
Ab Initio Studies of Mechanical, Dynamical, and Thermodynamic Properties of Fe-Pt Alloys


Abstract
1. Introduction
2. Computational Procedure
3. Results and Discussion
3.1. Mechanical Properties
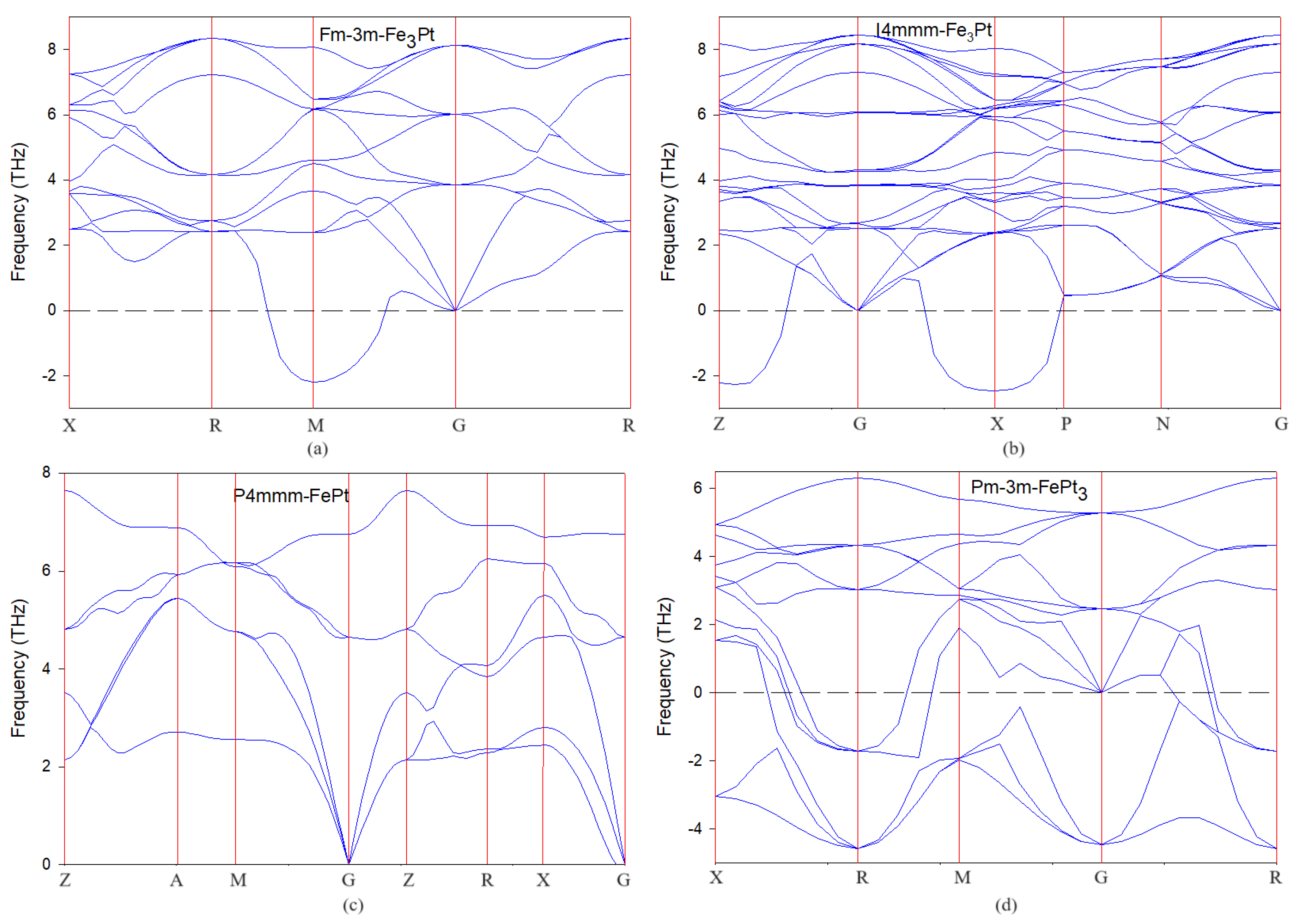
3.2. Phonon Dispersion Curves
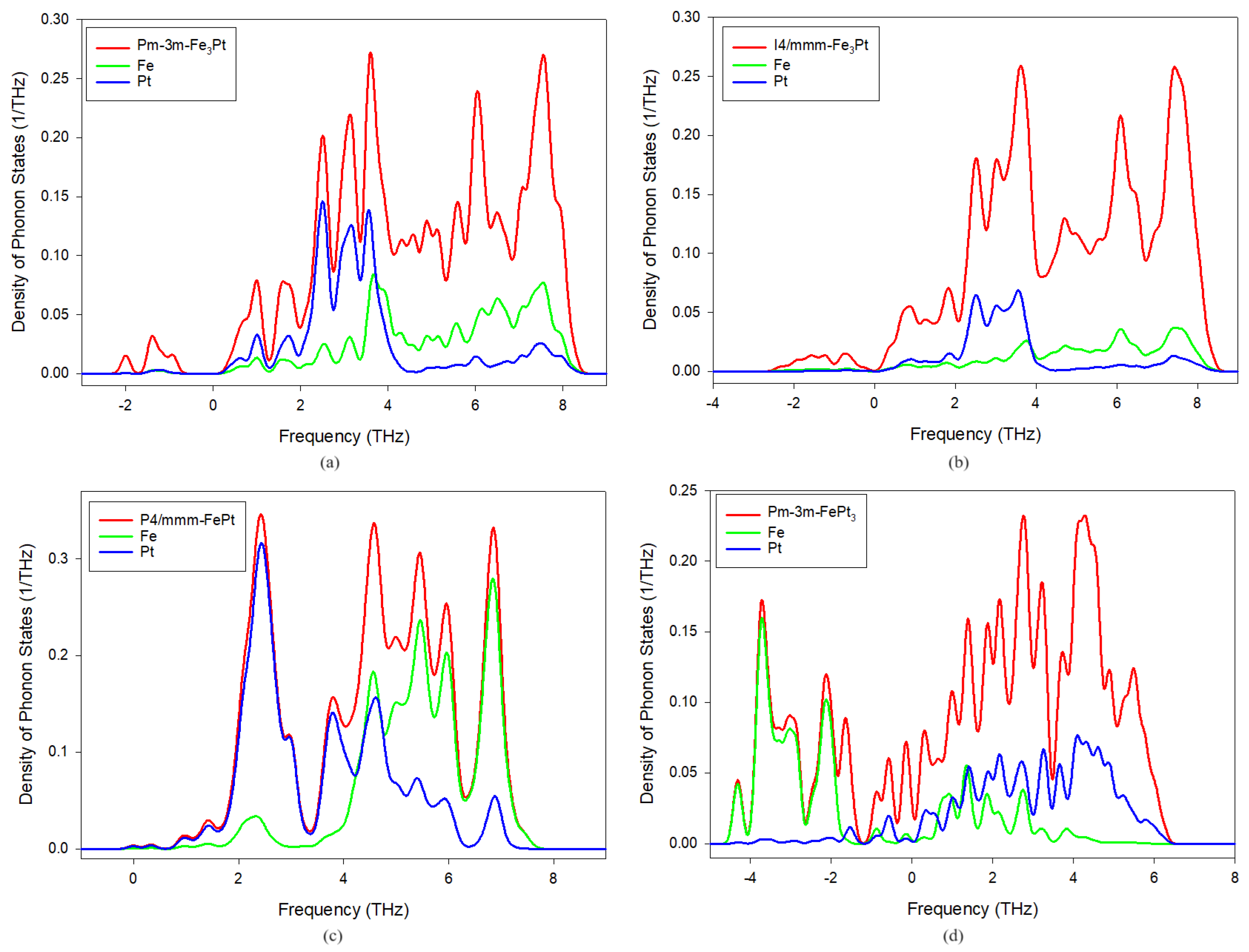
3.3. Phonon Density of States
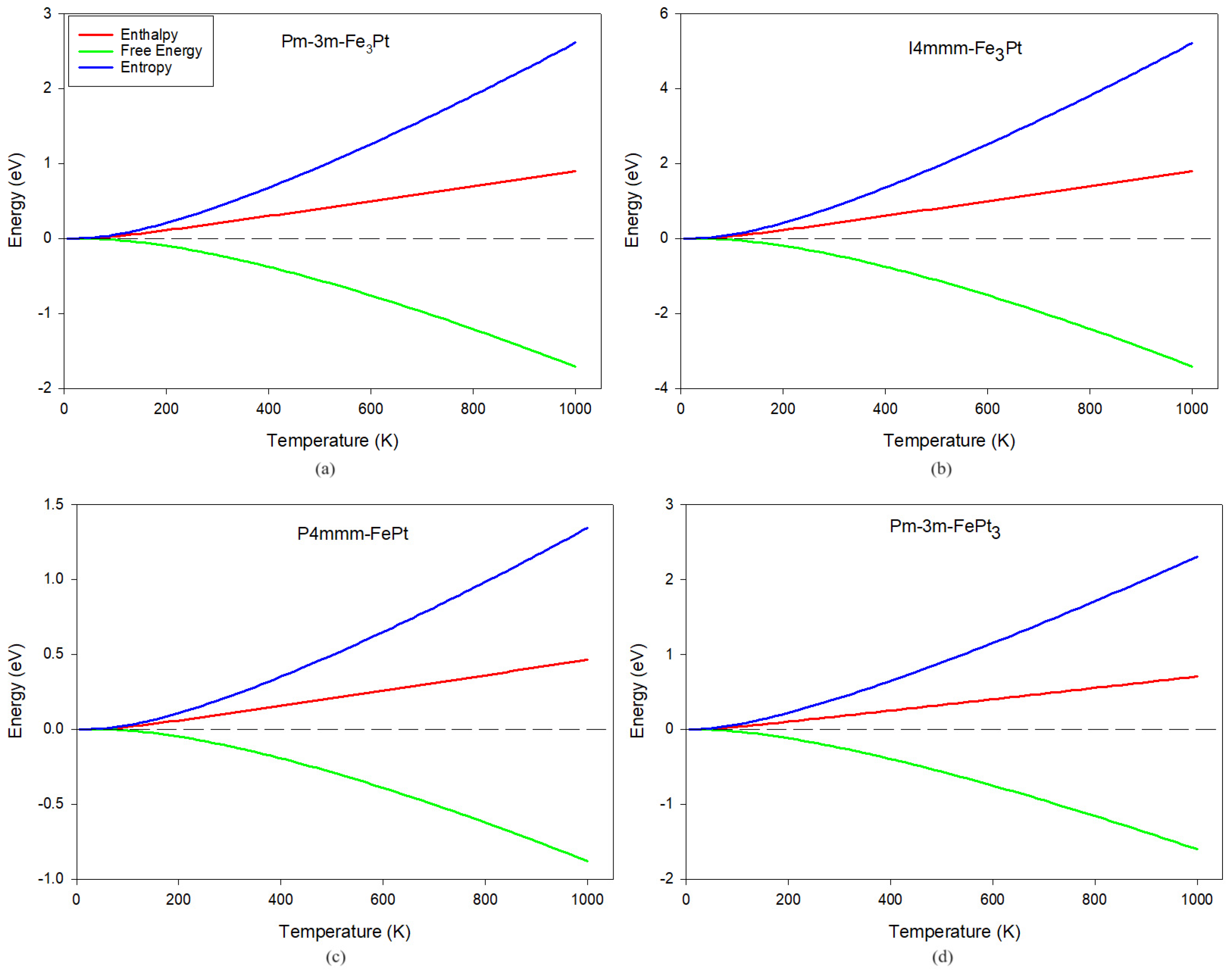
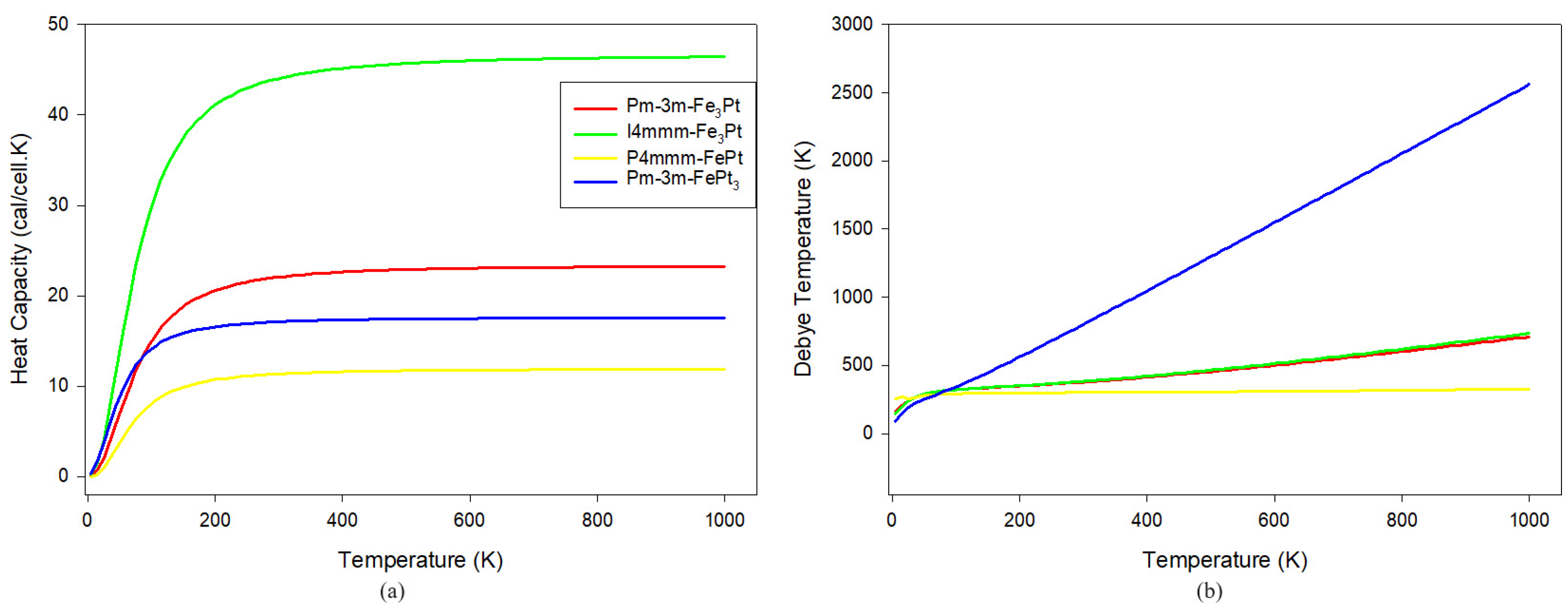
3.4. Thermodynamic Properties
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, J.P. FePt Magnetic Nanoparticles and Their Assembly for Future Magnetic Media. Proc. IEEE 2008, 96, 1847. [Google Scholar] [CrossRef]
- Alsaad, A.; Ahmad, A.A.; Obeidat, T.S. Structural, electronic and magnetic properties of the ordered binary FePt, MnPt, and CrPt3 alloys. Heliyon 2020, 6, e03545. [Google Scholar] [CrossRef] [PubMed]
- Lethole, N.; Ngoepe, P.; Chauke, H. Compositional Dependence of Magnetocrystalline Anisotropy, Magnetic Moments, and Energetic and Electronic Properties on Fe-Pt Alloys. Materials 2022, 15, 5679. [Google Scholar] [CrossRef] [PubMed]
- Laureti, S.; D’Acapito, F.; Imperatori, P.; Patrizi, E.; Varvaro, G.; Puri, A.; Cannas, C.; Capobianchi, A. Synthesis of highly ordered L10 MPt alloys (M = Fe, Co, Ni) from crystalline salts: An in situ study of the pre-ordered precursor reduction strategy. J. Mater. Chem. C 2023, 11, 16661. [Google Scholar] [CrossRef]
- Crisan, A.D.; Bednarcik, J.; Michalik, Š.; Crisan, O. In situ monitoring of disorder–order A1–L10 FePt phase transformation in nanocomposite FePt-based alloys. J. Alloys Compd. 2014, 615, S188. [Google Scholar] [CrossRef]
- Müller, M.; Erhart, P.; Albe, K. Thermodynamics of L10 ordering in FePt nanoparticles studied by Monte Carlo simulations based on an analytic bond-order potential. Phys. Rev. B 2007, 76, 155412. [Google Scholar] [CrossRef]
- Müller, M.; Albe, K. Lattice Monte Carlo simulations of FePt nanoparticles: Influence of size, composition, and surface segregation on order-disorder phenomena. Phys. Rev. B 2005, 72, 094203. [Google Scholar] [CrossRef]
- Yu, G.L.; Cheng, T.; Zhang, X. Effect of pressure on the magnetic, mechanical, and dynamical properties of L10-FePt alloy. J. Appl. Phys. 2023, 134, 085902. [Google Scholar] [CrossRef]
- Whang, S.H.; Feng, Q.; Gao, Y.Q. Ordering, deformation and microstructure in L10 type FePt. Acta Mater. 1998, 46, 6485. [Google Scholar] [CrossRef]
- Crisan, O.; Crisan, A.D.; Randrianantoandro, N. Temperature-Dependent Phase Evolution in FePt-Based Nanocomposite Multiple-Phased Magnetic Alloys. Nanomaterials 2022, 12, 4122. [Google Scholar] [CrossRef]
- Zotov, N.; Ludwig, A. First-principles calculations of the elastic constants of FeePt alloys. Intermetallics 2008, 16, 113. [Google Scholar] [CrossRef]
- Cabri, L.J.; Oberthür, T.; Schumann, D. The Mineralogy of Pt-Fe alloys and phase relations in the Pt–Fe binary system. Can. Miner. 2022, 60, 331. [Google Scholar] [CrossRef]
- Yu, G.; Cheng, T.; Zhang, X. Exploring the structural stability and related physical properties of FePt2 alloys. Phys. B Condens. Matter. 2024, 680, 415833. [Google Scholar] [CrossRef]
- Yu, G.; Cheng, T.; Zhang, X. Exploration of novel structures and related physical properties of Fe2Pt ordered alloys. Solid. State Sci. 2023, 146, 107380. [Google Scholar] [CrossRef]
- Pierron-Bohnes, V.; Montsouka, R.V.P.; Goyhenex, C.; Mehaddene, T.; Messad, L.; Bouzar, H.; Numakura, H.; Tanaka, K.; Hennion, B. Atomic migration in bulk and thin film L1 0 alloys: Experiments and molecular dynamics simulations. Defect Diffus. Forum 2007, 1, 263. [Google Scholar]
- Noda, Y.; Endoh, Y.; Katano, S.; Iizumi, M. Lattice dynamics of FePt alloys of AB3 type ordered structure. Phys. B 1983, 120, 317. [Google Scholar] [CrossRef]
- Sternik, M.; Couet, S.; Łazewski, J.; Jochym, P.T.; Parlinski, K.; Vantomme, A.; Temst, K.; Piekarz, P. Dynamical properties of ordered FeePt alloys. J. Alloys Compd. 2015, 651, 528. [Google Scholar] [CrossRef]
- Phasha, M.J.; Ngoepe, P.E.; Chauke, H.R.; Pettifor, D.G.; Nguyen-Mann, D. Link between structural and mechanical stability of fcc- and bcc-based ordered MgeLi alloys. Intermetallics 2010, 18, 2083. [Google Scholar] [CrossRef]
- Botha, L.M.; Ouma, C.N.M.; Obodo, K.O.; Bessarabov, D.G.; Sharypin, D.L.; Varyushin, P.S.; Plastinina, E.I. Ab Initio Study of Structural, Electronic, and Thermal Properties of Pt/Pd-Based Alloys. Condens. Matter. 2023, 8, 76. [Google Scholar] [CrossRef]
- Diale, R.G.; Ngoepe, P.E.; Moema, J.S.; Phasha, M.J.; Moller, H.; Chauke, H.R. A computational study of the thermodynamic and magnetic properties of Co alloyed MnPt. MRS Adv. 2023, 8, 651. [Google Scholar] [CrossRef]
- Al-Essa, S.; Essaoud, S.S.; Bouhemadou, A.; Ketfi, M.E.; Omran, S.B.; Chik, A.; Radjai, M.; Allali, D.; Khenata, R.; Al-Douri, Y. A Comprehensive Ab Initio Study of the Recently Synthesized Zintl Phase CsGaSb2 Structural, Dynamical Stability, Elastic and Thermodynamic Properties. J. Inorg. Organomet. Polym. Mater. 2024. [Google Scholar] [CrossRef]
- Kim, J.; Koo, Y.; Lee, B.J. Modified embedded-atom method interatomic potential for the Fe–Pt alloy system. J. Mater. Res. 2006, 21, 199. [Google Scholar] [CrossRef]
- Tajima, K.; Endoh, Y.; Ishikawa, Y.; Stirling, W.G. Acoustic-Phonon Softening in the Invar Alloy Fe3Pt. Phys. Rev. Lett. 1976, 37, 519. [Google Scholar] [CrossRef]
- Kastner, J.; Neuhaus, J.; Wassermann, E.; Petry, W.; Hennion, B.; Bach, H. TA1 [110] phonon dispersion and martensitic phase transition in ordered alloys Fe3Pt. Eur. Phys. J. B 1999, 11, 75. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Krist. Cryst. Mater. 2005, 220, 567. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Hao, D.; He, X.; Roitberg, A.E.; Zhang, S.; Wang, J. Development and Evaluation of Geometry Optimization Algorithms in Conjunction with ANI Potentials. J. Chem. Theory Comput. 2022, 18, 978. [Google Scholar] [CrossRef] [PubMed]
- Packwood, D.; Kermode, J.; Mones, L.; Bernstein, N.; Wooley, J.; Gould, N.; Ortner, C.; Csanyi, G. A universal preconditioner for simulating condensed phase materials. J. Chem. Phys. 2016, 144, 164109. [Google Scholar] [CrossRef] [PubMed]
- Togo, A. First-principles Phonon Calculations with Phonopy and Phono3py. J. Phys. Soc. Jpn. 2023, 92, 012001. [Google Scholar] [CrossRef]
- Togo, A.; Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 2015, 108, 1. [Google Scholar] [CrossRef]
- Chen, H.S. Elastic Anisotropy of Metal; Metallurgy Industry Press: Beijing, China, 1996. [Google Scholar]
- Fast, L.; Wills, J.M.; Johansson, B.; Eriksson, O. Elastic constants of hexagonal transition metals: Theory. Phys. Rev. B 1995, 51, 17431. [Google Scholar] [CrossRef] [PubMed]
- Karki, B.B.; Ackland, G.J.; Crain, J. Elastic instabilities in crystals from ab initio stress—Strain relations. J. Phys. Condens. Matter. 1997, 9, 8579. [Google Scholar] [CrossRef]
- Kittel, C. Introduction to Solid State Physics, 8th ed.; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2005. [Google Scholar]
- Born, M.; Huang, K. Dynamical Theory of Crystal Lattices; Clarendon: Oxford, UK, 1956. [Google Scholar]
- Ranganathan, S.I.; Ostoja-Starzewski, M. Universal Elastic Anisotropy Index. Phys. Rev. Lett. 2008, 101, 055504. [Google Scholar] [CrossRef] [PubMed]
- Voigt, W. Lehrbuch der Kristallphysik; Taubner: Leipzig, Germany, 1928. [Google Scholar]
- Reuss, A. Calculation of the flow limits of mixed crystals on the basis of the plasticity of monocrystals. Z. Angew. Math. Mech. 1929, 9, 55. [Google Scholar]
- Hill, R. The Elastic Behaviour of a Crystalline Aggregate. Proc. Phys. Soc. A 1952, 65, 349. [Google Scholar] [CrossRef]
- Pugh, S.F. XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. The London, Edinburgh, and Dublin Philosophical Mag. J. Sci. 1954, 45, 823. [Google Scholar]
- Frantsevich, I.N.; Voronov, F.F.; Bokuta, S.A. Elastic Constants and Elastic Moduli of Metals and Insulators; Frantsevich, I.N., Ed.; Naukova Dumka: Kiev, Ukraine, 1983; p. 60. [Google Scholar]
- Murtaza, G.; Gupta, S.K.; Seddik, T.; Khenata, R.; Alahmed, Z.A.; Ahmed, R.; Khachai, H.; Jha, P.K.; Omran, S.B. Structural, electronic, optical and thermodynamic properties of cubic REGa3 (RE = Sc or Lu) compounds: Ab initio study. J. Alloys Compd. 2014, 597, 36. [Google Scholar] [CrossRef]
- Haines, J.; Léger, J.M.; Bocquillon, G. Synthesis and Design of Superhard Materials. Annu. Rev. Mater. 2001, 31, 1. [Google Scholar] [CrossRef]
- Kiejna, A.; Wojciechowski, K.F. The surface of real metals. In Metal Surface Electron Physic; Alden Press: Oxford, UK, 1996; p. 19. [Google Scholar]
- Jiang, D.; Xiao, W.; Liu, D.; Liu, S. Structural stability, electronic structures, mechanical properties and debye temperature of W-Re alloys: A first-principles study. Fusion Eng. Des. 2021, 162, 112081. [Google Scholar] [CrossRef]
- Tohei, T.; Kuwabara, A.; Oba, F.; Tanaka, I. Debye temperature and stiffness of carbon and boron nitride polymorphs from first principles calculations. Phys. Rev. B 2006, 73, 064304. [Google Scholar] [CrossRef]
- Tian, Y.; Xu, B.; Zhao, Z. Microscopic theory of hardness and design of novel superhard crystals. Int. J. Refract. Hard. Met. 2012, 33, 93. [Google Scholar] [CrossRef]
- Chen, X.Q.; Niu, H.; Li, D.; Li, Y. Modeling hardness of polycrystalline materials and bulk metallic glasses. Intermetallics 2011, 19, 1275. [Google Scholar] [CrossRef]
- Zerrougui, Z.; Bouferrache, K.; Ghebouli, M.A.; Slimani, Y.; Chihi, T.; Ghebouli, B.; Fatmi, M.; Benlakhdar, F.; Mouhammad, S.A.; Algethami, N.; et al. Study of structural, elastic, mechanical, electronic and magnetic properties of FeX (X=Pt, Pd) austenitic and martensitic phases. Solid State Sci. 2023, 141, 107211. [Google Scholar] [CrossRef]
- Piekarz, P.; Lazewski, J.; Jochym, P.T.; Sternik, M.L.; Parlinski, K. Vibrational properties and stability of FePt nanoalloys. Phys. Rev. B 2017, 95, 134303. [Google Scholar] [CrossRef]
- Baroni, S.; de Gironcoli, S.; Corso, A.D.; Giannozzi, P. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 2001, 73, 515. [Google Scholar] [CrossRef]
- Dima, R.S.; Maleka, P.M.; Maluta, N.E.; Maphanga, R.R. Structural, Electronic, Mechanical, and Thermodynamic Properties of Na Deintercalation from Olivine NaMnPO4: First-Principles Study. Materials 2022, 15, 5280. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.; Wang, T.; Qi, W.; Li, Y. Debye temperature for binary alloys and its relationship with cohesive energy. Phys. B Condens. Matter. 2018, 531, 95. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | -Fe3Pt | -Fe3Pt | -FePt | -FePt3 |
|---|---|---|---|---|
| Lattice Parameters (Å) | ||||
| a (Å) | 3.735 (3.72) [10] | 5.276 | 2.725 (2.728) | 3.914 (3.87) [22] |
| c (Å) | - | - | 3.784 (3.85) [22] | - |
| Bond Length (Å) [4] | ||||
| Fe-Pt | 2.641 | 2.638 | 2.700 (2.667) | 2.768 |
| Fe-Fe | 2.641 | 2.638 | - | - |
| Pt-Pt | - | - | 2.768 | |
| Elastic Constants (GPa) [22] | ||||
| 206.344 (238.8) | 259.280 | 346.778 (304.2) | 301.055 (325.8) | |
| 209.513 | 292.106 (242.0) | |||
| 85.957 (90.46) | 68.757 | 113.855 (106.5) | 105.108 (90.4) | |
| 35.522 | 48.38190 (40.8) | |||
| 170.625 (184.1) | 90.790 | 73.457 (222.6) | 183.094 (230.1) | |
| 152.871 | 161.090 (197.3) | |||
| 17.859 | 84.245 | 136.66 | 58.981 | |
| 182.531 | 168.908 | 197.102 | 222.415 | |
| 46.379 | 51.670 | 87.423 | 83.360 | |
| 151.612 | 134.461 | 138.820 | 166.842 | |
| 128.274 | 140.667 | 228.488 | 222.306 | |
| 0.383 | 0.361 | 0.307 | 0.333 | |
| 1.624 | 1.082 | 0.891 | 0.412 | |
| 276.697 | 298.023 | 330.790 | 291.904 | |
| 2.931 | 3.907 | 8.650 | 6.906 | |
| 0.254 | 0.306 | 0.444 | 0.375 | |
| Enthalpy | |
| Free energy | |
| Entropy | |
| Heat capacity | |
| Debye temperature |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lethole, N.L.; Mukumba, P. Ab Initio Studies of Mechanical, Dynamical, and Thermodynamic Properties of Fe-Pt Alloys. Materials 2024, 17, 3879. https://doi.org/10.3390/ma17153879
Lethole NL, Mukumba P. Ab Initio Studies of Mechanical, Dynamical, and Thermodynamic Properties of Fe-Pt Alloys. Materials. 2024; 17(15):3879. https://doi.org/10.3390/ma17153879
Chicago/Turabian StyleLethole, Ndanduleni Lesley, and Patrick Mukumba. 2024. "Ab Initio Studies of Mechanical, Dynamical, and Thermodynamic Properties of Fe-Pt Alloys" Materials 17, no. 15: 3879. https://doi.org/10.3390/ma17153879
APA StyleLethole, N. L., & Mukumba, P. (2024). Ab Initio Studies of Mechanical, Dynamical, and Thermodynamic Properties of Fe-Pt Alloys. Materials, 17(15), 3879. https://doi.org/10.3390/ma17153879