Abstract
Camellia nitidissima Chi, is a rare and endangered plant that is narrowly distributed in South China and North Vietnam. In this study, seven polymorphic microsatellite markers were used to investigate the genetic diversity, recent population bottlenecks as well as population structure of twelve remnant populations of the plant. Our results indicated that, despite their severely fragmented natural range, C. nitidissima remnants maintained a moderate level of genetic variability, and only a bottlenecked population was detected by the clear evidences. No significant correlation was found between genetic diversity and population size. Significantly high genetic differences among populations were found, and the twelve populations could be classified into two distinct genetic groups. AMOVA indicated that 16.14% (16.73%, after one suspected artificial population was excluded) of the molecular variation was attributable to regional divergences (between Nanning and Fangcheng), and the majority of genetic variation existed within populations which were 69.24% (70.63%, after one suspected artificial population was excluded). For conservation management plans, the genetic resources of the two distinct groups are of equal importance for conservation, separate management unit for each of them should be considered. Given that all remnant populations are small and isolated, and many plants are illegally dug out for commercial purposes, management efforts in terms of habitat protection and legal protection, as well as transplantations and reintroductions, would be necessary for this species.
1. Introduction
In nature, rare and/or endangered species typically exist in small and geographically isolated populations [1]. Empirical studies have shown that, for a number of plant species, being in isolation or in small populations reduced genetic variation and fitness [2,3,4,5,6,7,8]. In principle, small population size and increased isolation tend to restrict the exchange of pollen and seed, thereby reducing interpopulation gene flow. Additionally, small populations are more prone to inbreeding and genetic drifts [9,10,11]. It is believed that restricted gene flow, inbreeding, and genetic drift can cause the loss of genetic diversity of populations and subsequently reduce a population’s ability to adapt to changing environments while increasing its susceptibility to disease and pests [12]. Sometimes, however, small and isolated populations may have normal or even enhanced gene flow without suffering from genetic erosion [13,14]; hence, inferences regarding genetic variation in rare or endangered species must be made with caution. Genetic diversity patterns are attributable to many factors, such as a species’ mating system as well as its demographic or life history [15,16]. Therefore, understanding genetic factors that increase the risks of extinction for particular species is critically important for their conservation [15,17].
Camellia nitidissima C. W. Chi (Theaceae) is a rare and endangered evergreen tree that is narrowly distributed in both the Guangxi Province of South China, as well as in North Vietnam [18]. It is a diploid tree (2 n = 30) [19] that grows in moist, shady habitats, but it tends to avoid ones with strong direct sunlight. It also usually grows to 2 to 3 m tall (Supplementary Figure S1a online), although it can reach up to 6 m [20]. It produces large (diameter: 1.2–2.3 cm), scented single axillary flowers which are cup-shaped, with many yellow petals and dozens of stamens with orange anthers (Supplementary Figure S1c online). The flowers, which blossom from November to March [21], are insect-pollinated, and pollination is primarily by bees [22]. It is likely that the plant relies on the bright yellow color of its petals to attract insects in order to promote allogamy. In South China, fruits of C. nitidissima set in the spring and ripen from October to December [21,23], while its large and heavy seeds (1.73–2.16 cm long and 1.94–2.5 cm in diameter, 2.3–3.5 g in weight) [21] have thick pericarps and seedcoats that help them avoid desiccation in dry climates and insulate them from frost in cold winters [23]. These seeds are mainly dispersed by gravity and occasionally by water (personal observation). This species has serious reproductive disadvantages, such as low seed productivity and low germination rates (c. 30%, when seeds are on the soil surface) [23].
C. nitidissima was first discovered in Fangcheng County in 1933, but, despite being known to the public since 1948, it initially received no attention from the public or horticulturists until the early 1960s when it was again found in Yongning County [24]. It is one of several camellias with yellow flowers, and, with its big size, golden color, and the transparent waxy appearance of its flowers, this species was honored as “the queen of camellia” [25]. As a result, it was introduced as an ornamental plant in many countries, where it has attracted the attention of gardeners worldwide [26,27].
In China, natural populations of camellias, including C. nitidissima, have been extensively investigated for many years [28], and, so far, only two disjunctive areas with C. nitidissima have been found in Guangxi, with the first being at the junction of Fushu, Longan, and Fusui, near the city of Nanning, and the other one located in Fangcheng, to the south of Mount Shiwan [18]. Most of the populations of C. nitidissima exist in residual forests or secondary forests [29]. Although the historic distribution range and population size of C. nitidissima remain unknown, it is clear that the species experienced a rapid decline due to increasing anthropogenic pressures on its natural habitat over the last few decades [23,30]. Moreover, despite its threatened status, C. nitidissima remains popular for horticultural trades. As a result, large numbers of its seeds are illegally collected, and its plants are also illegally dug out in the wild. Recently, C. nitidissima has been included in the checklist of the State Protection Category I in China [31].
To support conservation and management programs for C. nitidissima, information about its genetic variability and population structure in natural populations is necessary, and it has led to several molecular marker-based genetic studies to investigate the genetic variation of its remnant populations [21,28]. Combined analysis of RAPD and AFLP markers showed that C. nitidissima populations could be classified into two major genetic groups corresponding to the Nanning and Fangcheng areas, while Mantel tests revealed significant correlations between the genetic and geographical distances of C. nitidissima populations [28]. More recently, a population genetic study using ISSR markers indicated a low level of genetic diversity at both the species and population levels but a relatively high degree of differentiation among natural populations [21]. Detailed information on population genetics (e.g., breeding system, genetic bottlenecks), therefore, remained unresolved by these studies. Additionally, these previous studies may have underestimated the genetic consequences of past demographic events as they used traditional standard approaches for assessing population structure [32,33]. In contrast, nuclear microsatellite markers (SSR) are powerful tools for the study of population genetics because of their high polymorphism, codominant transmission, and presumably neutral and extensive genome coverage [34]. The polymorphic microsatellite markers pre-selected for C. nitidissima by Wei et al. [35] have now allowed us to conduct a detailed study of the population genetics of the species.
Here we use seven polymorphic microsatellite markers which have been pre-selected for C. nitidissima by Wei et al. [35] to conduct the detailed study of the population genetics of the species. The main objectives were as follows: (1) to examine the levels and patterns of genetic diversity of the species. Given its small population size and restricted geographic distribution, we expected C. nitidissima populations to be genetically impoverished; (2) to test whether the species has experienced genetic bottleneck; (3) to assess the genetic structure among populations in order to identify management units. Such information can have important implications for assessing the suitability of current-available conservation and management programs for this endangered species as well as for devising new conservation strategies.
2. Materials and Methods
2.1. Sample Collection
Leaf samples from 385 C. nitidissima individuals were collected between August and October 2008 from twelve geographically isolated extant populations across the entire distribution range of the species in Guangxi, China (Table 1 and Figure 1a). The NZS population consisted of three subpopulations which were distributed along the same slope but at different altitudes of mount Nazi (Table 1). However, the geographical coordinates (latitude and longitude locations) are not provided here because of the species’ threatened status and economic value. Overall, these populations could be divided into two regions separated by about 117–148 km [21], with four populations being to the west of Nanning and the remaining eight located around Fangcheng (Table 1). Three of the twelve populations (Table 1) were located in the protected area where C. nitidissima is currently preserved, and, within each region, the populations were separated from each other by 3 to 40 km. Population sizes were estimated in the field by inspection, and, depending on the results as well as accessibility to the areas, the sample sizes ranged from 11 for ZD and to 89 for NZS (30, 29, and 30 individuals respectively sampled for NZS-1, NZS-2, and NZS-3) (Table 1). After sample collection, all leaves were dried in silica gel in sealed polyethylene bags prior to storage at room temperature until genomic DNA was extracted.

Table 1.
Details of remnant populations of Camellia nitidissima in South China.
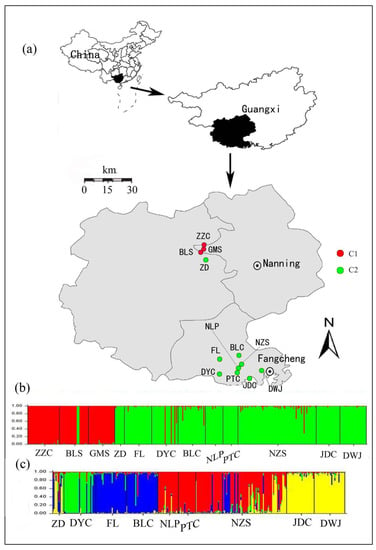
Figure 1.
Map showing the distribution of samples and Bayesian-based clusters for Camellia nitidissima performed using STRUCTURE. (a) The geographical distributions of the 12 C. nitidissima population; (b) Bayesian clustering of all individuals in the 12 populations (Names are shown in Table 1). The color bars represent the probability of assigning an individual to a particular cluster. The populations could be divided into two genetic groups (C1 and C2); (c) The assignment results for the nine populations in C2.
2.2. DNA Extraction and Microsatellite Analysis
Total DNA was extracted by following the CTAB method of Doyle [36]. The quality and quantity of the DNA were determined by electrophoresis on 1% agarose gels, while microsatellite genotyping was performed according to the method of Wei et al. [35] at seven loci (CamsinM3, CamsinM4, CamsinM5, MSCjaF37, MSCjaH38, MSCjaH46, and P12). Briefly, this involved polymerase chain reaction (PCR)-based amplifications which were performed in 10 µL reaction mixtures consisting of 5 ng of template DNA, 50 mM KCl, 20 mM Tris-HCl (pH 8.0), 1.5 mM MgCl2, 0.5 µM of each primer, 0.2 mM of each dNTP, and 1U of Taq DNA polymerase (Takara). The amplification procedure was carried out as described by Wei et al. [35] (4 min at 94 °C, followed by 35 cycles of 94 °C for 30 s, 51–63 °C depending on locus annealing temperature for 30 s, and 72 °C for 45 s, followed by 10 min at 72 °C), and PCR was performed on a LabCycler Gradient thermocycler (SensoQuest, Gottingen, Germany). PCR products were then resolved on 4% denaturing polyacrylamide gels and visualized by silver staining. In each polyacrylamide gel electrophoresis (PAGE), the same ladders plus the same 5 samples were used for all PAGEs as references. For the samples whose alleles were different by one base pair and were not confirmed in a run, they were chosen to be put together to run in the same PAGE.
2.3. Data Analysis
All conversions of the format of the genetic data were performed using the software CONVERT 1.3 [37] and GenALEx 6.3 [38]. The Ewens–Watterson test for neutrality of polymorphic markers was then conducted using Manly’s algorithm [39] as implemented in PopGene 1.31 [40] with 1000 simulated samples. The observed number of alleles (NA), the number of effective alleles (NE), the number of private alleles (NP), the proportion of polymorphic loci (P), the observed heterozygosity (HO), Nei’s unbiased expected heterozygosity (UHE) [41], and Wright’s inbreeding coefficient (FIS) [42] per (sub)population were subsequently calculated using the program GenALEx 6.3 [38] before testing the Hardy–Weinberg equilibrium (HWE) per (sub)population by using FSTAT 2.9.3 [43]. The FSTAT 2.9.3 [43] was also used to estimate allelic richness (AR). In addition, FIS per population, while considering the frequency of null alleles, was estimated with INEST 2.0 [44], applying the default Bayesian approach using 300,000 steps, sampling every 100 steps, and discarding the first 30,000 steps as burn-in. Then, FREENA [45] was used to estimate null allele frequencies for each population and locus while the (sub)population differentiation was measured by FST [46] for each pair of (sub)populations with the excluding null allele (ENA) correction. The software was then also used to estimate the global FST [46], both with and without ENA correction, in order to determine the influence of null alleles. The ENA correction method corrects for the positive bias introduced by the presence of null alleles. Correlation analyses between population size and parameters of genetic variation were eventually conducted using SPSS 19.0 for Windows (SPSS lnc., Chicago, IL, USA), and two-tailed analyses of correlation were performed using Pearson’s tests. The software was also used to test for significant differences between the mean Fst values within each region and between them, and the data were analyzed by performing one-way analysis of variance (ANOVA) followed by Duncan’s post-hoc test.
In addition, using BOTTLENECK 1.2.02 [47], Wilcoxon tests (two-tailed) for heterozygote excess were performed under the Infinite Allele Model (IAM), the Stepwise Mutation Model (SMM), and the Two-Phase Model (TPM) in order to examine potential bottlenecks. The IAM considers any point mutation along a stretch of DNA within a locus to constitute a new allele, whereas the SMM counts new alleles along a stretch of DNA with respect to the addition or subtraction of particular subsets of DNA motifs [48]. Moreover, the TPM has been proposed as an “intermediate” model that provides a more realistic picture of how some DNA sequences evolve [49]. Under the TPM, 70% and 30% of the mutations were assumed to occur under the SMM and the IAM, respectively. For each mutational model, 10,000 replicates were performed. By using the same software, the mode-shift indicator test was also used for detecting potential bottlenecks. The non-bottlenecked populations that are near mutation-drift equilibrium have a large proportion of alleles at low frequencies. In this test, the microsatellite alleles are grouped into ten frequency classes to investigate whether the distribution followed the normal L-shaped form, where alleles with low frequencies are the most numerous.
To evaluate the relationships among populations, a Bayesian cluster analysis, implemented in STRUCTURE 2.3.1 [50], was performed to assign individuals into clusters based on their multilocus genotypes. Using an admixture model with correlated allele frequencies among populations, 10 independent runs were performed for each K (putative cluster numbers, from 1 to 12), with 106 iterations after a burn-in period of 106 steps. However, since Ln Pr(X|K) does not reliably identify the optimal number of clusters, another ad hoc criterion, namely the ΔK [51], was calculated to determine the optimal K. CLUMPP 1.1.2 [52] was further used to calculate the average membership coefficient for each individual by aligning and converging the results from the above 10 runs. As all individuals could be assigned to two genetic groups (the red cluster and the green cluster in Figure 1b), as identified by STRUTURE at the highest hierarchical level, and, at the same time, nine populations were completely assigned to the green cluster (Figure 1b), we eventually repeated the above analyses for this cluster with K from 1 to 9 in order to understand the genetic structure in detail.
Furthermore, hierarchical analysis of molecular variance (AMOVA) was carried out with the software Arlequin 3.0 [53] to quantify the partitioning of genetic variance between regional groups (Nanning and Fangcheng), among populations within regional groups, as well as within populations.
Mantel tests were carried out using GenALEx 6.3 [38] to test for the significance of isolation by distance. First, a matrix of pairwise FST/(1 − FST) (FST was corrected by the ENA) values between populations was generated, and subsequently, it was compared against a matrix of geographic distance [54]. In this case, geographical distances between pairs of populations were calculated based on longitudes and latitudes using the Spheroidal Distance function in the Mathematica software (Wolfram Research, Champaign, IL, USA). This software generates the distance between two points on the earth (in km) based on the spheroidal model of the planet. Evidence of significant isolation by distance was then indicated by a significant positive correlation between pairwise FST/(1 − FST) values and the geographic distances.
It should, however, be noted that since the ZD population anomalously clustered with the group from the Fangcheng region (see results) (Figure 1b), it was suspected to have been artificially introduced. Thus, all of the above genetic parameters were also analyzed after excluding the ZD population from the dataset.
3. Results
3.1. Test of Neutrality
The neutrality test for all loci showed no significant differences (data not shown), both including or excluding the ZD population, hence indicating that the allele distribution was in accordance with the assumption of selective neutrality.
3.2. Genetic Diversity
A total of 130 alleles at 7 microsatellite loci were identified in the 385 C. nitidissima individuals. The number of observed alleles per population ranged from 4.286 in PTC to 11.857 in NZS, with a mean number of 6.583. Twenty-seven private alleles were also detected in 10 of the 12 populations (Table 2), and about 20.24% of the null allele frequencies were greater than 0.1 for each population and locus. The values of allelic richness among populations ranged from 2.478 to 3.679, with an average value of 3.033 (Table 2). It was also found that the value of genetic diversity was highest in the ZD population (UHE = 0.757) but lowest in the BLS population (UHE = 0.482), while three small populations, namely FL, DYC and JDC (Table 1), showed moderate UHE values when comparing with other populations (Table 2). In terms of the inbreeding coefficient, FIS values calculated in GenALEx 6.3 [38] (FIS-GenALEx) for the populations ranged from −0.031 to 0.324, with an average value of 0.126 (Table 2). In particular, eight of the twelve populations, along with three subpopulations of NZS, deviated significantly from HWE, and the FIS-GenALEx values of all these deviations were positive (Table 2), indicating heterozygote deficiencies. FIS estimates in INEST 2.0 [44] (FIS-INest) varied from 0.064 to 0.289, and the values of their 95% HDPI were positive and did not include zero (Table 2). Furthermore, when the ZD population was excluded, the mean values for HO, UHE, and FIS-GenALEx were 0.539, 0.608, and 0.107, respectively (Table 2). Correlation analyses also found that observed heterozygosity, unbiased expected heterozygosity, and inbreeding coefficient, both with and without the ZD population, were not related to the population size (with the ZD population: R = 0.286, p = 0.368 for HO, R = 0.192, p = 0.551 for UHE, R = −0.032, p = 0.922 for FIS-GenALEx; after excluding the ZD population: R = 0.249, p = 0.461 for HO, R = 0.506, p = 0.112 for UHE, R = 0.204, p = 0.547 for FIS-GenALEx. NZS population was not divided into three subpopulations in both cases).
Table 2.
Genetic variation and test of bottleneck effects for Camellia nitidissima.
3.3. Test for Bottleneck Effects
A significant excess of heterozygosity was detected in five and four (sub)populations under the assumptions of the IAM and SMM models, respectively, while similar results were obtained in only one population under that of the TPM model (Table 2). None of the (sub)populations simultaneously displayed significant excess of heterozygosity in all three models (Table 2).
As a second method to detect potential bottlenecks, the mode-shift indicator test indicated the abundance of low frequency (<0.10) alleles for every (sub)population (Supplementary Table S1 and Figure S2 online) along with a normal L-shaped graph for every (sub)population but PTC (Supplementary Figure S2 online).
3.4. Genetic Structure
The global measure of FST with and without the ZD population changed from 0.199 to 0.190 and 0.203 to 0.194, respectively, after excluding null allele (ENA) correction (Table 2). The corrected pairwise FST values were all significantly high (p < 0.05, Table 3). More specifically, three populations (ZZC, BLS, and GMS) from the Nanning region showed higher genetic differentiation when compared with each other and other populations. Similarly, significantly higher genetic differentiation was also found between populations from Nanning and Fangcheng regions, especially after excluding the ZD population, unlike the case when populations within the regions were compared (Table 3).
Table 3.
Pairwise estimated values of FST among (sub)populations of Camellia nitidissima.
With increasing K numbers from 1 to 12 in STRUCTURE, the value of ln Pr(X|K) increased continuously without forming a plateau, but ΔK showed a large peak at K = 2 (Supplementary Figure S3 online). At this value of K (K = 2), most individuals from the Nanning region, as well as several ones from the DYC population, were assigned to one cluster (C1, red), while those from the region of Fangcheng except several individuals, along with the ZD population and several individuals of BLS population from Nanning region, clustered separately (C2, green) (Figure 1b). Furthermore, within the BLC and NZS populations, some individuals showed a certain degree of genetic admixture (Figure 1b). Further analyses indicated that the nine populations within the C2 could be further divided into four clusters (optimal K = 4, Supplementary Figure S4 online), namely the green cluster (C2-I), the blue cluster (C2-II), the red cluster (C2-III) and the yellow cluster (C2-IV), and genotypes from different populations are merged together in the NZS population (Figure 1c).
AMOVA results, summarized in Table 4, indicated that 16.14% and 16.73% of molecular variation (after testing with and without the ZD population, respectively) could be attributed to regional differences between the regions of Nanning and Fangcheng. However, most of the molecular variance (69.24 and 70.63% corresponding to test results with and without the ZD population, respectively) occurred within populations. In addition, results of Mantel tests, both with and without the ZD population, indicated that there was a significant “isolation by distance” pattern when all the populations were analyzed together (R = 0.525, p = 0.040 with the ZD population and R = 0.818, p = 0.010 after excluding the ZD population) (Figure S5a,b online). A similar pattern was observed when the analysis was performed for the populations of Fangcheng region (R = 0.473, p = 0.030) (Figure S5c online); however, it was not observed for the populations of the Nanning region (R = 0.485, p = 0.180 with the ZD population and R = 0.038, p = 0.470 after excluding the ZD population) (Figure S5d,e online).
Table 4.
Results of hierarchical AMOVA testing for Camellia nitidissima populations.
4. Discussion
4.1. Genetic Variation of C. nitidissima
Some studies have demonstrated that threatened and endangered species tend to possess low levels of genetic diversity [33,55,56,57,58], while others did not support similar conclusions [9,17,59,60,61]. In the case of this study, results from microsatellite analysis showed that C. nitidissima maintained a moderate level of genetic diversity, with a mean UHE value of 0.620 (0.608 after the ZD population was excluded) and more than 69% (70%, after the ZD population was excluded) of the genetic variation occurring among individuals within populations. Previously reported genetic diversities in C. nitidissima have been based on dominant molecular markers (RAPD, AFLP, ISSR), which yielded mean HE values of 0.1069, 0.1288, and 0.0831 for RAPD, AFLP, and ISSR markers, respectively [21,28]. These values were much lower in comparison to our results, but such inconsistencies based on microsatellites and dominant markers were also reported for another endangered species, Changiostyrax dolichocarpa, where its mean HE was 0.64 for microsatellites [62] and 0.13 for ISSR markers [63]. Fundamental differences between microsatellites and dominant markers likely account for these different estimates. To overcome such differences due to markers, comparative studies using the same molecular markers are therefore necessary to study genetic variations in endangered species. In the present study, our data showed that the genetic diversity in C. nitidissima (mean UHE = 0.620 (0.608, after the ZD population was excluded) Table 2) was relatively higher than that of many gravity-dispersed (mean HE = 0.500) [64] and narrowly distributed species (mean HE = 0.560) [64] but similar to that of its widespread congener C. sinensis (HE = 0.620) [65] which was studied using the same molecular markers. Thus, it is reasonable to believe that, despite severe fragmentation, C. nitidissima remnants maintained a moderate level of genetic diversity even though this result was not consistent with the hypothesis that C. nitidissima is genetically impoverished.
In fact, many tree species are probably resilient to habitat fragmentation within one or two generations after fragmentation because they already contain high genetic diversity [62,66,67,68]. Although the natural population size of C. nitidissima has declined greatly in recent decades, the current fragmented populations are probably remnants of the old generation of the population. Long generation time actually helps to keep these ancient genetic variations for long time periods. This is why for the small populations (FL, BLC, and JDC), we did not find reduced genetic variations compared to the larger ones (Table 1 and Table 2). However, since for small-sized populations the likelihood of inbreeding and genetic drift increases [69], plant species with small and isolated populations tend to be vulnerable to demographic, environmental, and genetic stochasticity [9]. To support this view, it would be interesting to point out that during our field survey, poor fruit set was found in most individuals of this species, with this being indicative of potential issues with inbreeding depression.
Mutations in microsatellites generally do not appear consistently with either the IAM or the SMM [70], and the TPM model fits better for most of the microsatellites [71]. Hence, the results from the TPM should be considered to be more reliable for this study and revealed that only the PTC population had undergone bottleneck (Table 2). It was also confirmed by the mode-shift indicator test that all (sub) populations but PTC have a normal L-shaped graph (Supplementary Figure S2 online), which showed that only the PTC population is bottlenecked population. In addition, allelic richness is an alternative criterion for measuring genetic diversity, and it is more sensitive to bottlenecks than expected heterozygosity [72,73]. In this study, the allelic richness of PTC population (2.662) was somewhat lower than those of all (sub) populations (2.749–3.588), but BLS (2.478) (Table 2), it, therefore, reflected the possibility of the bottleneck of the PTC population.
Heterozygote deficiencies were also found in several C. nitidissima populations (Table 2). Heterozygote deficits can arise from a number of factors, especially through null alleles, biparental inbreeding, or population substructure (i.e., the Wahlund effect [74]) [75,76]. In the present study, we found that the global FST value showed a negligible change after correcting for the occurrence of null alleles, thus indicating that the influence of null alleles was negligible in our data. Furthermore, the fact that the FIS-GenALEx values of 4 of the 12 populations along with one subpopulation of NZS were higher than that of species in mixed breeding system (mean FIS = 0.15, it was calculated with the formula (HE − HO)/HE, and HO and HE from Nybom [64]) suggested possible inbreeding in C. nitidissima. Then, the FIS-INest values of their 95% HDPI for all populations were positive and did not include zero (Table 2), which indicated significant inbreeding in all these populations. In addition, results from pairwise comparisons of FST values, AMOVA, and bayesian cluster analyses indicated the presence of significant population structuring within the sampling regions of C. nitidissima (Table 3, Table 4, and Figure 1), and significant genetic structure within the fragmented populations can cause a potential Wahlund effect [77]. Therefore, we suspected that these deficiencies were primarily due to biparental inbreeding or population substructures.
4.2. Genetic Differentiation and Population Genetic Structure
The corrected pairwise FST values were all significant at p < 0.05 (Table 3), indicating high genetic differences among populations. While the overall FST of 0.190 (0.194, after the ZD population was excluded) was below the average value normally observed for plants in general (0.26) [64], narrow (0.23) [64], and outcrossing (0.22) [64], it still indicated a possible geographically-restricted gene flow among the remnant populations. A similar conclusion could be drawn from the fact that significant genetic structure was found in the sampling regions (Figure 1), and the private alleles were detected in 10 of the 12 populations. These observations could be linked to the geographical distances that were no less than 3 km between the populations, while mountain ranges (400–1400 m in elevation) also contributed to the separation. Moreover, C. nitidissima was found to be mainly pollinated by bees [22], with eighteen bee species reported as having a maximum flight distance from 540 to 2050 m [78]. Although there was no information about the flight range of other bee species, it is likely that the presence of the above geographical barriers could have hampered the movement of bees from one population to another, thus promoting pollen exchange only between nearby aggregations. In addition, with its large and heavy seeds, interpopulation seed dispersal was unlikely for C. nitidissima.
Significant divergences between the two regions were also observed after the ZD population was excluded. This genetic divergence has been interpreted as either the result of localized selection processes [79,80,81,82,83,84] or due to seeds/pollen dispersal [85,86]. However, in the present study, the neutrality tests showed that there was no evidence of selection. In fact, these two regions were separated by more than 100 km of low mountains (400–1400 m in elevation), and as such, it was almost impossible to exchange seed/pollen between them. Simulation studies previously showed that population substructures develop rapidly under “isolation by distance” models without spatial heterogeneous selection [87,88]. Therefore, the geographically-restricted gene flow between the two regions could have been the cause of the observed regional divergence of this species. However, it is noteworthy that the ZD population was the most distinct from the other populations of the Nanning region as it clustered with those of the Fangcheng region irrespective of its geographical isolation (Figure 1) (which is in accordance with the previous finding by Wei et al. [21]; it was named L population in that paper). Moreover, a few individuals that were genetically clustered with populations from other regions, irrespective of geographical isolation, were also found in BLS and DYC (Figure 1b). There was a high probability that all individuals from the ZD population, as well as several ones from the BLS population (green), could be migrants or offspring of migrants from the Fangcheng region, while several individuals from DYC population (red) were opposite, they could be migrants or offspring of migrants from the Nanning region (Figure 1b). Furthermore, the geographic distances between ZD and other populations of the Nanning region are less than 16 km, and these populations were located in the same climate zone. We, therefore, suspected that other factors, such as human-mediated gene flow, could have probably influenced the overall genetic structure of C. nitidissima. As an ornamental plant, C. nitidissima dispersal pathways were unavoidably influenced by humans, with the anomalous clustering likely to have been caused by the artificial introduction of some individual plants.
Interestingly, the NZS population harbor all genotypes of different populations of the Fangcheng region (Figure 1c), as well as low genetic differentiation between NZS and these populations were found (Table 2), suggesting a possibility that the NZS population was of hybridogenous origin and shared genetic stock with other populations in Fangcheng region, and therefore NZS could be a local “genetic melting-pot”.
4.3. Conservation Implications for C. nitidissima
The present results indicated that C. nitidissima remnants maintained a moderate level of genetic variability despite their severely fragmented natural range. However, since all extant populations are small and isolated, large numbers of their seeds are illegally collected, and many plants are illegally dug out for commercial purposes, the species face an uncertain future.
In 1986, in order to protect this valuable genetic resource, one natural reserve was established in Fangcheng, which is one of the primary habitats of the species in China, yet germplasm of the other nine populations (Table 1), especially ZZC, GMS, ZD, FL, BLC, DWJ, DYC, and JDC which harbor private alleles were never included. Since the unprotected populations can be quite vulnerable, the protection of their habitats is particularly urgent, and for this purpose, given the significant genetic divergence between the regions of Nanning and Fangcheng, a separate management unit could be considered for each of them. Moreover, as the populations of the Fangcheng region were divided into four genetically distinct groups, for each of these four groups, a separate management subunit based on genetic differentiation, different population sizes, as well as the presence of private alleles could also be considered. Finally, the NZS population needs to be prioritized for conservation in the Fangcheng region, as almost all regional genotypes were present in this population (Figure 1c).
An increased legal framework will undoubtedly be required to protect the species against the illegal harvesting of seeds and the digging up of plants from natural populations, especially to allow the plants to persist. In addition, management efforts in the form of transplantations and reintroductions may also play an important role in recovering threatened populations [89]. Given that the present population size of C. nitidissima is generally small and that seedling recruitment is halted in some populations, augmentation of each population by artificially propagated progenies of local individuals is recommended to increase effective population size. It is also worth noting that plants recruited from local seed sources are more likely to exhibit increased fitness over non-local genotypes in specific environments. Genetic theory predicts that the transfer of individuals from one population to another could result in outbreeding depression and reduced fitness [9,12,90]. However, for some populations that lack seed sources, artificially increasing the number of individuals by using the closest genetic neighbors could be justified. This could be the case for the FL, DYC, and JDC populations that not only consist of an extremely low number of individuals (39, 32, and 37, respectively) but also lack seed sources. Thus, reinforcement by this method could be considered. Finally, given that significant genetic divergences exist between the populations of Nanning and Fangcheng, the transfer of individuals from one region to another should be carried out with caution.
In the 1980s, an ex situ conservation program for this species was started at Guilin Botanical Garden, in which C. nitidissima seeds and seedlings collected from Yongning, Fangcheng, Dongxing, and Fusui counties were introduced. Today, there are about 800 C. nitidissima individuals in the garden, and most of them blossom and set fruits [21]; however, according to the sampling record, the germplasm of the Long’an County (ZZC) has not been included. Therefore, the collection of seeds from ZZC populations, along with the enhancement of current ex situ conservation, should be conducted. For C. nitidissima, the extant twelve populations of the two regions may have evolved into locally adapted ecotypes. Hence, care should be taken not to cross the Nanning stock and the Fangcheng stock until it is known whether outbreeding depression could be a problem.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/f13101662/s1, Table S1. The proportion of alleles in different allelic frequency classes for different (sub)populations of Camellia nitidissima; Figure S1. Plant photos of Camellia nitidissima; Figure S2. Graphic representation of proportion of alleles in different allelic frequency classes for different (sub)populations of Camellia nitidissima; Figure S3. Criteria for selecting the optimal K for assignment of all individuals during the Bayesian cluster analysis; Figure S4. Criteria for selecting the optimal K for assigning individuals in C2 during the Bayesian cluster analysis; Figure S5. Relationship between pairwise FST/(1 − FST) and geographic distance among populations of Camellia nitidissima.
Author Contributions
Conceptualization, Z.C., X.H. and X.W.; data processing and formal analyses, Z.C., J.W., J.T. and Z.W.; writing—original draft, Z.C., X.H. and X.W.; review and editing, Z.C., J.W., J.T. and Z.W.; funding acquisition, S.C. and X.W.; overall investigation, Z.C., Z.W. and X.H. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (NO: 32060248), the Key Research and Development Project of Guangxi (NO: GuiKeAB22080044), and the Central Guidance on Local Science and Technology Development Fund (NO: ZY21195035).
Institutional Review Board Statement
The sample collection was permitted by the Guangxi Fangcheng Management Division of Yellow Camellia National Nature Reserve, and the sampling process was followed by the Regulations of the People’s Republic of China on the Protection of Wild Plants.
Informed Consent Statement
Not applicable.
Data Availability Statement
The raw data is available from the first author for any further information/queries.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ellstrand, N.C. Gene Flow by Pollen: Implications for Plant Conservation Genetics. Oikos 1992, 63, 77. [Google Scholar] [CrossRef]
- Raijmann, L.E.; Van Leeuwen, N.C.; Kersten, R.; Oostermeijer, J.G.B.; Nijs, H.C.D.; Menken, S.B. Genetic Variation and Outcrossing Rate in Relation to Population Size in Gentiana pneumonanthe L. Conserv. Biol. 1994, 8, 1014–1026. [Google Scholar] [CrossRef]
- Heschel, M.S.; Paige, K.N. Inbreeding depression, environmental stress, and population size variation in Scarlet Gilia (Ip-omopsis aggregate). Conserv. Biol. 1995, 9, 126–133. [Google Scholar] [CrossRef]
- Sun, M. Effects of Population Size, Mating System, and Evolutionary Origin on Genetic Diversity in Spiranthes sinensis and S. hongkongensis. Conserv. Biol. 1996, 10, 785–795. [Google Scholar] [CrossRef]
- Buza, L.; Young, A.; Thrall, P. Genetic erosion, inbreeding and reduced fitness in fragmented populations of the endangered tetraploid pea Swainsona recta. Biol. Conserv. 2000, 93, 177–186. [Google Scholar] [CrossRef]
- Kery, M.; Matthies, D.; Spillmann, H.-H. Reduced fecundity and offspring performance in small populations of the declining grassland plants Primula veris and Gentiana lutea. J. Ecol. 2000, 88, 17–30. [Google Scholar] [CrossRef]
- Oostermeijer, J.G.B. Population viability analysis of the rare Gentiana pneumonanthe: Importance of genetics, demography, and reproductive biology. In Genetics, Demography and Viability of Fragmented Populations; Young, A.G., Clarke, G.M., Eds.; Cambridge University Press: Cambridge, UK, 2000; pp. 313–334. [Google Scholar]
- Cascante, A.; Quesada, M.; Lobo, J.J.; Fuchs, E.A. Effects of Dry Tropical Forest Fragmentation on the Reproductive Success and Genetic Structure of the Tree Samanea saman. Conserv. Biol. 2002, 16, 137–147. [Google Scholar] [CrossRef]
- Ellstrand, N.C.; Elam, D.R. Population genetic consequences of small population size: Implications for plant conservation. Annu. Rev. Ecol. Syst. 1993, 24, 217–242. [Google Scholar] [CrossRef]
- Lennon, J.; Kunin, W.E.; Gaston, K.J. The Biology of Rarity: Causes and Consequences of Rare-Common Differences. J. Anim. Ecol. 1997, 66, 916. [Google Scholar] [CrossRef]
- Lande, R. Extinction risks from anthropogenic, ecological, and genetic factors. In Genetics and the Extinction of Species: DNA and the Conservation of Biodiversity; Landweber, L.F., Dobson, A.P., Eds.; Princeton University Press: Princeton, NJ, USA, 1999; pp. 1–22. [Google Scholar]
- Barrett, S.C.H.; Kohn, J. Genetic and evolutionary consequences of small population size in plants: Implications for conservation. In Genetics and Conservation of Rare Plants; Falk, D.A., Holsinger, K.E., Eds.; Oxford University Press: Oxford, UK, 1991; pp. 3–30. [Google Scholar]
- Young, A.; Boyle, T.; Brown, T. The population genetic consequences of habitat fragmentation for plants. Trends Ecol. Evol. 1996, 11, 413–418. [Google Scholar] [CrossRef]
- White, G.M.; Boshier, D.H.; Powell, W. Increased pollen flow counteracts fragmentation in a tropical dry forest: An example from Swietenia humilis Zuccarini. Proc. Natl. Acad. Sci. USA 2002, 99, 2038–2042. [Google Scholar] [CrossRef] [PubMed]
- Hamrick, J.L.; Godt, M.J.W. Conservation genetics of endemic plant species. In Conservation Genetics: Case Histories from Nature; Avise, J.C., Hamrick, J.L., Eds.; Chapman & Hall: London, UK, 1996; pp. 281–304. [Google Scholar]
- Charlesworth, B.; Charlesworth, D.; Barton, N.H. The Effects of Genetic and Geographic Structure on Neutral Variation. Annu. Rev. Ecol. Evol. Syst. 2003, 34, 99–125. [Google Scholar] [CrossRef]
- Godt, M.; Hamrick, J. Allozyme diversity in the endangered pitcher plant Sarracenia rubra ssp. Alabamensis (Sarraceniaceae) and its close relative S. rubra ssp. rubra. Am. J. Bot. 1998, 85, 802–810. [Google Scholar] [PubMed]
- Su, Z.M.; Mo, X.L. Geographic distribution of Camellia section Chrysantha from China. Guihaia 1988, 8, 75–81, (In Chinese with English Abstract). [Google Scholar]
- Huang, P.J.; Zhou, Q.L. Karyotype study on Camellia nitidissima. Guihaia 1982, 2, 15–16, (In Chinese with English abstract). [Google Scholar]
- Wei, X.; Jiang, S.Y.; Jiang, Y.S.; Tang, H.; Cao, H.L. Research progress of Camellia nitidissima, a rare and endangered plant. J. Fujian For. Sci. Technol. 2006, 33, 169–174, (In Chinese with English abstract). [Google Scholar]
- Wei, X.; Cao, H.L.; Jiang, Y.S.; Ye, H.W.; Ge, X.J.; Li, F. Population genetic structure of Camellia nitidissima (Theaceae) and conservation implications. Bot. Stud. 2008, 49, 147–153. [Google Scholar]
- Cheng, J.H.; Chen, J.Y.; Zhao, S.W. Interspecific cross breeding for new yellow camellias. J. Beijing Forest Univ. 1994, 16, 55–59, (In Chinese with English abstract). [Google Scholar]
- Yang, H.Q.; Wei, X.; Zeng, X.L.; Ye, H.W.; Yin, X.J.; Wang, Z.M.; Jiang, Y.S. Seed biology and germination ecophysiology of Camellia nitidissima. Forest Ecol. Manag. 2008, 255, 113–118. [Google Scholar] [CrossRef]
- Deng, G.Y.; Yang, Z.D.; Lu, T.L. A brief review of research on yellow camellia in China. J. Guangxi Agr. Biol. Sci. 2000, 19, 126–130, (In Chinese with English abstract). [Google Scholar]
- Liang, S.Y. Golden Camellia; China Forest Press: Beijing, China, 1993. [Google Scholar]
- Parks, C.R. Breeding Progress with Yellow Camellias; American Camellia Yearbook: Ft. Valley, GA, USA, 2000; pp. 9–10. [Google Scholar]
- Nishimoto, S.I.; Hashimoto, F.; Shimizu, K.; Sakata, Y. Petal coloration of interspecific hydrids between Camellia chysantha × C. japonica. J. Jpn Soc. Hortic. Sci. 2004, 73, 189–191. [Google Scholar] [CrossRef][Green Version]
- Tang, S.; Bin, X.; Wang, L.; Zhong, Y. Genetic Diversity and Population Structure of Yellow Camellia (Camellia nitidissima) in China as Revealed by RAPD and AFLP Markers. Biochem. Genet. 2006, 44, 444–456. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.M. A preliminary study on the population ecology of Camellia sect. nitidissima. Guangxi Sci. 1994, 1, 31–36, (In Chinese with English abstract). [Google Scholar]
- Tan, W.F.; Li, D.Q. Analysis on protection gap of sect. Chrysantha Chang. Guangxi Forest Sci. 2010, 39, 53–54. (In Chinese) [Google Scholar]
- Fu, L.G. China Plant Red Data Book; Science Press: Beijing, China, 1992. [Google Scholar]
- Selkoe, K.A.; Toonen, R.J. Microsatellites for ecologists: A practical guide to using and evaluating microsatellite markers. Ecol. Lett. 2006, 9, 615–629. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.; Wang, J.; Huang, H. Demographic bottlenecks and low gene flow in remnant populations of the critically endangered Berchemiella wilsonii var. pubipetiolata (Rhamnaceae) inferred from microsatellite markers. Conserv. Genet. 2008, 9, 191–199. [Google Scholar] [CrossRef]
- Powell, W.; Machray, G.C.; Provan, J. Polymorphism revealed by simple sequence repeats. Trends Plant Sci. 1996, 1, 215–222. [Google Scholar] [CrossRef]
- Wei, J.Q.; Chen, Z.Y.; Wang, Z.F.; Tang, H.; Jiang, Y.S.; Wei, X.; Li, X.Y.; Qi, X.X. Isolation and characterization of polymorphic microsatellite loci in Camellia nitidissima Chi (Theaceae). Am. J. Bot. 2010, 97, e89–e90. [Google Scholar] [CrossRef]
- Doyle, J. DNA protocols for plants—CTAB total DNA isolation. In Molecular Techniques in Taxonomy; Hewitt, G.M., Johnston, A., Eds.; Springer: Berlin, Germany, 1991; pp. 283–293. [Google Scholar]
- Glaubitz, J.C. CONVERT: A user-friendly program to reformat diploid genotypic data for commonly used population genetic software packages. Mol. Ecol. Notes 2004, 4, 309–310. [Google Scholar] [CrossRef]
- Peakall, R.O.D.; Smouse, P.E. genalex 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Manly, B.F.J. The Statistics of Natural Selection; Chapman and Hall: London, UK, 1985. [Google Scholar]
- Yeh, F.C.; Yang, R.C.; Boyle, T. POPGENE Version 1.31. Microsoft Window-based Freeware for Population Genetic Analysis Quick User Guide; University of Alberta and Centre for International Forestry Research: Edmonton, AB, Canada; Bogor, Indonesia, 1999. [Google Scholar]
- Nei, M. Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 1978, 89, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Wright, S. Evolution and the Genetics of Populations. Vol 4: Variability within and among Natural Populations; University of Chicago Press: Chicago, IL, USA, 1978. [Google Scholar]
- Goudet, J. FSTAT, a Program to Estimate and Test Gene Diversities and Fixation Indices (Version 2.9.3); ScienceOpen, Inc.: Burlington, MA, USA, 2001. [Google Scholar]
- Chybicki, I.J. INEST 2.0: The User Manual. Poland: Department of Genetics; Kazimierz Wielki University: Bydgoszcz, Poland, 2015. [Google Scholar]
- Chapuis, M.P.; Estoup, A. Microsatellite null alleles and estimation of population differentiation. Mol. Biol. Evol. 2007, 24, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Weir, B.S. Genetic Data Analysis; Sinauer Associates: Sunderland, MA, USA, 1996. [Google Scholar]
- Piry, S.; Luikart, G.; Cornuet, J.M. BOTTLENECK: A computer program for detecting recent reductions in the effective population size using allele frequency data. J. Heredity 1999, 90, 502–503. [Google Scholar] [CrossRef]
- Lawler, R.R. Testing for a historical population bottleneck in wild Verreaux’s sifaka (Propithecus verreauxi verreauxi) using microsatellite data. Am. J. Primatol. 2008, 70, 990–994. [Google Scholar] [CrossRef] [PubMed]
- Di Rienzo, A.; Peterson, A.C.; Garza, J.C.; Valdes, A.M.; Slatkin, M.; Freimer, N.B. Mutational processes of simple sequence repeat loci in human populations. Proc. Natl. Acad. Sci. USA 1994, 91, 3166–3170. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Wen, X.; Falush, D. STRUCTURE Version 2.3.; University of Chicago: Chicago, IL, USA, 2009. [Google Scholar]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software structure: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef]
- Jakobsson, M.; Rosenberg, N.A. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 2007, 23, 1801–1806. [Google Scholar] [CrossRef]
- Excoffier, L.; Laval, G.; Schneider, S. Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol. Bioinform. 2005, 1, 47–50. [Google Scholar] [CrossRef]
- Rousset, F. Genetic Differentiation and Estimation of Gene Flow from F-Statistics Under Isolation by Distance. Genetics 1997, 145, 1219–1228. [Google Scholar] [CrossRef]
- Ge, S.; Hong, D.-Y.; Wang, H.-Q.; Liu, Z.-Y.; Zhang, C.-M. Population Genetic Structure and Conservation of an Endangered Conifer, Cathaya argyrophylla (Pinaceae). Bot. Gaz. 1998, 159, 351–357. [Google Scholar] [CrossRef]
- Li, Y.; Chen, X.-Y.; Zhang, X.; Wu, T.-Y.; Lu, H.-P.; Cai, Y.-W. Genetic Differences between Wild and Artificial Populations of Metasequoia glyptostroboides: Implications for Species Recovery. Conserv. Biol. 2005, 19, 224–231. [Google Scholar] [CrossRef]
- Kaneko, S.; Isagi, Y.; Nobushima, F. Genetic differentiation among populations of an oceanic island: The case of Metrosideros boninensis, an endangered endemic tree species in the Bonin Islands. Plant Spec. Biol. 2008, 23, 119–128. [Google Scholar] [CrossRef]
- Moreira, R.G.; McCauley, R.A.; Cortés-Palomec, A.C.; Fernandes, G.W.; Oyama, K. Spatial genetic structure of Coccoloba cereifera (Polygonaceae), a critically endangered microendemic species of Brazilian rupestrian fields. Conserv. Genet. 2009, 11, 1247–1255. [Google Scholar] [CrossRef]
- Gitzendanner, M.A.; Soltis, P.S. Patterns of genetic variation in rare and widespread plant congeners. Am. J. Bot. 2000, 87, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Chen, X.-Y.; Zhang, X.; Li, Y.-Y.; Fu, C.-X.; Qiu, Y.-X. Genetic variation of Ginkgo biloba L. (Ginkgoaceae) based on cpDNA PCR-RFLPs: Inference of glacial refugia. Heredity 2005, 94, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Straub, S.C.K.; Doyle, J.J. Conservation genetics of Amorpha georgiana (Fabaceae), an endangered legume of the Southeastern United States. Mol. Ecol. 2009, 18, 4349–4365. [Google Scholar] [CrossRef]
- Yao, X.; Ye, Q.; Kang, M.; Huang, H. Microsatellite analysis reveals interpopulation differentiation and gene flow in the endangered tree Changiostyrax dolichocarpa (Styracaceae) with fragmented distribution in central China. New Phytol. 2007, 176, 472–480. [Google Scholar] [CrossRef]
- Cao, P.-J.; Yao, Q.-F.; Ding, B.-Y.; Zeng, H.-Y.; Zhong, Y.-X.; Fu, C.-X.; Jin, X.-F. Genetic diversity of Sinojackia dolichocarpa (Styracaceae), a species endangered and endemic to China, detected by inter-simple sequence repeat (ISSR). Biochem. Syst. Ecol. 2006, 34, 231–239. [Google Scholar] [CrossRef]
- Nybom, H. Comparison of different nuclear DNA markers for estimating intraspecific genetic diversity in plants. Mol. Ecol. 2004, 13, 1143–1155. [Google Scholar] [CrossRef]
- Wang, X.; Dong, L.-J.; Duan, J.-H.; Li, S.-J.; Zhang, S.-G. Genetic diversity and relationship of 84 tea cultivars (Camellia sinensis (L.) O. Kuntze) by SSR markers. J. HUNAN Agric. Univ. 2011, 37, 260–266. [Google Scholar] [CrossRef]
- Collevatti, R.G.; Grattapaglia, D.; Hay, J.D. High resolution microsatellite based analysis of the mating system allows the detection of significant biparental inbreeding in Caryocar brasiliense, an endangered tropical tree species. Heredity 2001, 86, 60–67. [Google Scholar] [CrossRef] [PubMed]
- White, G.M.; Boshier, D.H.; Powell, W. Genetic variation within a fragmented population of Swietenia humilis Zucc. Mol. Ecol. 1999, 8, 1899–1909. [Google Scholar] [CrossRef] [PubMed]
- Young, A.G.; Merriam, H.G.; Warwick, S.I. The effects of forest fragmentation on genetic variation in Acer saccharum Marsh. (sugar maple) populations. Heredity 1993, 71, 277–289. [Google Scholar] [CrossRef][Green Version]
- Hamrick, J. Response of forest trees to global environmental changes. For. Ecol. Manag. 2004, 197, 323–335. [Google Scholar] [CrossRef]
- Kusza, S.; Priskin, K.; Ivankovic, A.; Jedrzejewska, B.; Podgorski, T.; Jávor, A.; Mihók, S. Genetic characterization and population bottleneck in the Hucul horse based on microsatellite and mitochondrial data. Biol. J. Linn. Soc. 2013, 109, 54–65. [Google Scholar] [CrossRef]
- Kataria, R.S.; Kathiravan, P.; Bulandi, S.S.; Pandey, D.; Mishra, B.P. Microsatellite-based genetic monitoring to detect cryptic demographic bottleneck in Indian riverine buffaloes (Bubalus bubalis). Trop Anim Health Prod. 2010, 42, 849–855. [Google Scholar] [CrossRef]
- Rajora, O.P.; Rahman, M.H.; Buchert, G.P.; Dancik, B.P. Microsatellite DNA analysis of genetic effects of harvesting in old-growth eastern white pine (Pinus strobus) in Ontario, Canada. Mol. Ecol. 2000, 9, 339–348. [Google Scholar] [CrossRef]
- Caballero, A.; Rodríguez-Ramilo, S.T.; Ávila, V.; Fernández, J. Management of genetic diversity of subdivided populations in conservation programmes. Conserv. Genet. 2010, 11, 409–419. [Google Scholar] [CrossRef]
- Wahlund, S. Zusammensetzung von Populationen und Korrelationserscheinungen vom Standpunkt der Vererbungslehre aus Betrachtet. Hereditas 1928, 11, 65–106. [Google Scholar] [CrossRef]
- Paxton, R.J.; Thorén, P.A.; Gyllenstrand, N.; Tengö, J. Microsatellite DNA analysis reveals low diploid male production in a communal bee with inbreeding. Biol. J. Linn. Soc. 2000, 69, 483–502. [Google Scholar] [CrossRef]
- Wang, Z.F.; Hamrick, J.L.; Godt, M.J.W. High genetic diversity in Sarracenia leucophylla (Sarraceniaceae), a carnivorous wet-land herb. J. Hered. 2004, 95, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Jump, A.S.; Peñuelas, J. Genetic effects of chronic habitat fragmentation in a wind-pollinated tree. Proc. Natl. Acad. Sci. USA 2006, 103, 8096–8100. [Google Scholar] [CrossRef] [PubMed]
- Araújo, E.D.; Costa, M.; Chaud-Netto, J.; Fowler, H.G. Body size and flight distance in stingless bees (Hymenoptera: Meliponini): Inference of flight range and possible ecological implications. Braz. J. Biol. 2004, 64, 563–568. [Google Scholar] [CrossRef]
- Hamrick, J.L.; Allard, R.W. Microgeographical Variation in Allozyme Frequencies in Avena barbata. Proc. Natl. Acad. Sci. USA 1972, 69, 2100–2104. [Google Scholar] [CrossRef] [PubMed]
- Lack, A.J.; Kay, Q.O.N. Allele frequencies, genetic relationships and heterozygosity in Polygala vulgaris populations from contrasting habitats in southern Britain. Biol. J. Linn. Soc. 1988, 34, 119–147. [Google Scholar] [CrossRef]
- Van Rossum, F.; Vekemans, X.; Meerts, P.; Gratia, E.; Lefebvre, C. Allozyme variation in relation to ecotypic differentiation and population size in marginal populations of Silene nutans. Heredity 1997, 78, 552–560. [Google Scholar] [CrossRef]
- Li, Y.C.; Fahima, T.; Beiles, A.; Korol, A.B.; Nevo, E. Microclimatic stress and adaptive DNA differentiation in wild emmer wheat, Triticum dicoccoides. Theor. Appl. Genet. 1999, 98, 873–883. [Google Scholar] [CrossRef]
- Owuor, E.D.; Fahima, T.; Beharav, A.; Korol, A.; Nevo, E. RAPD divergence caused by microsite natural selection. Genetica 1999, 105, 177–192. [Google Scholar] [CrossRef]
- Qiu, Y.; Liu, Y.; Kang, M.; Yi, G.; Huang, H. Spatial and temporal population genetic variation and structure of Nothotsuga longibracteata (Pinaceae), a relic conifer species endemic to subtropical China. Genet. Mol. Biol. 2013, 36, 598–607. [Google Scholar] [CrossRef]
- Schaal, B.A. Population Structure and Local Differentiation in Liatris cylindracea. Am. Nat. 1975, 109, 511–528. [Google Scholar] [CrossRef]
- Waser, N.M. Spatial genetic heterogeneity in a population of the montane perennial herb Delphinium nelsonii. Heredity 1987, 58, 249–256. [Google Scholar] [CrossRef]
- Turner, M.E.; Stephens, J.C.; Anderson, W.W. Homozygosity and patch structure in plant populations as a result of near-est-neighbor pollination. Proc. Natl. Acad. Sci. USA 1982, 79, 2903–2907. [Google Scholar] [CrossRef] [PubMed]
- Sokal, R.R.; Wartenburg, D.E. A test of spatial autocorrelation analysis using an isolation-by-distance model. Genetics 1983, 105, 219–237. [Google Scholar] [CrossRef] [PubMed]
- Barnaud, A.; Houliston, G.J. Population genetics of the threatened tree daisy Olearia gardneri (Asteraceae), conservation of a critically endangered species. Conserv. Genet. 2009, 11, 1515–1522. [Google Scholar] [CrossRef]
- Booy, G.; Hendriks, R.J.J.; Smulders, M.J.M.; Groenendael, J.M.; Vosman, B. Genetic Diversity and the Survival of Populations. Plant Biol. 2000, 2, 379–395. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).