

Electrochemical Characterization of Electrodeposited Copper in Amine CO2 Capture Media



Abstract
1. Introduction
2. Materials and Methods
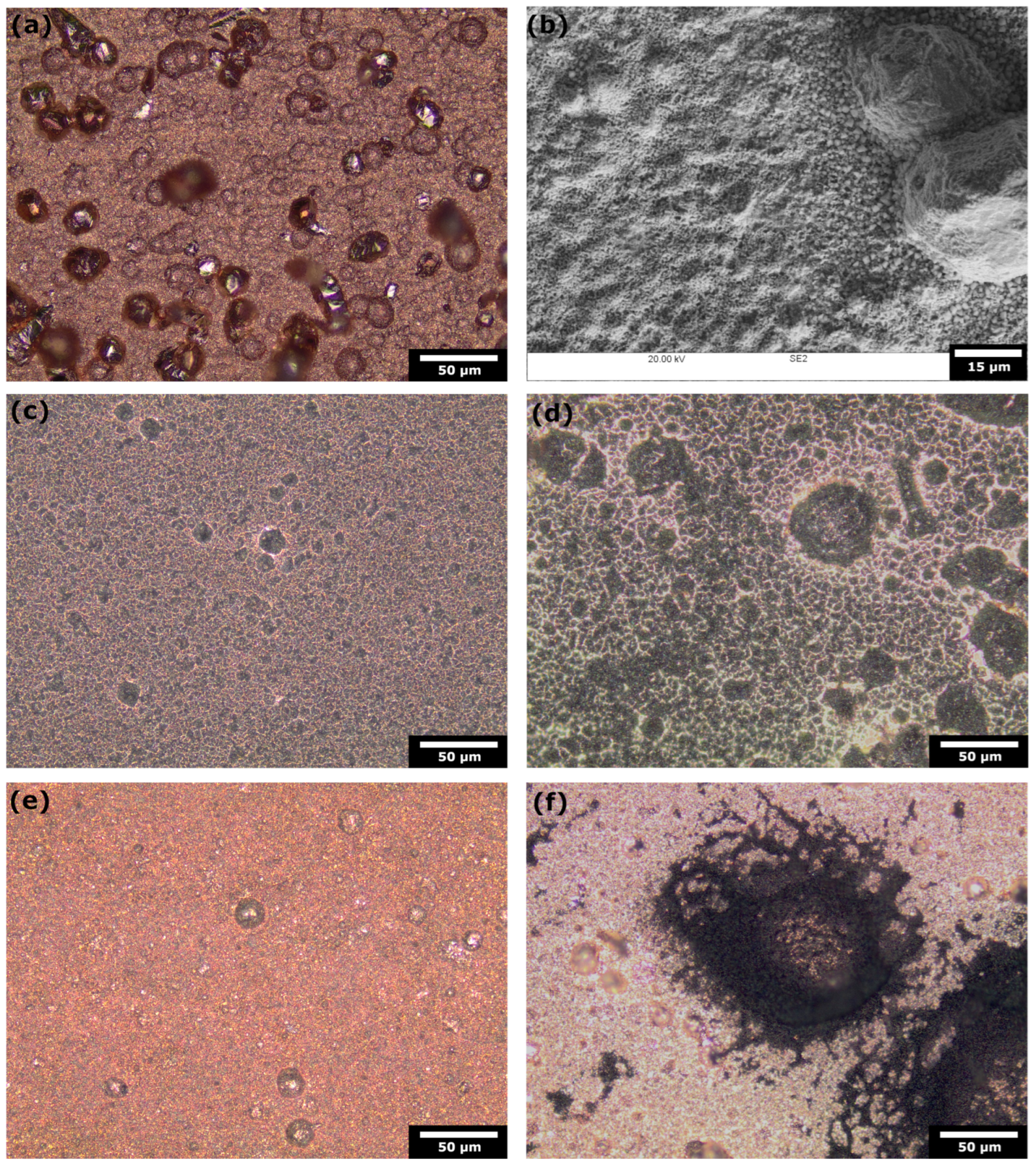
2.1. Specimen Preparation and Characterisation
2.2. Amine Media
- 30 wt.% Monoethanolamine (MEA), CAS: 141-43-5, a primary amine extensively utilized for CO2 capture [2].
- 37 wt.% Methyldiethanolamine (MDEA) CAS: 105-59-9, a tertiary amine.
- 30 wt.% MDEA + 21 wt.% piperazine (PZ), CAS: 110-85-0, generally used to improve capture kinetics [35].
- 30 wt.% 2-Amino-2-methyl-1-propanol (AMP), CAS: 124-68-5, a sterically hindered primary amine known for its elevated CO2 absorption capacity [36].
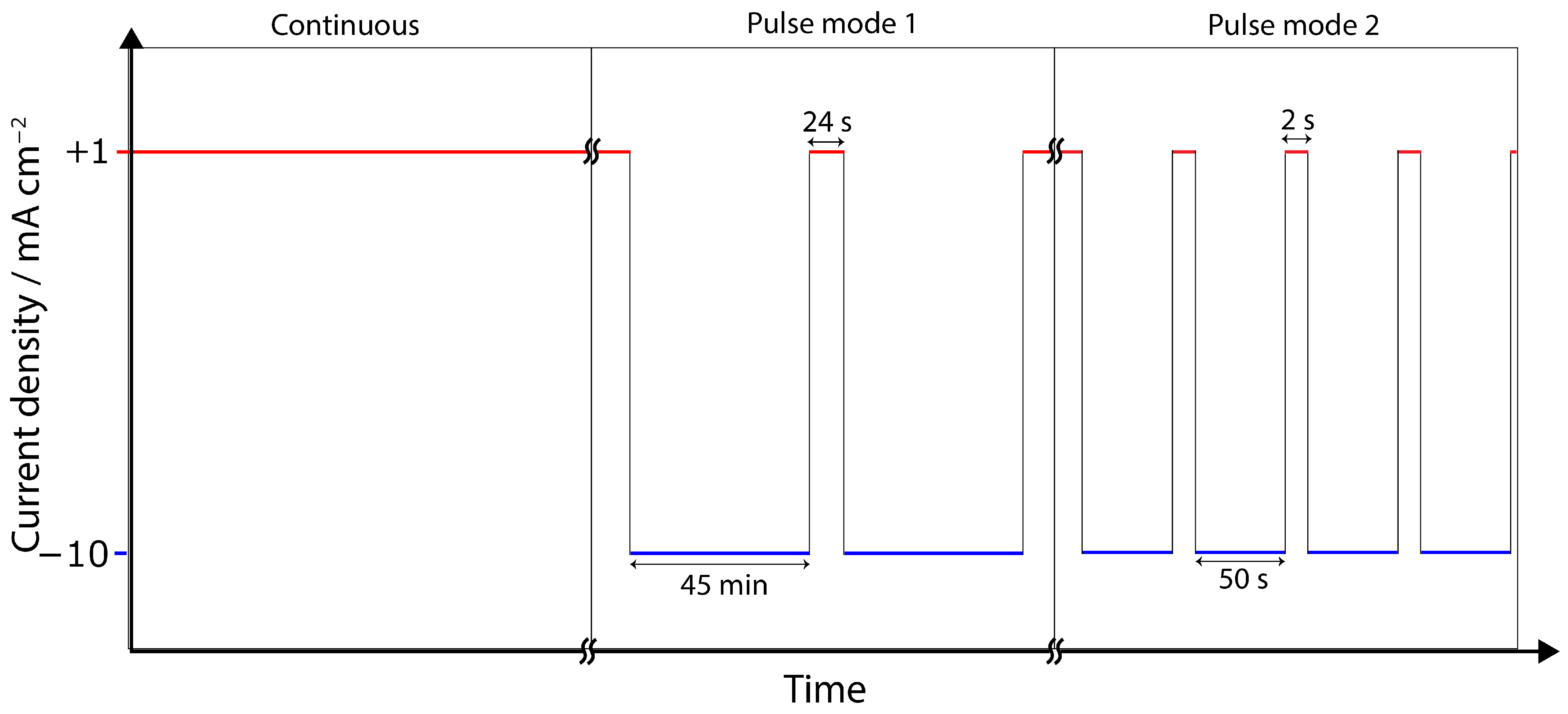
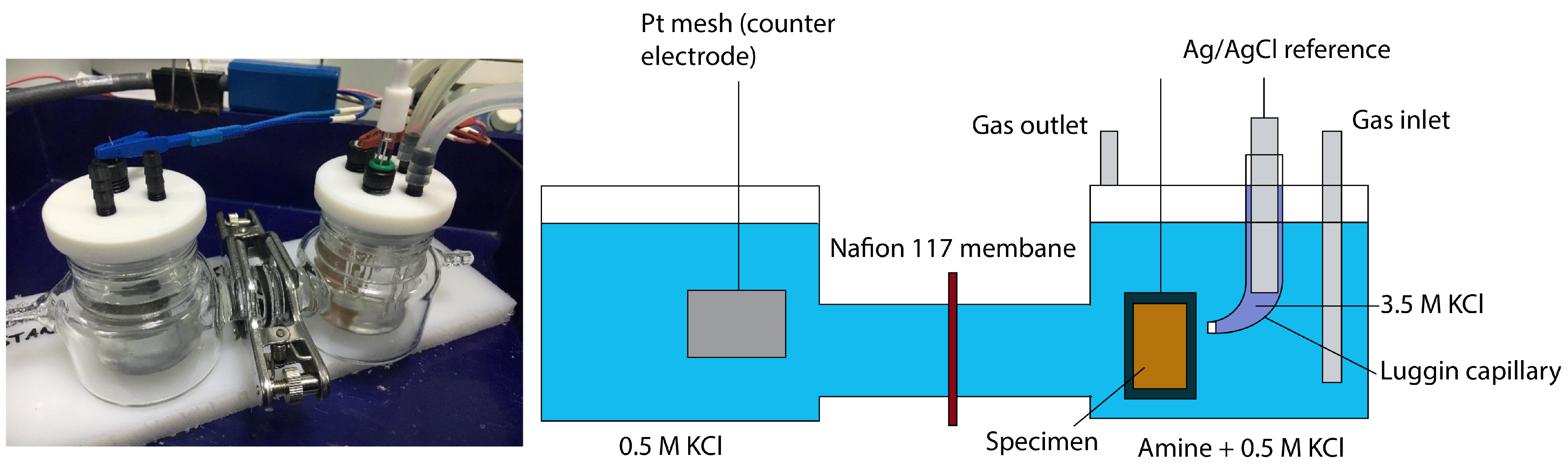
2.3. Electrochemical Tests
3. Results
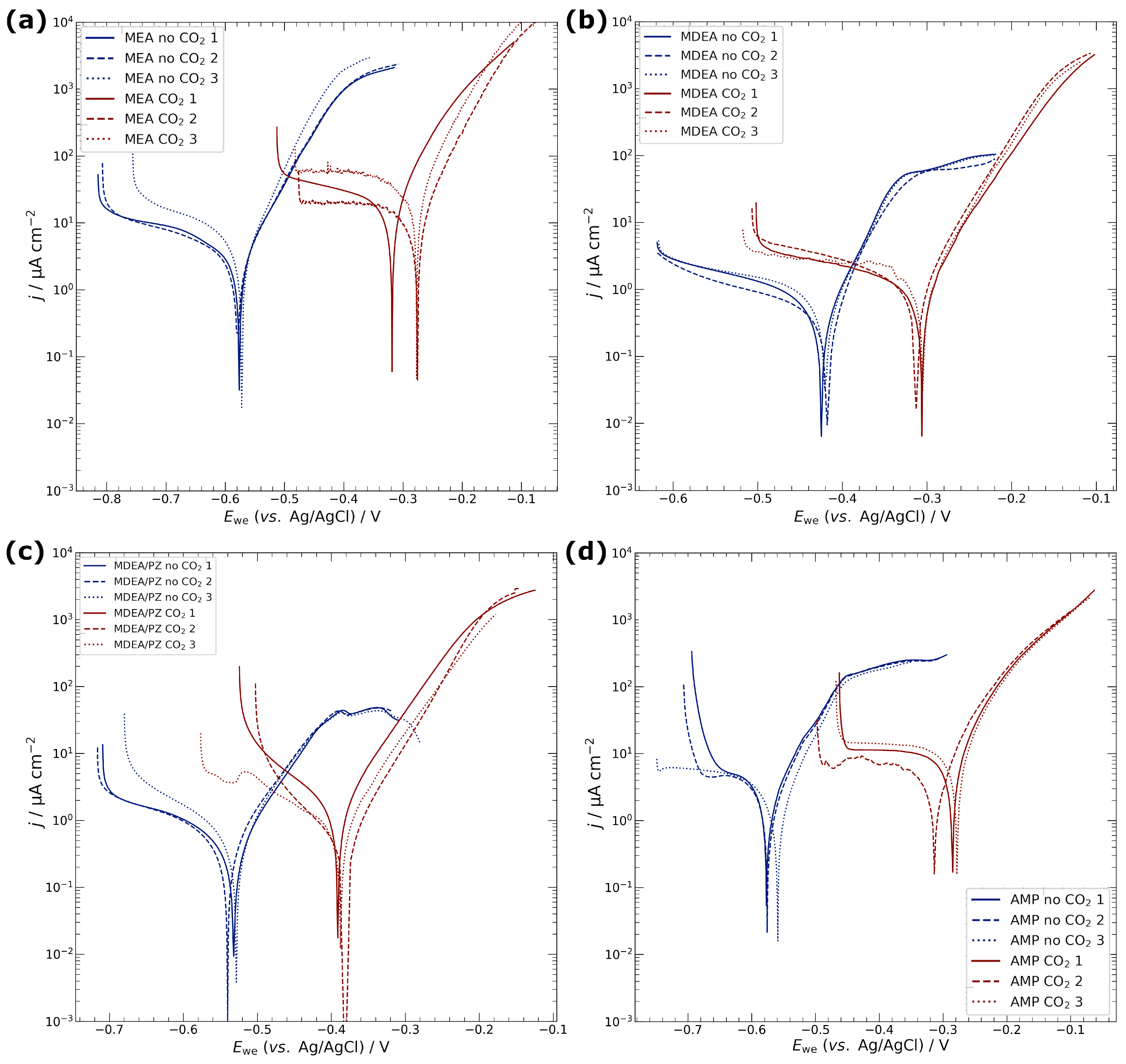
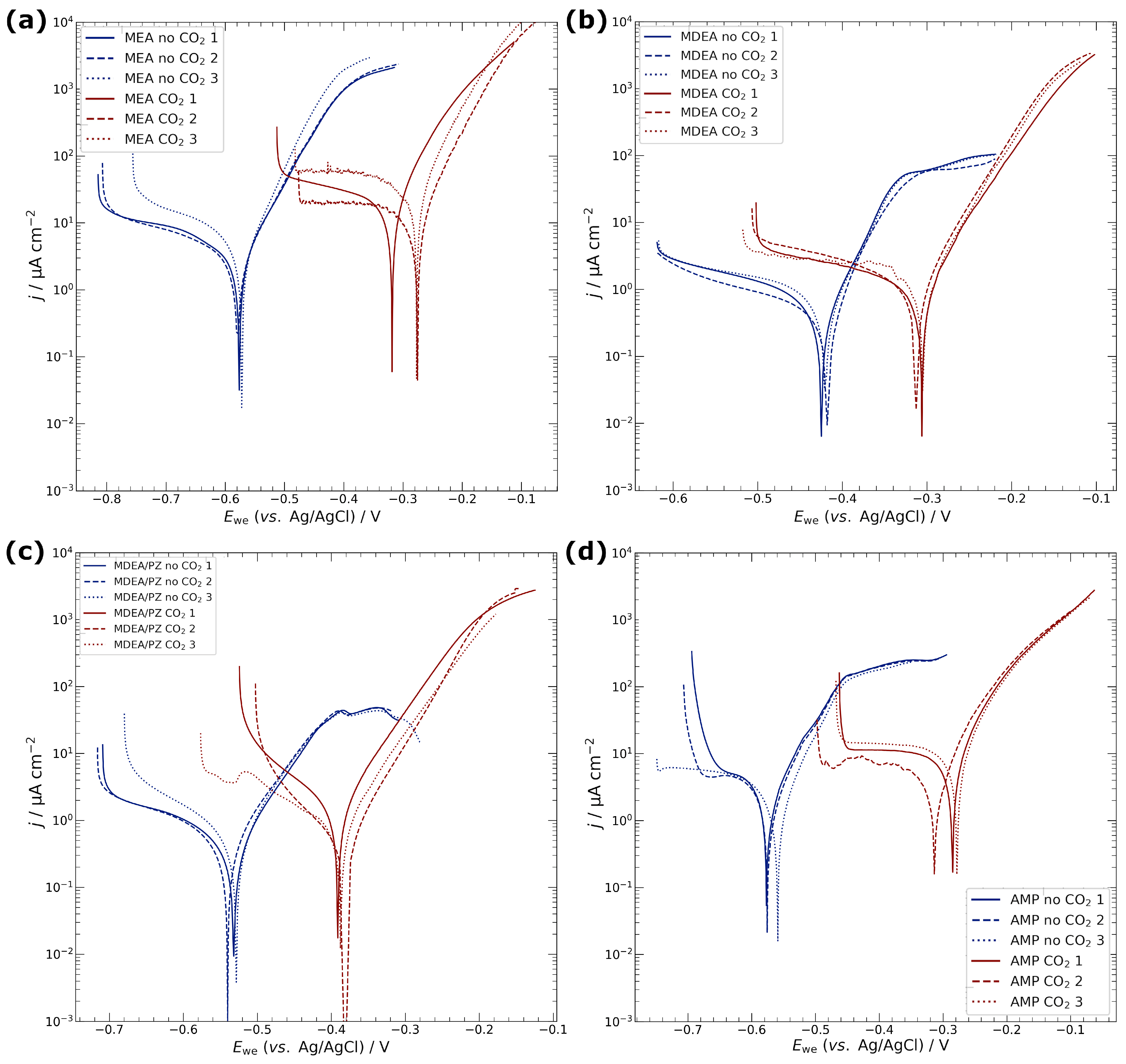
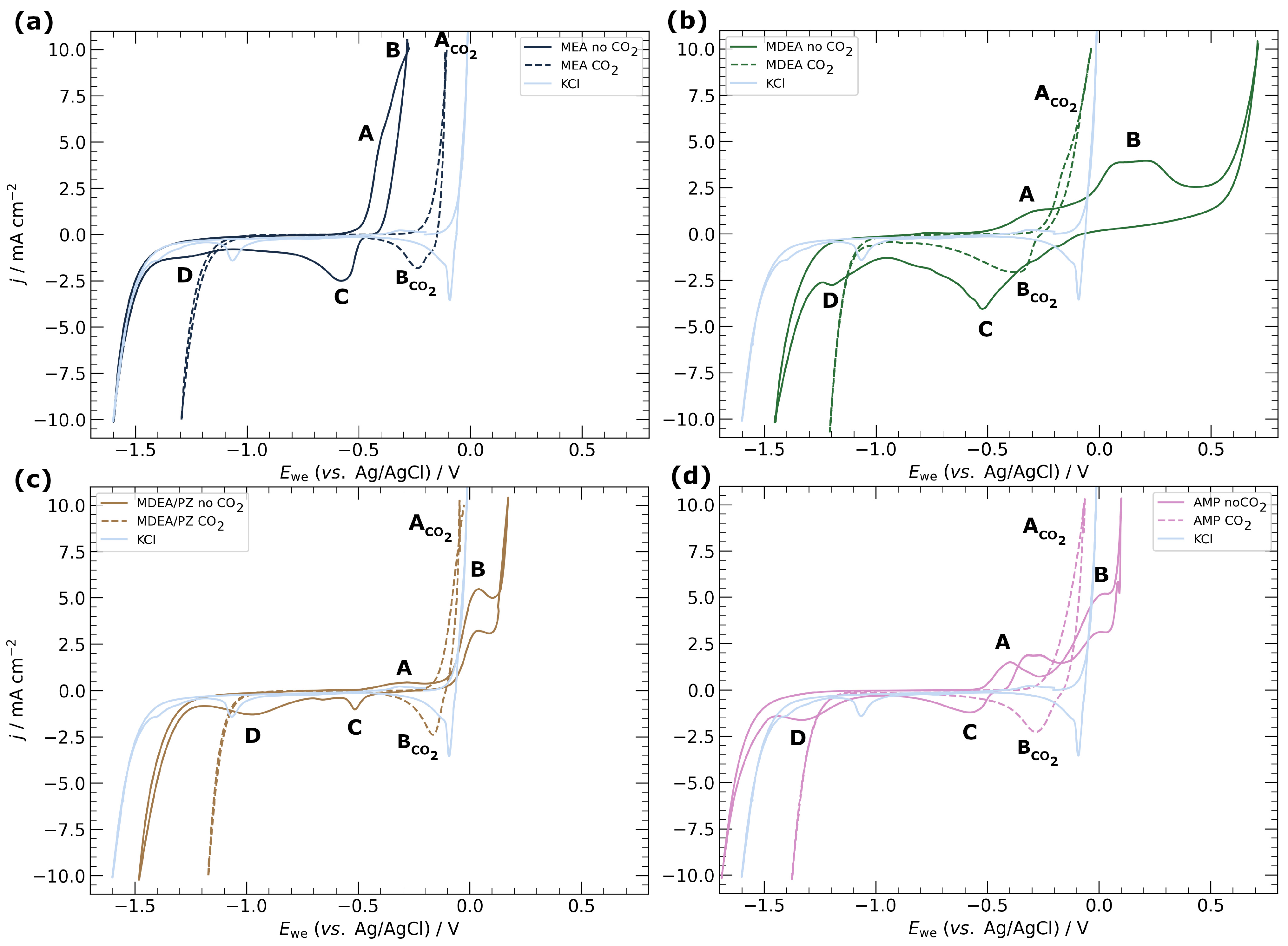
3.1. Electrochemical Characterisation of Electrodeposited Copper in Amine Media
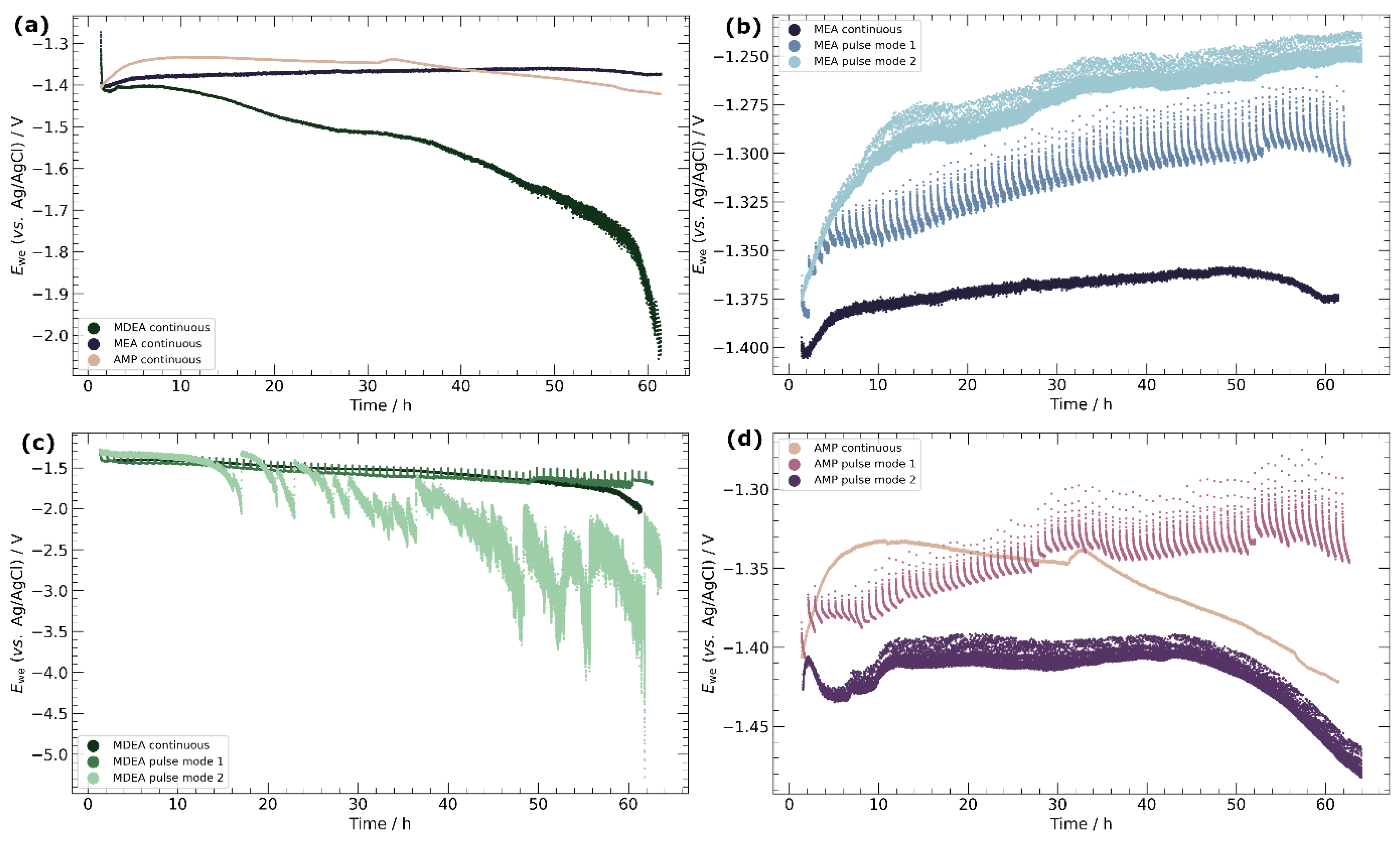
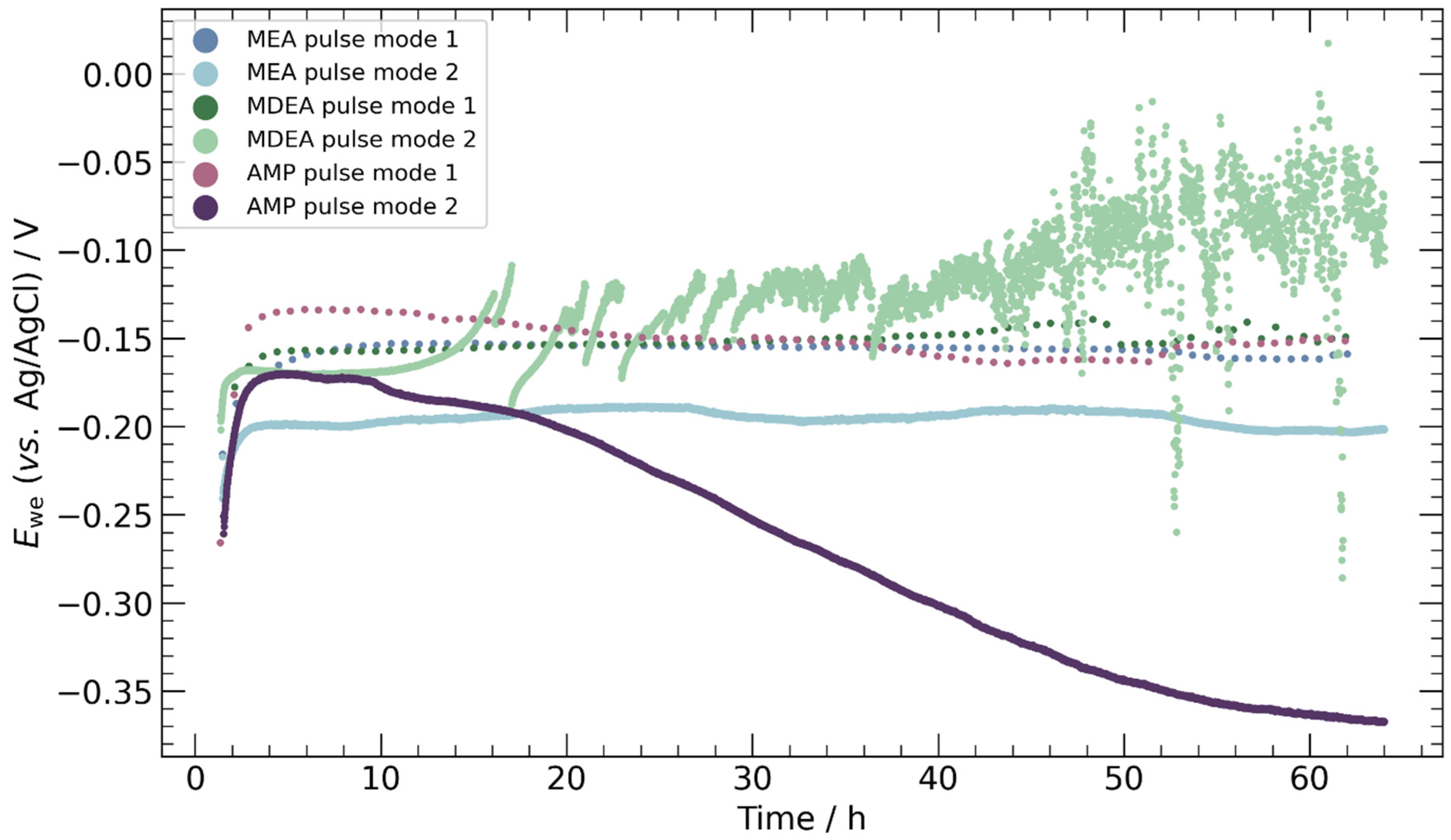
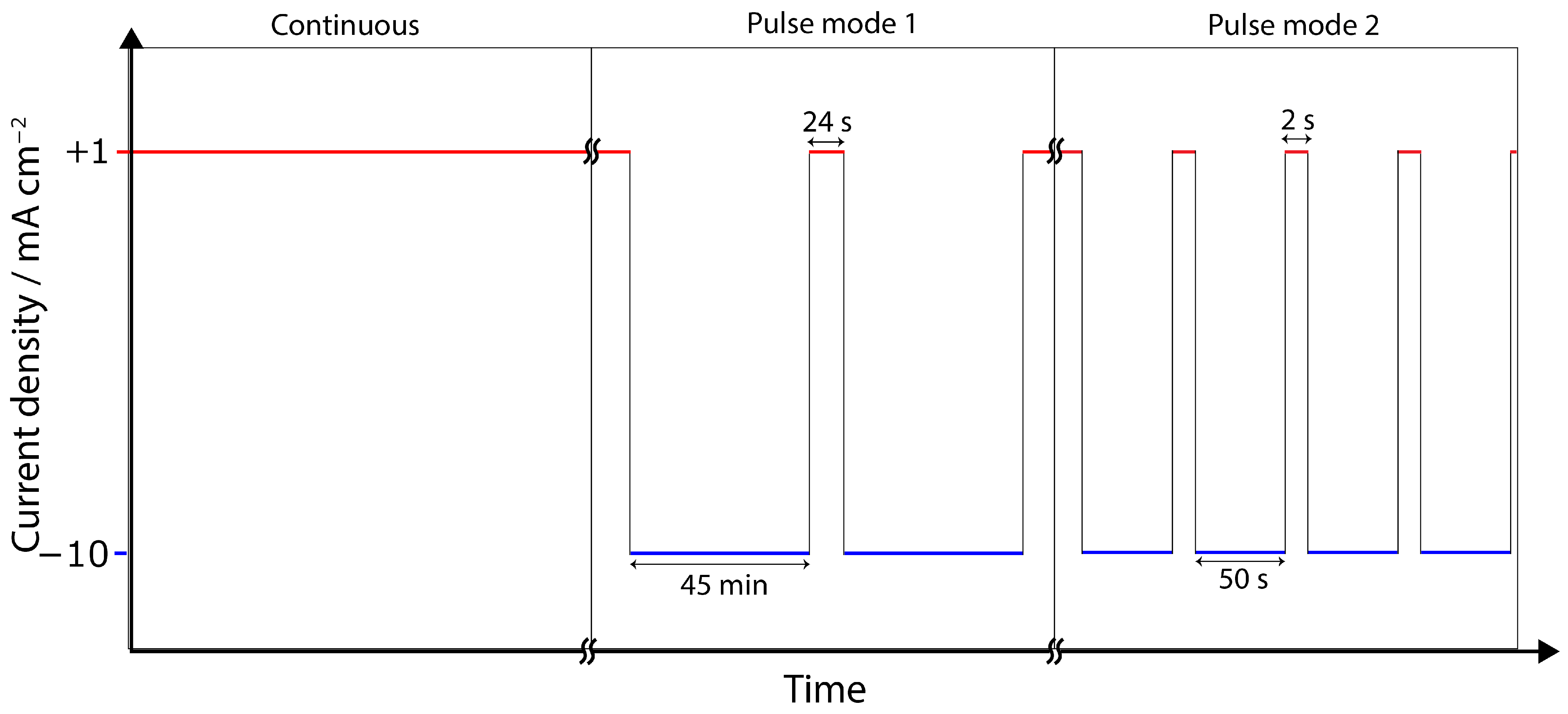
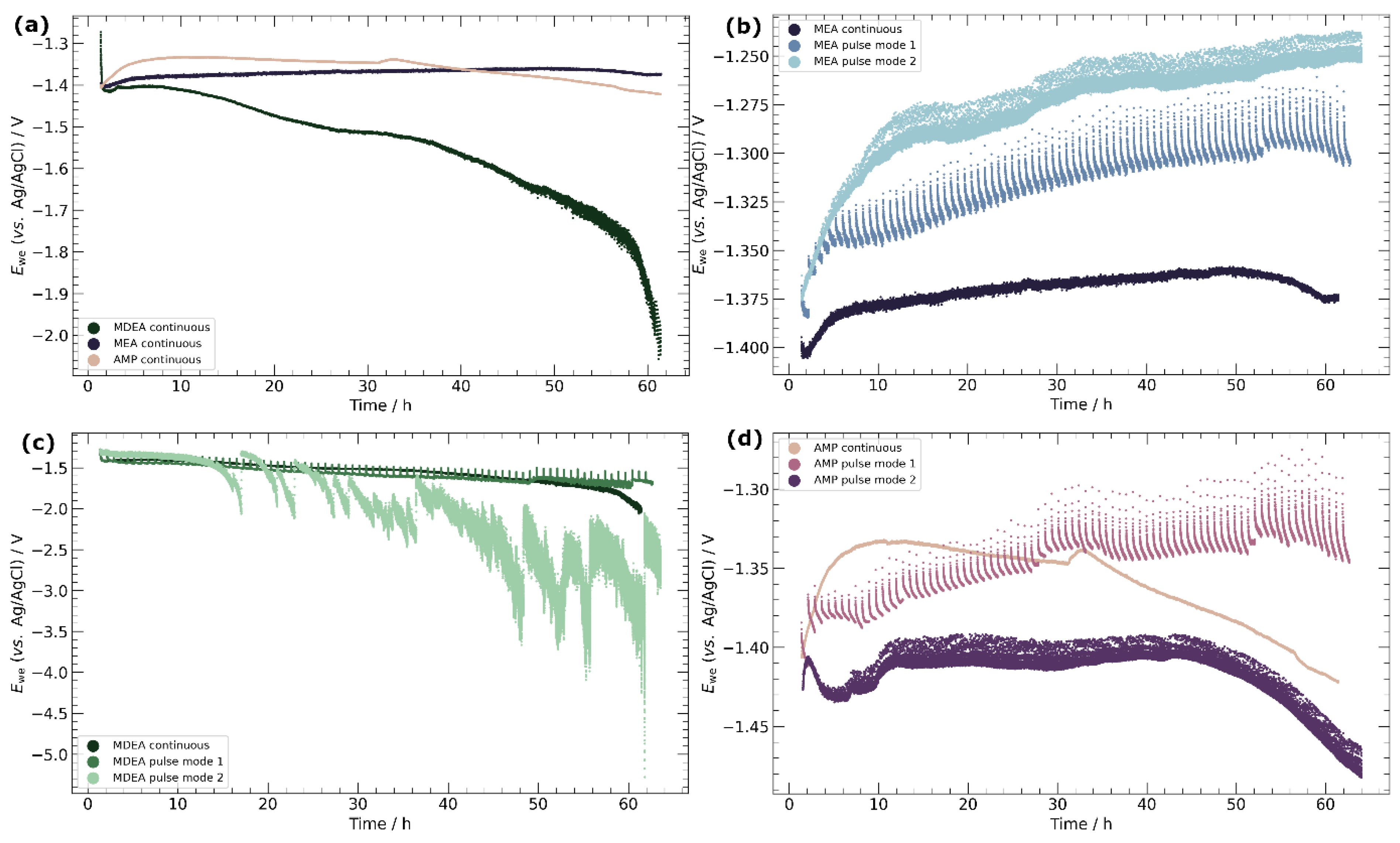
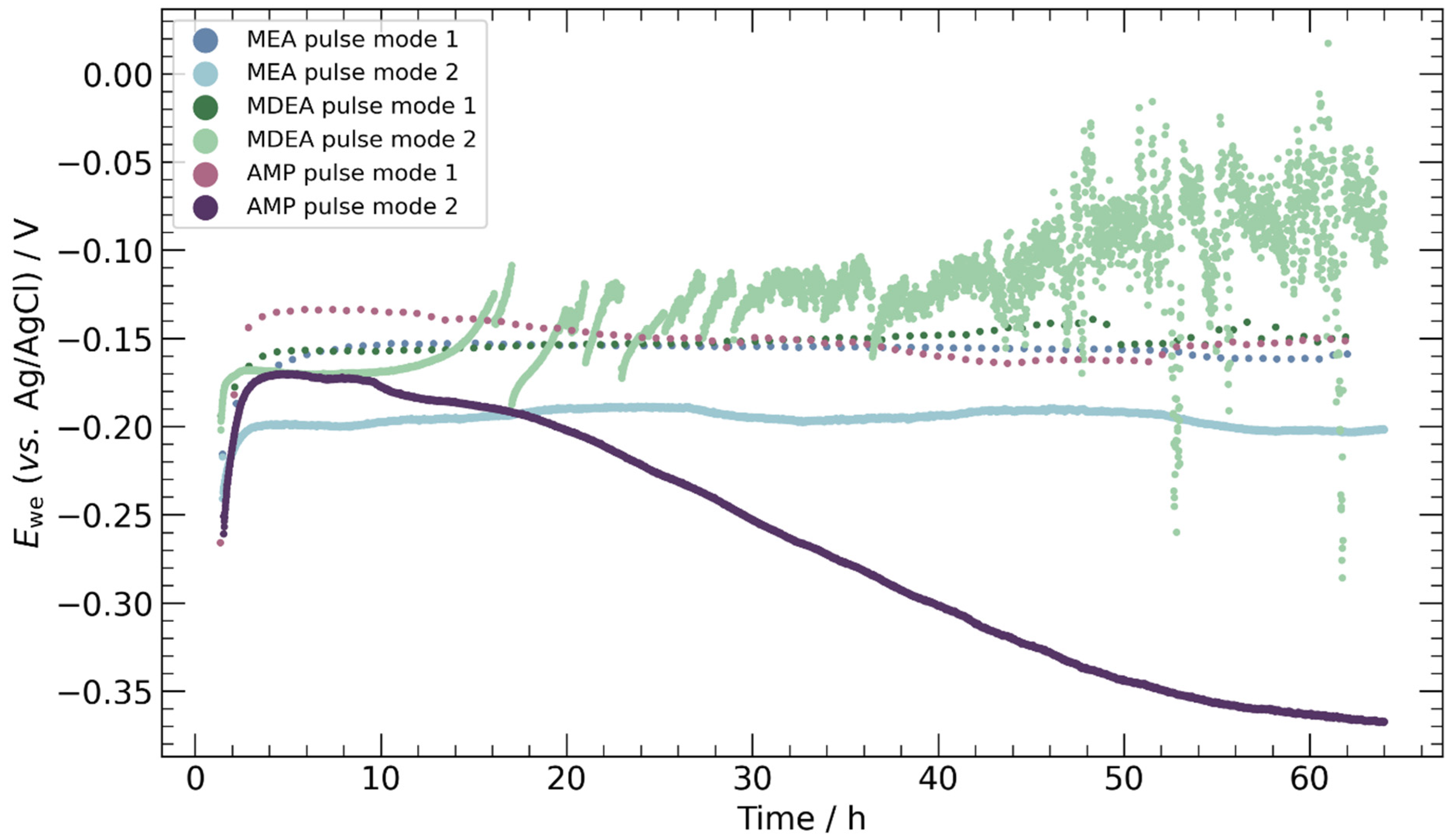
3.2. Chronopotentiometry
4. Discussion
4.1. Corrosion of Copper Catalyst in Amine Media
4.2. Choice of Amine Capture Media for ECR
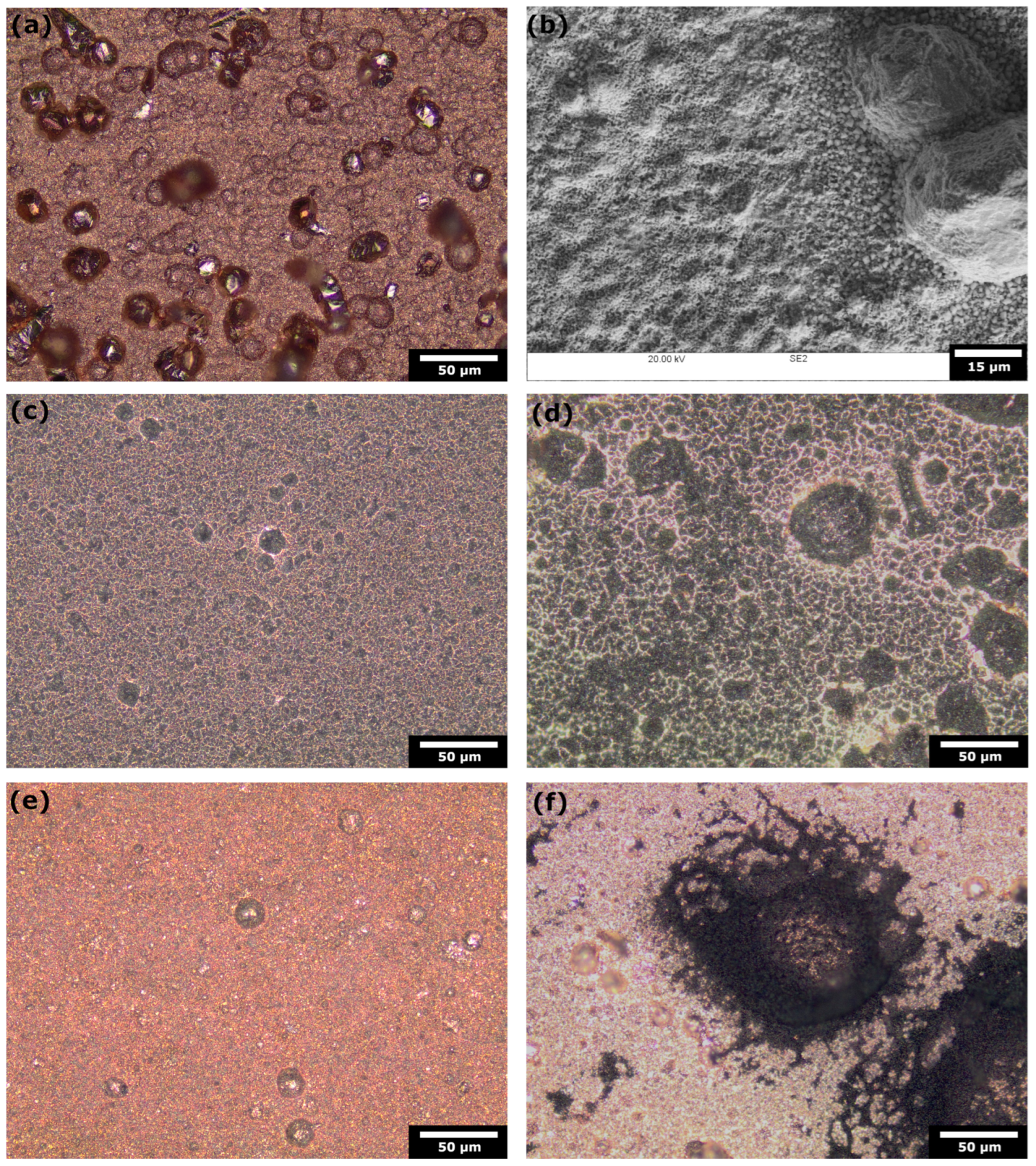
4.3. Pulse ECR in Amine Media
5. Conclusions
- Corrosion is not a significant impediment to the catalyst’s longevity in amine media. Both computational modeling and experimental data corroborate that the inherent corrosion of copper in these conditions does not critically limit the operational lifespan of the catalyst. This insight alleviates concerns regarding the durability of copper catalysts in practical ECR applications.
- Primary amines, particularly MEA, demonstrate higher compatibility with ECR processes, characterized by the absence of carbonate salt precipitation and more stable potentials over time. This observation emphasizes the importance of considering the amine type in optimizing ECR performance and underscores the potential for tailored catalyst–amine combinations to improve efficiency.
- Pulse ECR demonstrated significant potential in improving ECR stability, manifested by a shift in cathodic potential and effective mitigation of deposits on the catalyst surface through periodic oxidation. This highlights the importance of exploring innovative operational strategies to augment the stability and efficiency of ECR processes.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- van Soest, H.L.; den Elzen, M.G.J.; van Vuuren, D.P. Net-Zero Emission Targets for Major Emitting Countries Consistent with the Paris Agreement. Nat. Commun. 2021, 12, 2140. [Google Scholar] [CrossRef] [PubMed]
- Rochelle, G.T. Amine Scrubbing for CO2 Capture. Science 2009, 325, 1652–1654. [Google Scholar] [CrossRef] [PubMed]
- Boot-Handford, M.E.; Abanades, J.C.; Anthony, E.J.; Blunt, M.J.; Brandani, S.; Mac Dowell, N.; Fernández, J.R.; Ferrari, M.-C.; Gross, R.; Hallett, J.P.; et al. Carbon Capture and Storage Update. Energy Environ. Sci. 2014, 7, 130–189. [Google Scholar] [CrossRef]
- Jakobsen, J.B.; Rønne, M.H.; Daasbjerg, K.; Skrydstrup, T. Are Amines the Holy Grail for Facilitating CO2 Reduction? Angew. Chem. Int. Ed. 2021, 60, 9174–9179. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, I.; Goryachev, A.; Digdaya, I.A.; Li, X.; Atwater, H.A.; Vermaas, D.A.; Xiang, C. Coupling Electrochemical CO2 Conversion with CO2 Capture. Nat. Catal. 2021, 4, 952–958. [Google Scholar] [CrossRef]
- Wakerley, D.; Lamaison, S.; Wicks, J.; Clemens, A.; Feaster, J.; Corral, D.; Jaffer, S.A.; Sarkar, A.; Fontecave, M.; Duoss, E.B.; et al. Gas Diffusion Electrodes, Reactor Designs and Key Metrics of Low-Temperature CO2 Electrolysers. Nat. Energy 2022, 7, 130–143. [Google Scholar] [CrossRef]
- Nitopi, S.; Bertheussen, E.; Scott, S.B.; Liu, X.; Engstfeld, A.K.; Horch, S.; Seger, B.; Stephens, I.E.L.; Chan, K.; Hahn, C.; et al. Progress and Perspectives of Electrochemical CO2 Reduction on Copper in Aqueous Electrolyte. Chem. Rev. 2019, 119, 7610–7672. [Google Scholar] [CrossRef] [PubMed]
- Kibria, M.G.; Edwards, J.P.; Gabardo, C.M.; Dinh, C.-T.; Seifitokaldani, A.; Sinton, D.; Sargent, E.H. Electrochemical CO2 Reduction into Chemical Feedstocks: From Mechanistic Electrocatalysis Models to System Design. Adv. Mater. 2019, 31, 1807166. [Google Scholar] [CrossRef] [PubMed]
- García de Arquer, F.P.; Dinh, C.-T.; Ozden, A.; Wicks, J.; McCallum, C.; Kirmani, A.R.; Nam, D.-H.; Gabardo, C.; Seifitokaldani, A.; Wang, X.; et al. CO2 Electrolysis to Multicarbon Products at Activities Greater than 1 A Cm−2. Science 2020, 367, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, J.; Zheng, G. Designing Copper-Based Catalysts for Efficient Carbon Dioxide Electroreduction. Adv. Mater. 2021, 33, 2005798. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Zhang, J. Architectural Design for Enhanced C2 Product Selectivity in Electrochemical CO2 Reduction Using Cu-Based Catalysts: A Review. ACS Nano 2021, 15, 7975–8000. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Xue, S.; Barber, J.; Zhou, Y.; Meng, J.; Ke, X. An Overview of Cu-Based Heterogeneous Electrocatalysts for CO2 Reduction. J. Mater. Chem. A Mater. 2020, 8, 4700–4734. [Google Scholar] [CrossRef]
- Popović, S.; Smiljanić, M.; Jovanovič, P.; Vavra, J.; Buonsanti, R.; Hodnik, N. Stability and Degradation Mechanisms of Copper-Based Catalysts for Electrochemical CO2 Reduction. Angew. Chem. Int. Ed. 2020, 59, 14736–14746. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Hörmann, N.; Oveisi, E.; Loiudice, A.; De Gregorio, G.L.; Andreussi, O.; Marzari, N.; Buonsanti, R. Potential-Induced Nanoclustering of Metallic Catalysts during Electrochemical CO2 Reduction. Nat. Commun. 2018, 9, 3117. [Google Scholar] [CrossRef]
- Simon, G.H.; Kley, C.S.; Roldan Cuenya, B. Potential-Dependent Morphology of Copper Catalysts During CO2 Electroreduction Revealed by In Situ Atomic Force Microscopy. Angew. Chem. Int. Ed. 2021, 60, 2561–2568. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Arán-Ais, R.M.; Jeon, H.S.; Roldan Cuenya, B. Rational Catalyst and Electrolyte Design for CO2 Electroreduction towards Multicarbon Products. Nat. Catal. 2019, 2, 198–210. [Google Scholar] [CrossRef]
- Grosse, P.; Gao, D.; Scholten, F.; Sinev, I.; Mistry, H.; Cuenya, B.R. Dynamic Changes in the Structure, Chemical State and Catalytic Selectivity of Cu Nanocubes during CO2 Electroreduction: Size and Support Effects. Angew. Chem. Int. Ed. 2018, 57, 6192–6197. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Lee, S.Y.; Lee, C.W.; Cho, M.K.; Won, D.H.; Kim, C.; Oh, H.-S.; Min, B.K.; Hwang, Y.J. Electrochemical Fragmentation of Cu2O Nanoparticles Enhancing Selective C–C Coupling from CO2 Reduction Reaction. J. Am. Chem. Soc. 2019, 141, 4624–4633. [Google Scholar] [CrossRef] [PubMed]
- Popovic, S.; Bele, M.; Hodnik, N. Reconstruction of Copper Nanoparticles at Electrochemical CO2 Reduction Reaction Conditions Occurs via Two-Step Dissolution/Redeposition Mechanism. ChemElectroChem 2021, 8, 2634–2639. [Google Scholar] [CrossRef]
- Garg, S.; Xu, Q.; Moss, A.B.; Mirolo, M.; Deng, W.; Chorkendorff, I.; Drnec, J.; Seger, B. How Alkali Cations Affect Salt Precipitation and CO2 Electrolysis Performance in Membrane Electrode Assembly Electrolyzers. Energy Environ. Sci. 2023, 16, 1631–1643. [Google Scholar] [CrossRef]
- Xu, Y.; Edwards, J.P.; Liu, S.; Miao, R.K.; Huang, J.E.; Gabardo, C.M.; O’Brien, C.P.; Li, J.; Sargent, E.H.; Sinton, D. Self-Cleaning CO2 Reduction Systems: Unsteady Electrochemical Forcing Enables Stability. ACS Energy Lett. 2021, 6, 809–815. [Google Scholar] [CrossRef]
- Sassenburg, M.; Kelly, M.; Subramanian, S.; Smith, W.A.; Burdyny, T. Zero-Gap Electrochemical CO2 Reduction Cells: Challenges and Operational Strategies for Prevention of Salt Precipitation. ACS Energy Lett. 2023, 8, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Obasanjo, C.A.; Gao, G.; Khiarak, B.N.; Pham, T.H.; Crane, J.; Dinh, C.-T. Progress and Perspectives of Pulse Electrolysis for Stable Electrochemical Carbon Dioxide Reduction. Energy Fuels 2023, 37, 13601–13623. [Google Scholar] [CrossRef]
- Obasanjo, C.A.; Zeraati, A.S.; Shiran, H.S.; Nguyen, T.N.; Sadaf, S.M.; Kibria, M.G.; Dinh, C.-T. In Situ Regeneration of Copper Catalysts for Long-Term Electrochemical CO2 Reduction to Multiple Carbon Products. J. Mater. Chem. A Mater. 2022, 10, 20059–20070. [Google Scholar] [CrossRef]
- Zhang, X.-D.; Liu, T.; Liu, C.; Zheng, D.-S.; Huang, J.-M.; Liu, Q.-W.; Yuan, W.-W.; Yin, Y.; Huang, L.-R.; Xu, M.; et al. Asymmetric Low-Frequency Pulsed Strategy Enables Ultralong CO2 Reduction Stability and Controllable Product Selectivity. J. Am. Chem. Soc. 2023, 145, 2195–2206. [Google Scholar] [CrossRef] [PubMed]
- Jeon, H.S.; Timoshenko, J.; Rettenmaier, C.; Herzog, A.; Yoon, A.; Chee, S.W.; Oener, S.; Hejral, U.; Haase, F.T.; Roldan Cuenya, B. Selectivity Control of Cu Nanocrystals in a Gas-Fed Flow Cell through CO2 Pulsed Electroreduction. J. Am. Chem. Soc. 2021, 143, 7578–7587. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.N.; Chen, Z.; Zeraati, A.S.; Shiran, H.S.; Sadaf, S.M.; Kibria, M.G.; Sargent, E.H.; Dinh, C.-T. Catalyst Regeneration via Chemical Oxidation Enables Long-Term Electrochemical Carbon Dioxide Reduction. J. Am. Chem. Soc. 2022, 144, 13254–13265. [Google Scholar] [CrossRef] [PubMed]
- Yano, J.; Yamasaki, S. Pulse-Mode Electrochemical Reduction of Carbon Dioxide Using Copper and Copper Oxide Electrodes for Selective Ethylene Formation. J. Appl. Electrochem. 2008, 38, 1721–1726. [Google Scholar] [CrossRef]
- Cofell, E.R.; Park, Z.; Nwabara, U.O.; Harris, L.C.; Bhargava, S.S.; Gewirth, A.A.; Kenis, P.J.A. Potential Cycling of Silver Cathodes in an Alkaline CO2 Flow Electrolyzer for Accelerated Stress Testing and Carbonate Inhibition. ACS Appl. Energy Mater. 2022, 5, 12013–12021. [Google Scholar] [CrossRef]
- Abdinejad, M.; Mirza, Z.; Zhang, X.; Kraatz, H.-B. Enhanced Electrocatalytic Activity of Primary Amines for CO2 Reduction Using Copper Electrodes in Aqueous Solution. ACS Sustain. Chem. Eng. 2020, 8, 1715–1720. [Google Scholar] [CrossRef]
- Bohlen, B.; Daems, N.; Breugelmans, T. Electrochemical Production of Formate Directly from Amine-Based CO2 Capture Media. ECS Meet. Abstr. 2023, MA2023-01, 1722. [Google Scholar] [CrossRef]
- Hossain, M.N.; Ahmad, S.; da Silva, I.S.; Kraatz, H.-B. Electrochemical Reduction of CO2 at Coinage Metal Nanodendrites in Aqueous Ethanolamine. Chem.—Eur. J. 2021, 27, 1346–1355. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Li, F.; Zhang, Y.; Bentley, C.L.; Horne, M.; Bond, A.M.; Zhang, J. Electrochemical Reduction of Carbon Dioxide in a Monoethanolamine Capture Medium. ChemSusChem 2017, 10, 4109–4118. [Google Scholar] [CrossRef] [PubMed]
- Filotás, D.; Nagy, T.; Nagy, L.; Mizsey, P.; Nagy, G. Extended Investigation of Electrochemical CO2 Reduction in Ethanolamine Solutions by SECM. Electroanalysis 2018, 30, 690–697. [Google Scholar] [CrossRef]
- Frailie, P.T.; Rochelle, G.T. Kinetics of Aqueous Methyldiethanolamine/Piperazine for CO2 Capture. In Process Systems and Materials for CO2 Capture; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2017; pp. 137–152. ISBN 9781119106418. [Google Scholar]
- Sartori, G.; Savage, D.W. Sterically Hindered Amines for Carbon Dioxide Removal from Gases. Ind. Eng. Chem. Fundam. 1983, 22, 239–249. [Google Scholar] [CrossRef]
- Trethewey, K.R.; Chamberlain, J. Corrosion for Science and Engineering, 2nd ed.; NACE International: Houston, TX, USA, 1995. [Google Scholar]
- Beverskog, B.; Puigdomenech, I. Revised Pourbaix Diagrams for Copper at 25 to 300 °C. J. Electrochem. Soc. 1997, 144, 3476. [Google Scholar] [CrossRef]
- Abd el Haleem, S.M.; Ateya, B.G. Cyclic Voltammetry of Copper in Sodium Hydroxide Solutions. J. Electroanal. Chem. Interfacial Electrochem. 1981, 117, 309–319. [Google Scholar] [CrossRef]
- ASTM G102-89; Standard Practice for Calculation of Corrosion Rates and Related Information from Electrochemical Measurements. ASTM International: West Conshohocken, PA, USA, 2004.
- Li, C.W.; Kanan, M.W. CO2 Reduction at Low Overpotential on Cu Electrodes Resulting from the Reduction of Thick Cu2O Films. J. Am. Chem. Soc. 2012, 134, 7231–7234. [Google Scholar] [CrossRef]
- MacDowell, N.; Florin, N.; Buchard, A.; Hallett, J.; Galindo, A.; Jackson, G.; Adjiman, C.S.; Williams, C.K.; Shah, N.; Fennell, P. An Overview of CO2 Capture Technologies. Energy Environ. Sci. 2010, 3, 1645–1669. [Google Scholar] [CrossRef]
- Bougie, F.; Iliuta, M.C. Sterically Hindered Amine-Based Absorbents for the Removal of CO2 from Gas Streams. J. Chem. Eng. Data 2012, 57, 635–669. [Google Scholar] [CrossRef]
- Edali, M.; Aboudheir, A.; Idem, R. Kinetics of Carbon Dioxide Absorption into Mixed Aqueous Solutions of MDEA and MEA Using a Laminar Jet Apparatus and a Numerically Solved 2D Absorption Rate/Kinetics Model. Int. J. Greenh. Gas Control 2009, 3, 550–560. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Media | CO2 | Ecorr/V | jcorr/µA cm−2 | Rp/kΩ cm2 |
|---|---|---|---|---|
| MEA | no | −0.58 ± 0.01 | 2.4 ± 0.1 | 3.8 ± 0.7 |
| yes | −0.29 ± 0.02 | 14.1 ± 6.9 | 0.7 ± 0.2 | |
| MDEA | no | −0.42 ± 0.01 | 0.3 ± 0.1 | 19.0 ± 4.4 |
| yes | −0.33 ± 0.03 | 1.0 ± 0.1 | 10.4 ± 0.5 | |
| MDEA/PZ | no | −0.53 ± 0.01 | 0.4 ± 0.1 | 18.3 ± 6.3 |
| yes | −0.39 ± 0.01 | 0.9 ± 0.6 | 12.6 ± 11.3 | |
| AMP | no | −0.57 ± 0.01 | 1.5 ± 0.3 | 5.6 ± 1.1 |
| yes | −0.29 ± 0.02 | 7.7 ± 1.3 | 1.0 ± 0.6 | |
| KCl | no | −0.20 ± 0.03 | 2.4 ± 0.6 | 3.7 ± 1.1 |
| Media | CO2 | A/V | B/V | C/V | D/V |
|---|---|---|---|---|---|
| MEA | no | −0.5 | −0.4 | −0.48 | −1.2 |
| yes | −0.21 | −0.22 | - | - | |
| MDEA | no | −0.46 | −0.27 | −0.1 | −0.97 |
| yes | −0.27 | −0.27 | - | - | |
| MDEA/PZ | no | −0.44 | −0.15 | −0.47 | −0.75 |
| yes | −0.17 | −0.12 | - | - | |
| AMP | no | −0.52 | −0.27 | −0.51 | −1.1 |
| yes | −0.25 | −0.18 | - | - | |
| KCl | no | −0.4 | −0.1 | −0.06 | −0.95 |
| Dissolution into Cu(I) | Dissolution into Cu(II) | ||||
|---|---|---|---|---|---|
| Media | CO2 | CR/mm y−1 | Time/Days | CR/mm y−1 | Time/Days |
| MEA | no | 0.056 | 328 | 0.028 | 655 |
| yes | 0.327 | 56 | 0.164 | 112 | |
| MDEA | no | 0.007 | 2623 | 0.003 | 5242 |
| yes | 0.023 | 787 | 0.012 | 1573 | |
| MDEA/PZ | no | 0.009 | 1967 | 0.005 | 3931 |
| yes | 0.021 | 874 | 0.010 | 1747 | |
| AMP | no | 0.035 | 525 | 0.017 | 1048 |
| yes | 0.179 | 102 | 0.089 | 204 | |
| KCl | no | 0.056 | 328 | 0.028 | 655 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Penot, C.; Maniam, K.K.; Paul, S. Electrochemical Characterization of Electrodeposited Copper in Amine CO2 Capture Media. Materials 2024, 17, 1825. https://doi.org/10.3390/ma17081825
Penot C, Maniam KK, Paul S. Electrochemical Characterization of Electrodeposited Copper in Amine CO2 Capture Media. Materials. 2024; 17(8):1825. https://doi.org/10.3390/ma17081825
Chicago/Turabian StylePenot, Corentin, Kranthi Kumar Maniam, and Shiladitya Paul. 2024. "Electrochemical Characterization of Electrodeposited Copper in Amine CO2 Capture Media" Materials 17, no. 8: 1825. https://doi.org/10.3390/ma17081825
APA StylePenot, C., Maniam, K. K., & Paul, S. (2024). Electrochemical Characterization of Electrodeposited Copper in Amine CO2 Capture Media. Materials, 17(8), 1825. https://doi.org/10.3390/ma17081825