google Scholar] [CrossRef]
- Wang, S.; Dai, S.; Yan, H.; Ding, J.; Yuan, N. Conductive double-crosslinked network hydrogel with superior stretchability and self-healing ability. Mater. Res. Express 2019, 6, 105712. [Google Scholar] [CrossRef]
- Coșman, B.P.; Bucătariu, S.M.; Constantin, M.; Fundueanu, G. Temperature/pH-Sensitive Double Cross-Linked Hydrogels as Platform for Controlled Delivery of Metoclopramide. Gels 2022, 8, 824. [Google Scholar] [CrossRef]
- Cheng, Q.; Ding, S.; Zheng, Y.; Wu, M.; Peng, Y.Y.; Diaz-Dussan, D.; Shi, Z.; Liu, Y.; Zeng, H.; Cui, Z.; et al. Dual Cross-Linked Hydrogels with Injectable, Self-Healing, and Antibacterial Properties Based on the Chemical and Physical Cross-Linking. Biomacromolecules 2021, 22, 1685–1694. [Google Scholar] [CrossRef]
- Yuan, Y.; Shen, S.; Fan, D. A physicochemical double cross-linked multifunctional hydrogel for dynamic burn wound healing: Shape adaptability, injectable self-healing property and enhanced adhesion. Biomaterials 2021, 276, 120838. [Google Scholar] [CrossRef] [PubMed]
- Huin-Amargier, C.; Marchal, P.; Payan, E.; Netter, P.; Dellacherie, E. New physically and chemically crosslinked hyaluronate (HA)-based hydrogels for cartilage repair. J. Biomed. Mater. Res. 2006, 76A, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Huang, J.; Zhong, Y.; Li, K.; Zhang, L.; Cai, J. High-Strength and High-Toughness Double-Cross-Linked Cellulose Hydrogels: A New Strategy Using Sequential Chemical and Physical Cross-Linking. Adv. Funct. Mater. 2016, 26, 6279–6287. [Google Scholar] [CrossRef]
- Young, A.T.; White, O.C.; Daniele, M.A. Rheological Properties of Coordinated Physical Gelation and Chemical Crosslinking in Gelatin Methacryloyl (GelMA) Hydrogels. Macromol. Biosci. 2020, 20, 2000183. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.; Chu, C.C. Synthesis, characterization and biodegradation of poly(ester amide)s based hydrogels. Polymer 2010, 51, 4200–4210. [Google Scholar] [CrossRef]
- Müller, M.; Becher, J.; Schnabelrauch, M.; Zenobi-Wong, M. Nanostructured Pluronic hydrogels as bioinks for 3D bioprinting. Biofabrication 2015, 7, 035006. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Huang, C.; Han, X.; Li, S.; Wang, Z.; Huang, J.; Liu, H.; Chen, Z. Fabrication of aerogel scaffolds with adjustable macro/micro-pore structure through 3D printing and sacrificial template method for tissue engineering. Mater. Des. 2022, 217, 110662. [Google Scholar] [CrossRef]
- Min, Y.; Li, J.; Liu, F.; Yeow, E.K.L.; Xing, B. Near-Infrared Light-Mediated Photoactivation of a Platinum Antitumor Prodrug and Simultaneous Cellular Apoptosis Imaging by Upconversion-Luminescent Nanoparticles. Angew. Chem. Int. Ed. 2014, 53, 1012–1016. [Google Scholar] [CrossRef] [PubMed]
- Sortino, S. Photoactivated nanomaterials for biomedical release applications. J. Mater. Chem. 2012, 22, 301–318. [Google Scholar] [CrossRef]
- Zhou, D.; Dong, Q.; Liang, K.; Xu, W.; Zhou, Y.; Xiao, P. Photocrosslinked methacrylated poly(vinyl alcohol)/hydroxyapatite nanocomposite hydrogels with enhanced mechanical strength and cell adhesion. J. Polym. Sci. Part A Polym. Chem. 2019, 57, 1882–1889. [Google Scholar] [CrossRef]
- Boffito, M.; Gioffredi, E.; Chiono, V.; Calzone, S.; Ranzato, E.; Martinotti, S.; Ciardelli, G. Novel polyurethane-based thermosensitive hydrogels as drug release and tissue engineering platforms: Design and in vivo characterization. Polym. Int. 2016, 65, 756–769. [Google Scholar] [CrossRef]
- Boffito, M.; Torchio, A.; Tonda-Turo, C.; Laurano, R.; Gisbert-Garzarán, M.; Berkmann, J.C.; Cassino, C.; Manzano, M.; Duda, G.N.; Vallet-Regí, M.; et al. Hybrid Injectable Sol-Gel Systems Based on Thermo-Sensitive Polyurethane Hydrogels Carrying pH-Sensitive Mesoporous Silica Nanoparticles for the Controlled and Triggered Release of Therapeutic Agents. Front. Bioeng. Biotechnol. 2020, 8, 384. [Google Scholar] [CrossRef]
- Laurano, R.; Chiono, V.; Ceresa, C.; Fracchia, L.; Zoso, A.; Ciardelli, G.; Boffito, M. Custom-design of intrinsically antimicrobial polyurethane hydrogels as multifunctional injectable delivery systems for mini-invasive wound treatment. Eng. Regen. 2021, 2, 263–278. [Google Scholar] [CrossRef]
- Laurano, R.; Cassino, C.; Ciardelli, G.; Chiono, V.; Boffito, M. Polyurethane-based thiomers: A new multifunctional copolymer platform for biomedical applications. React. Funct. Polym. 2020, 146, 104413. [Google Scholar] [CrossRef]
- Torchio, A.; Cassino, C.; Lavella, M.; Gallina, A.; Stefani, A.; Boffito, M.; Ciardelli, G. Injectable supramolecular hydrogels based on custom-made poly(ether urethane)s and α-cyclodextrins as efficient delivery vehicles of curcumin. Mater. Sci. Eng. C Mater. Biol. Appl. 2021, 127, 112194. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Sheybani, N.; Yeudall, W.A.; Yang, H. The effect of photoinitiators on intracellular AKT signaling pathway in tissue engineering application. Biomater. Sci. 2015, 3, 250–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arkenberg, M.R.; Nguyen, H.D.; Lin, C.C. Recent advances in bio-orthogonal and dynamic crosslinking of biomimetic hydrogels. J. Mater. Chem. B 2020, 8, 7835–7855. [Google Scholar] [CrossRef] [PubMed]
- Sharifi, S.; Sharifi, H.; Akbari, A.; Chodosh, J. Systematic optimization of visible light-induced crosslinking conditions of gelatin methacryloyl (GelMA). Sci. Rep. 2021, 11, 23276. [Google Scholar] [CrossRef] [PubMed]
- Fu, A.; Gwon, K.; Kim, M.; Tae, G.; Kornfield, J.A. Visible light initated thiol-acrylate photo-polymerisation of heparin-based hydrogels. Biomacromolecules 2015, 16, 497–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, H.; Liu, H.Y.; Lin, C.C. Improving gelation efficiency and cytocompatibility of visible light polymerised thiol-norbornene hydrogels via addition of soluble tyrosine. Biomater. Sci. 2017, 5, 589–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurano, R.; Boffito, M. Thermosensitive Micellar Hydrogels as Vehicles to Deliver Drugs with Different Wettability. Front. Bioeng. Biotechnol. 2020, 8, 708. [Google Scholar] [CrossRef] [PubMed]
- Laurano, R.; Abrami, M.; Grassi, M.; Ciardelli, G.; Boffito, M.; Chiono, V. Using Poloxamer® 407 as Building Block of Amphiphilic Poly(ether urethane)s: Effect of its Molecular Weight Distribution on Thermo-Sensitive Hydrogel Performances in the Perspective of Their Biomedical Application. Front. Mater. 2020, 7, 594515. [Google Scholar] [CrossRef]
- Laurano, R.; Boffito, M.; Cassino, C.; Liberti, F.; Ciardelli, G.; Chiono, V. Design of Injectable Bioartificial Hydrogels by Green Chemistry for Mini-Invasive Applications in the Biomedical or Aesthetic Medicine Fields. Gels 2023, 9, 59. [Google Scholar] [CrossRef] [PubMed]
- Pradal, C.; Jack, K.S.; Grøndahl, L.; Cooper-White, J.J. Gelation kinetics and viscoelastic properties of pluronic and α-cyclodextrin-based psudopolyrotaxane hydrogels. Biomacromolecules 2013, 14, 3780–3792. [Google Scholar] [CrossRef]
- Laurano, R.; Boffito, M.; Torchio, A.; Cassino, C.; Chiono, V.; Ciardelli, G. Plasma Treatment of Polymer Powder as an Effective Tool to Functionalize Polymers: Case Study Application on an Amphiphilic Polyurethane. Polymers 2019, 11, 2109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakajima, N.; Ikada, Y. Mechanism of amide formation by carbodiimide for bioconjugation in aqueous media. Biocon. Chem. 1995, 5, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Bruckner, R. Electrophilic additions to the C=C double bond. In Organic Mechanism; Harmata, M., Ed.; Springer: Berlin, Germany, 2010; pp. 103–156. [Google Scholar]
- Hussain, K.; Aslam, Z.; Ullah, S.; Raza Shah, M. Synthesis of pH responsive, photocrosslinked gelatin-based hydrogel system for control release of ceftriaxone. Chem. Phys. Lipids 2021, 238, 105101. [Google Scholar] [CrossRef]
- Qin, X.; He, R.; Chen, H.; Fu, D.; Peng, Y.; Meng, S.; Chen, C.; Yang, L. Methacrylated pullulan/polyethylene (glycol) diacrylate composite hydrogel for cartilage tissue engineering. J. Biomater. Sci. Polym. Ed. 2021, 32, 1057–1071. [Google Scholar] [CrossRef]
- Qin, J.; Jiang, J.; Ye, S.; Wang, S.; Xiao, M.; Tao, Y.; Jie, G.; Meng, Y. High performance poly(urethane-co-amide) from CO2-based dicarbamate: An alternative to long chain polyamide. RCS Adv. 2019, 9, 26080–26090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McOscar, T.V.C.; Gramlich, W.M. Hydrogels from norbornene-functionalised carboxymethyl cellulose using a UV-initiated thiol-ene click reaction. Cellulose 2018, 25, 6351–6545. [Google Scholar] [CrossRef]
- Van Hoorick, J.; Gruber, P.; Markovic, M.; Rollot, M.; Graulus, G.J.; Vagenende, M.; Tromayer, M.; Van Erps, J.; Thienpont, H.; Martins, J.C.; et al. Highly reactive thiol-norbornene photo-click hydrogels: Toward improved processability. Macromol. Rapid Commun. 2018, 39, 1800181–1800188. [Google Scholar] [CrossRef] [PubMed]
- Mao, G.; Sukumaran, S.; Beaucage, G.; Saboungi, M.L.; Thiyagarajan, P. PEO-PPO-PEO Block Copolymer Micelles in Aqueous Electrolyte Solutions: Effect of Carbonate Anions and Temperature on the Micellar Structure and Interaction. Macromolecules 2001, 34, 552–558. [Google Scholar] [CrossRef]
- Laurano, R.; Boffito, M.; Abrami, M.; Grassi, M.; Zoso, A.; Chiono, V.; Ciardelli, G. Dual stimuli-responsive polyurethane-based hydrogels as smart drug delivery carriers for the advanced treatment of chronic skin wounds. Bioact. Mater. 2021, 6, 3013–3024. [Google Scholar] [CrossRef] [PubMed]
- Popielarz, R.; Vogt, O. Effect of coinitiator type on initiation efficiency of two-component photoinitiator systems based on eosin. J. Polym. Sci. A 2008, 46, 3519–3532. [Google Scholar] [CrossRef]
- Timpe, H.J.; Jockusch, S.; Koerner, K. Radiation Curing in Polymer Science and Technology; Fouassier, J.P., Rabek, J.F., Eds.; Elsevier: London, UK, 1993; Volume 2, pp. 575–602. [Google Scholar]
- Lin, C.C.; Seok Ki, C.; Shih, H. Thiol-norbornene photoclick hydrogels for tissue engineering applications. J. Appl. Polym. Sci. 2015, 1, 41563–41574. [Google Scholar] [CrossRef] [Green Version]
- Fairbanks, B.D.; Schwartz, M.P.; Halevi, A.E.; Nuttelman, C.R.; Bowman, C.N.; Anseth, K.S. A versatile synthetic extracellular matrix mimic via thiol-norbornene photopolymerisation. Adv. Mater. 2009, 21, 5005–5010. [Google Scholar] [CrossRef] [Green Version]
- Shih, H.; Lin, C.C. Crosslinking and degradation of step-growth hydrogels formed by thiol-ene photo-click chemistry. Biomacromolecules 2012, 13, 2003–2012. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Han, F.; He, Y.; Zhang, J.; Wu, H.; Tao, J.; Zhang, C.; Zhang, F.; Liu, J. Unraveling the Voltage Failure Mechanism in Metal Sulfide Anodes for Sodium Storage and Improving Their Long Cycle Life by Sulfur-Doped Carbon Protection. Adv. Funct. Mater. 2021, 31, 2007266. [Google Scholar] [CrossRef]
- Wang, L.; Li, L.; Wang, X.; Huang, D.; Yang, F.; Shen, H.; Li, Z.; Wu, D. UV-triggered thiol-disulfide exchange reaction towards tailored biodegradable hydrogels. Polym. Chem. 2016, 7, 1429–1438. [Google Scholar] [CrossRef]
- Van Damme, L.; Van Hoorick, J.; Blondeel, P.; Van Vlierberghe, S. Toward Adipose Tissue Engineering Using Thiol-Norbornene Photo-Crosslinkable Gelatin Hydrogels. Biomacromolecules 2021, 22, 2408–2418. [Google Scholar] [CrossRef] [PubMed]
- Salinas, C.N.; Anseth, K.S. Mixed mode thiol-acrylate photopolymerisation for the synthesis of PEG-peptide hydrogels. Macromolecules 2008, 41, 6019–6026. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Thiol-Acrylate formulations | |
| S-DHP407/A-DHP407_ctrl | S-DHP407 and A-DHP407 dissolved in PBS solution, mixed at 1:1 molar ratio (18% w/v overall concentration) and added with 1 mM EY |
| S-DHP407/A-DHP407_light | S-DHP407 and A-DHP407 dissolved in PBS solution, mixed at 1:1 molar ratio (18% w/v overall concentration), added with 1 mM EY and exposed to green light (525 nm) at 80,000 Lux for 10 min |
| S-DHP407/A-DHP407_TEOA_ctrl | S-DHP407 and A-DHP407 dissolved in PBS solution, mixed at 1:1 molar ratio (18% w/v overall concentration) and added with 1 mM EY and 7.5 mM TEOA |
| S-DHP407/A-DHP407_TEOA_light | S-DHP407 and A-DHP407 dissolved in PBS solution, mixed at 1:1 molar ratio (18% w/v overall concentration), added with 1 mM EY and 7.5 mM TEOA and exposed to green light (525 nm) at 80,000 Lux for 10 min |
| Thiol-Norbornene formulations | |
| S-DHP407/NB-DHP407_ctrl | S-DHP407 and NB-DHP407 dissolved in PBS solution, mixed at 1:1 molar ratio (18% w/v overall concentration) and added with 0.5 mM EY |
| S-DHP407/NB-DHP407_light | S-DHP407 and NB-DHP407 dissolved in PBS solution, mixed at 1:1 molar ratio (18% w/v overall concentration), added with 0.5 mM EY and exposed to green light (525 nm) at 80,000 Lux for 10 min |
| S-DHP407/NB-DHP407_TYR_ctrl | S-DHP407 and NB-DHP407 dissolved in PBS solution, mixed at 1:1 molar ratio (18% w/v overall concentration) and added with 0.5 mM EY and 0.1 mM TYR |
| S-DHP407/NB-DHP407_TYR_light | S-DHP407 and NB-DHP407 dissolved in PBS solution, mixed at 1:1 molar ratio (18% w/v overall concentration), added with 0.5 mM EY and 0.1 mM TYR and exposed to green light (525 nm) at 80,000 Lux for 10 min |
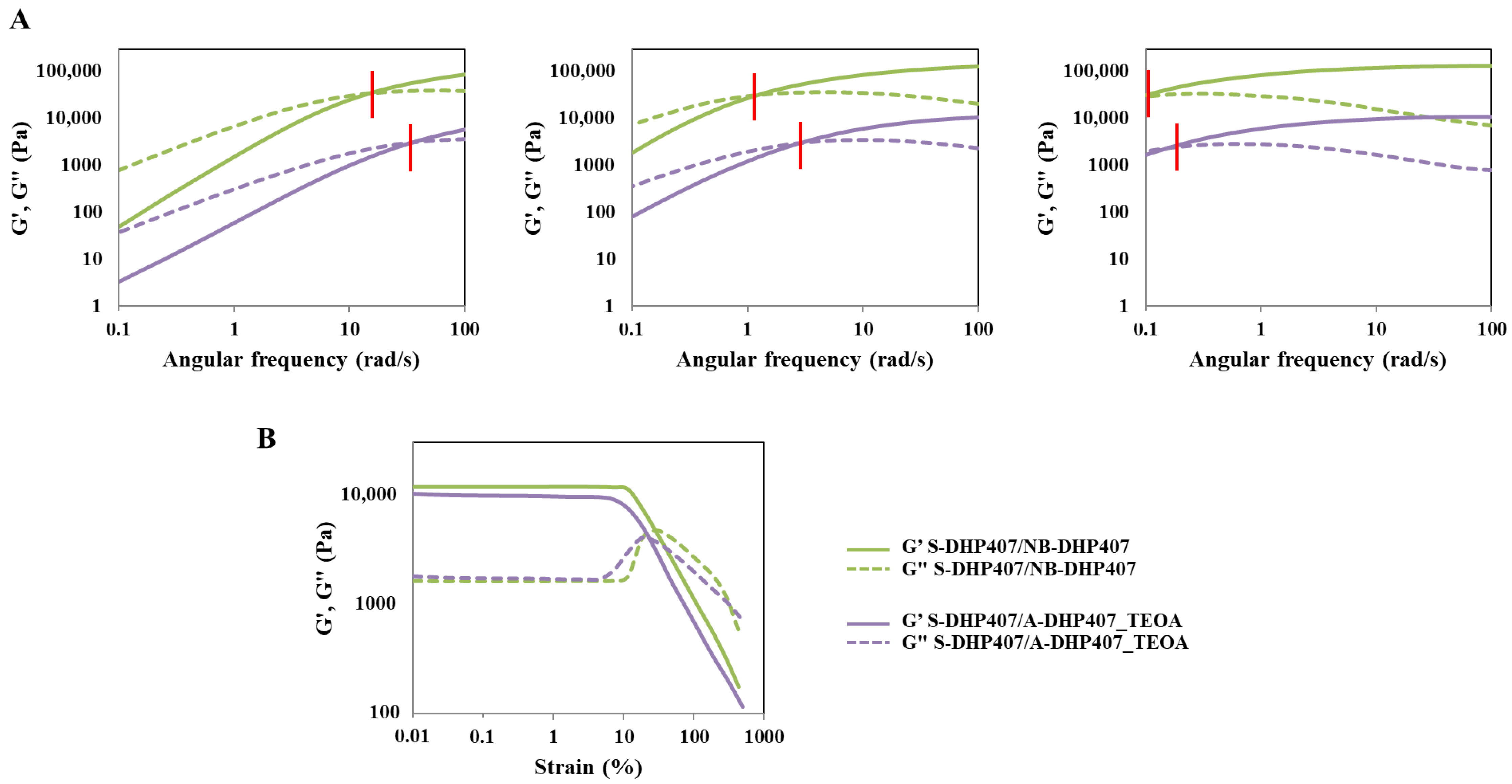
| ωcrossover (rad/s) | |||
|---|---|---|---|
| @ 25 °C | @ 30 °C | @ 37 °C | |
| S-DHP407/A-DHP407_TEOA_ctrl | >100 | 7.2 | 0.36 |
| S-DHP407/A-DHP407_TEOA_light | 34 | 2.8 | 0.16 |
| S-DHP407/NB-DHP407_TYR_ctrl | 80 | 6.0 | 0.32 |
| S-DHP407/NB-DHP407_TYR_light | 38 | 2.8 | 0.17 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laurano, R.; Boffito, M.; Cassino, C.; Midei, L.; Pappalardo, R.; Chiono, V.; Ciardelli, G. Thiol-Ene Photo-Click Hydrogels with Tunable Mechanical Properties Resulting from the Exposure of Different -Ene Moieties through a Green Chemistry. Materials 2023, 16, 2024. https://doi.org/10.3390/ma16052024
Laurano R, Boffito M, Cassino C, Midei L, Pappalardo R, Chiono V, Ciardelli G. Thiol-Ene Photo-Click Hydrogels with Tunable Mechanical Properties Resulting from the Exposure of Different -Ene Moieties through a Green Chemistry. Materials. 2023; 16(5):2024. https://doi.org/10.3390/ma16052024
Chicago/Turabian StyleLaurano, Rossella, Monica Boffito, Claudio Cassino, Ludovica Midei, Roberta Pappalardo, Valeria Chiono, and Gianluca Ciardelli. 2023. "Thiol-Ene Photo-Click Hydrogels with Tunable Mechanical Properties Resulting from the Exposure of Different -Ene Moieties through a Green Chemistry" Materials 16, no. 5: 2024. https://doi.org/10.3390/ma16052024