
Transcriptomic Determination of the Core Genes Regulating the Growth and Physiological Traits of Quercus mongolica Fisch. ex Ledeb

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Collection and Cultivation of Plant Materials
2.2. Determination of Q. mongolica Growth and Physiological Traits
2.3. Transcriptome Analysis
2.4. Weighted Gene Co-Expression Network Analysis
2.5. Verification of Core Genes by qRT-PCR
2.6. Statistical Analysis
3. Results
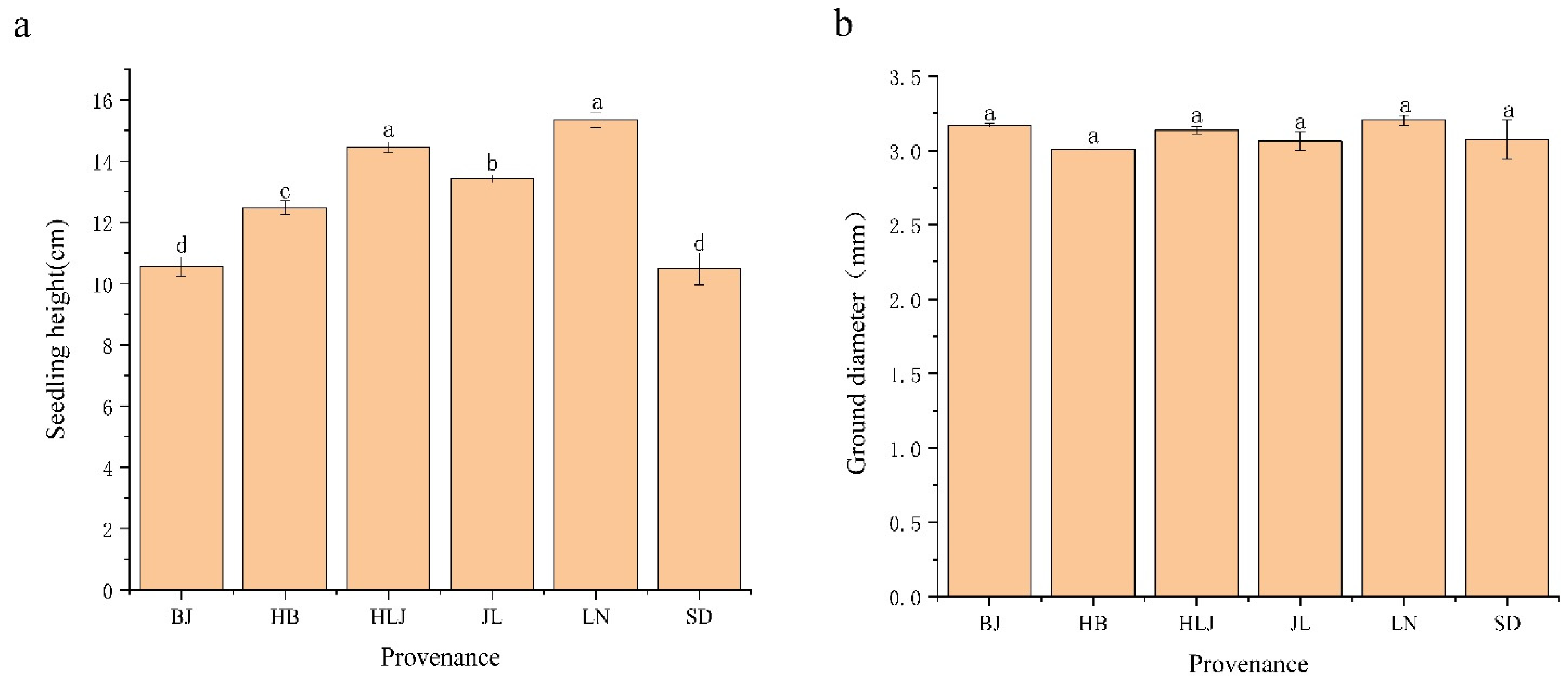
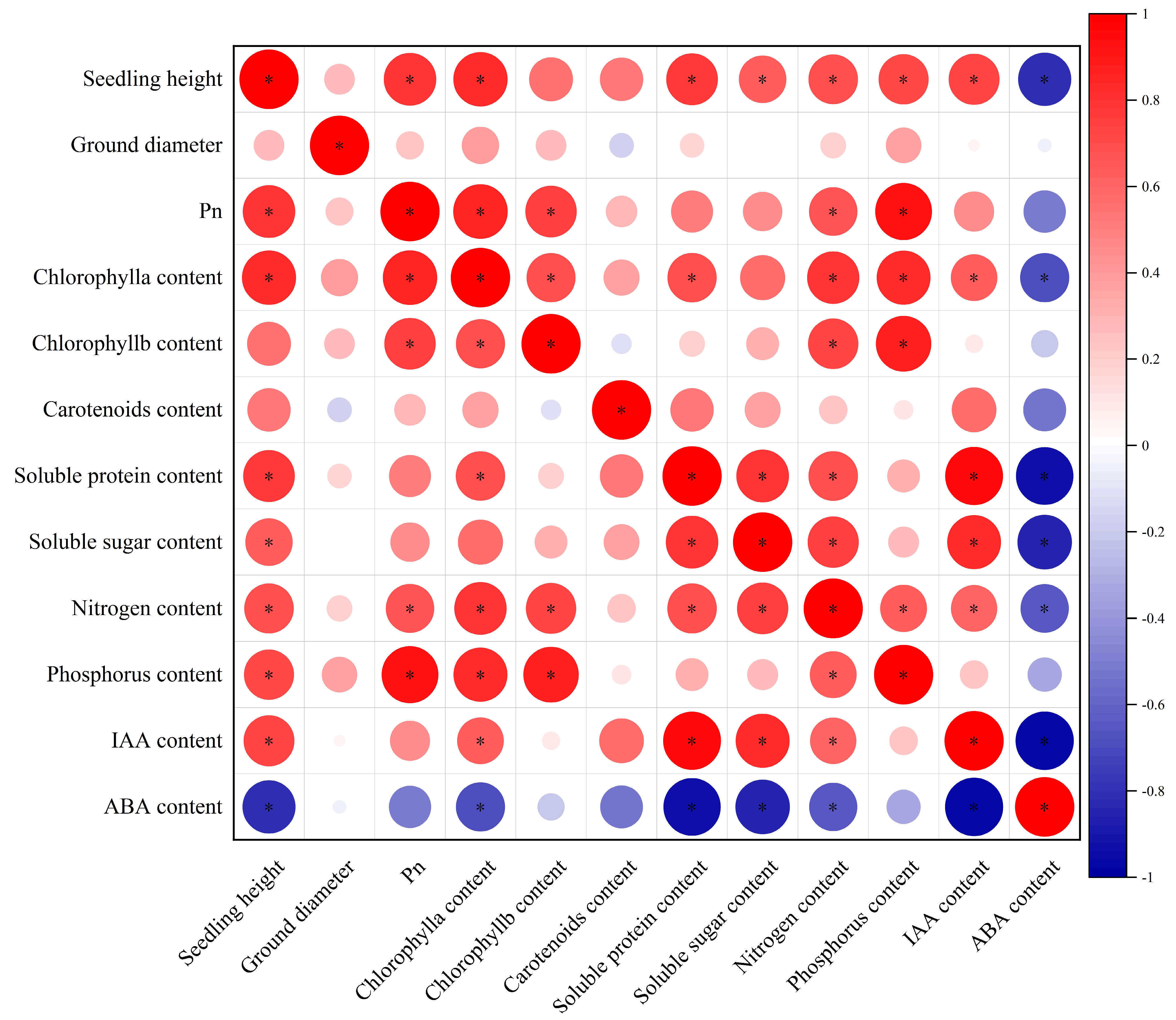
3.1. Growth and Physiological Biochemical Indicators of Q. mongolica of Different Provenances
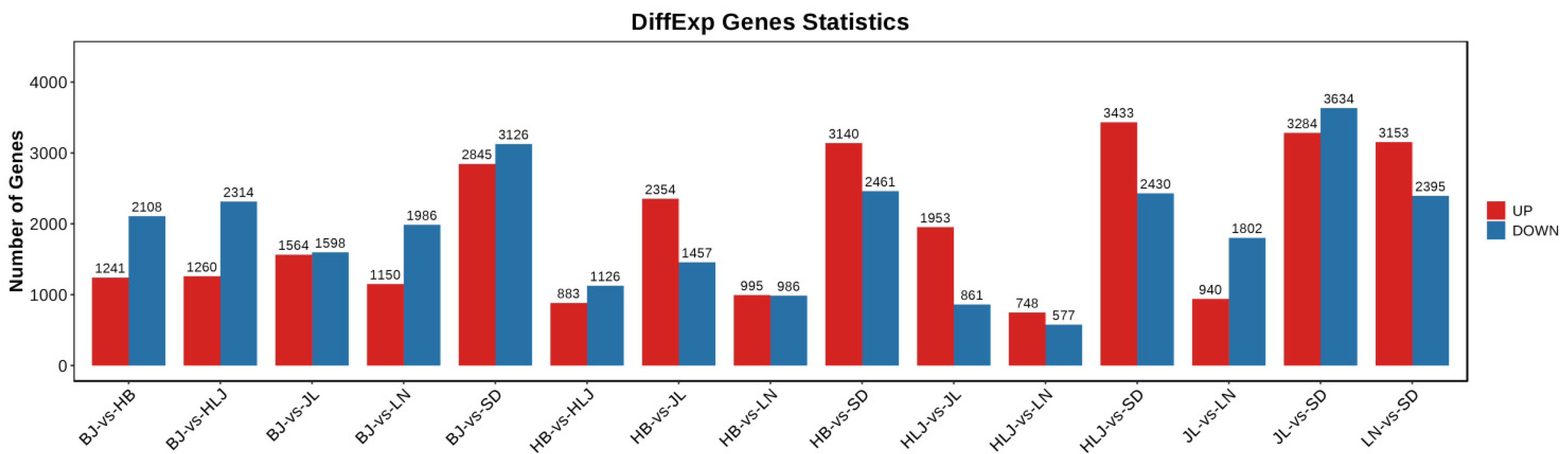
3.2. DEGs in the Leaves of Q. mongolica of Different Provenances
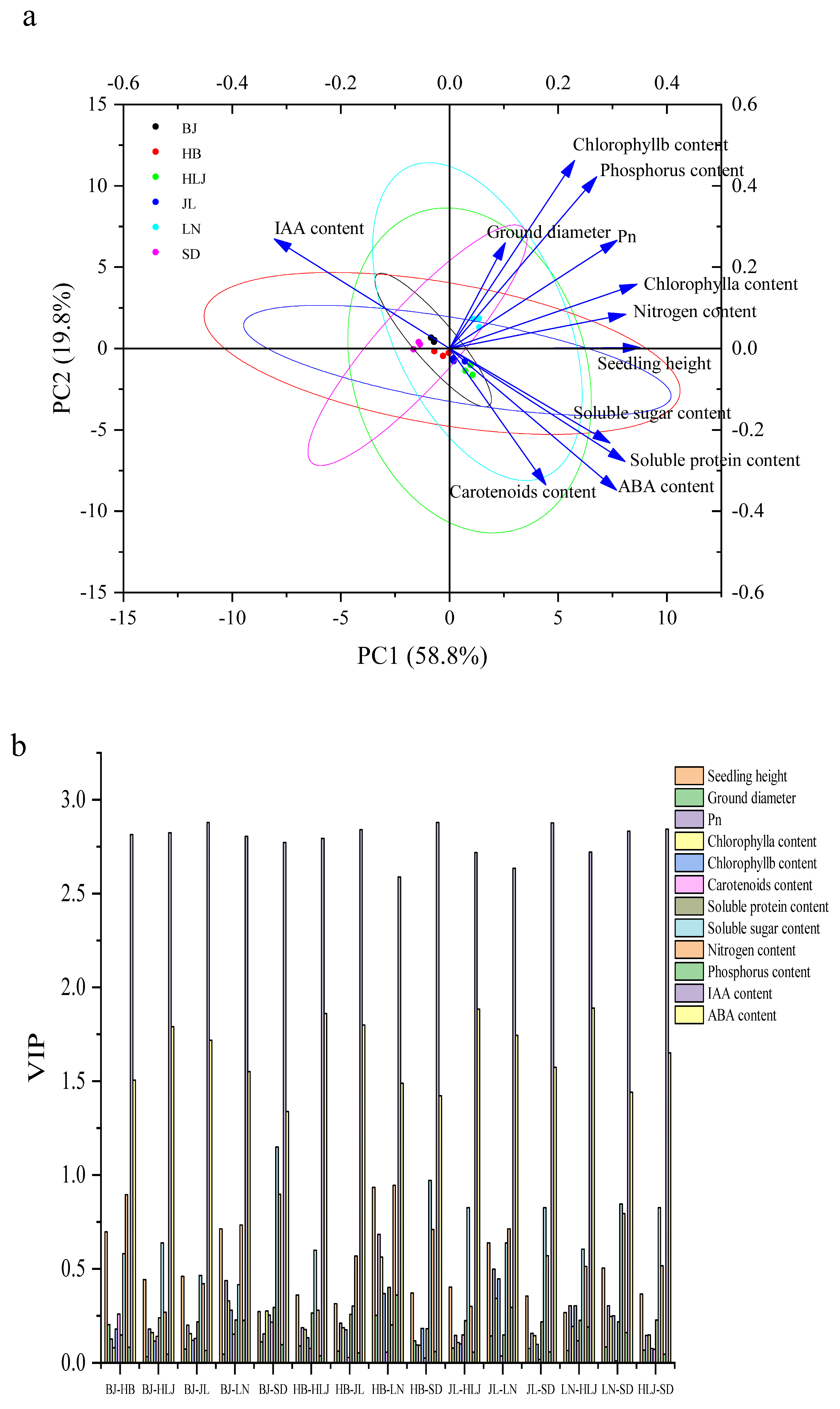
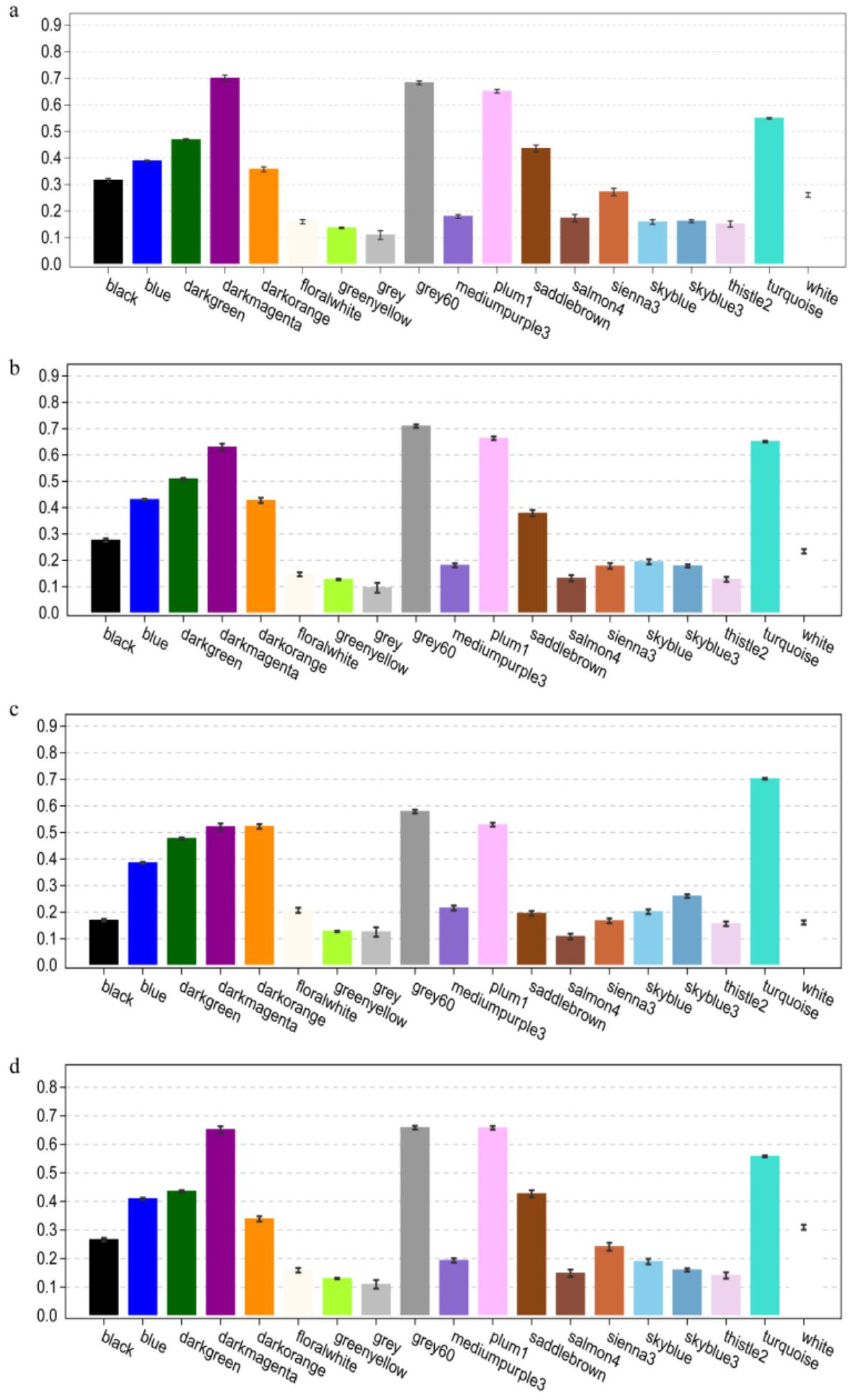
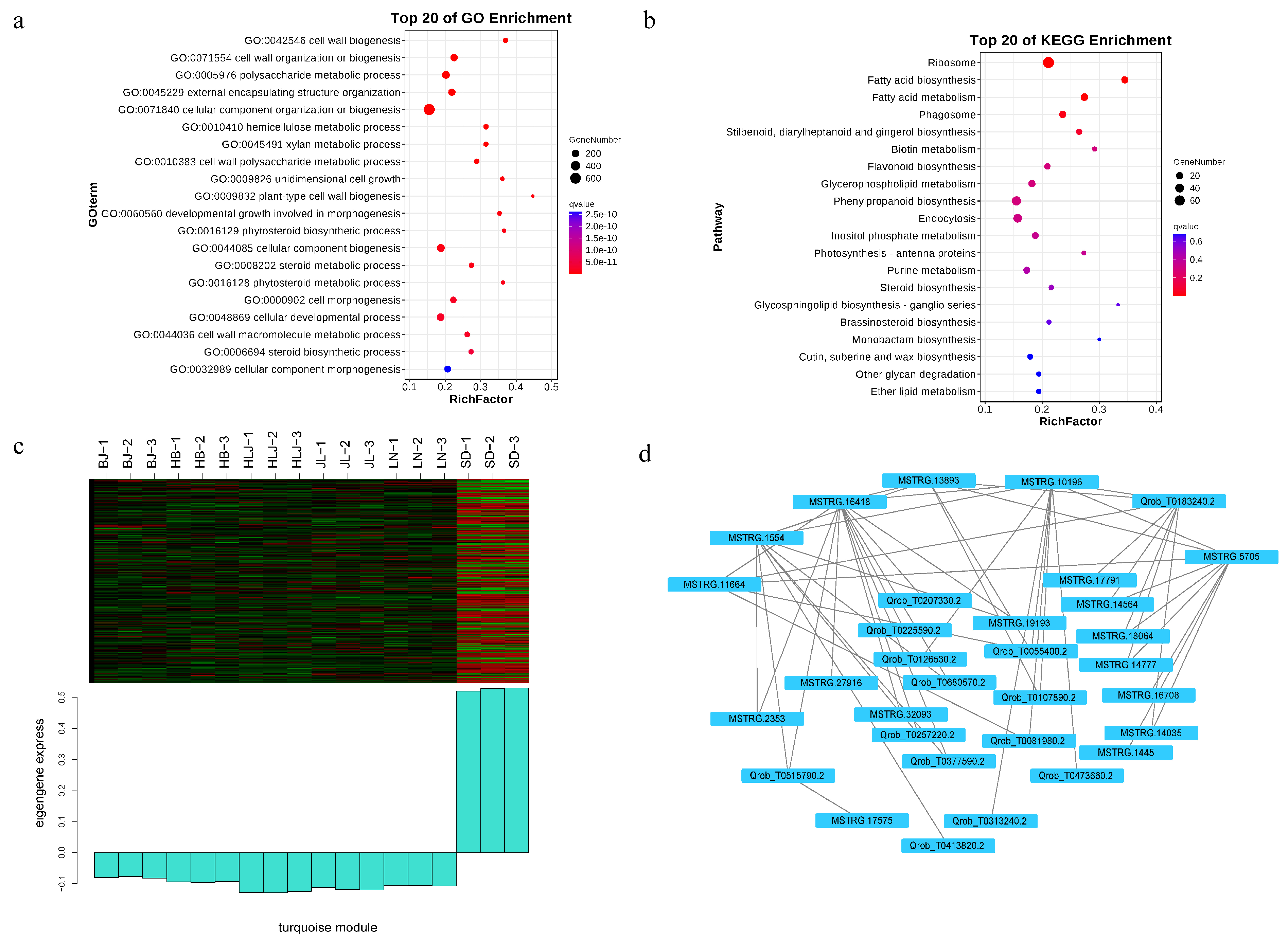
3.3. Co-Expression Network and Core Gene Mining Results
3.4. Core Genes Related to the Abscisic Acid Content
3.5. Core Genes Related to Indole-3-Acetic Acid Content
3.6. Core Genes Related to the Soluble Sugar Content
3.7. Core Genes Related to the Soluble Protein Content
3.8. Verification of Core Genes by qRT-PCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Daghlian, C.P.; Crepet, W.L. Oak catkins, leaves and fruits from the Oligocene Catahoula Formation and their evolutionary significance. Am. J. Bot. 1983, 70, 639–649. [Google Scholar] [CrossRef]
- Xu, X.H.; Yang, B.S.; Wang, H.; Cao, Y.A.; Li, K.; Gao, S.W. Temperature sensitivity of soil heterotrophic respiration is altered by carbon substrate along the development of Quercus mongolica forest in northeast China. Appl. Soil Ecol. 2019, 133, 52–61. [Google Scholar] [CrossRef]
- Shen, L.; Liu, P.X.; An, M.M.; Liang, R.N.; He, X.W.; Zhao, G.Z. Sphingomonas quercus sp. Nov., Isolated from Rhizosphere Soil of Quercus mongolica. Curr. Microbiol. 2022, 79, 122. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.Y.; Lu, X.M.; Zhou, G.S.; Ren, H.R. Spectral Characteristics during Leaf Flourishing Development of Quercus mongolica and Its Influencing Factors. Spectrosc. Spect. Anal. 2021, 41, 2924–2929. [Google Scholar]
- Lyu, S.N.; Wang, X.C.; Zhang, Y.D.; Li, Z.S. Different responses of Korean pine (Pinus koraiensis) and Mongolia oak (Quercus mongolica) growth to recent climate warming in northeast China. Dendrochronologia 2017, 45, 113–122. [Google Scholar] [CrossRef]
- Sun, Y.; Guo, J.L.; Zeng, X.R.; Chen, R.S.; Feng, Y.; Chen, S.; Yang, K. Chromosome-scale genome assembly of Castanopsis tibetana provides a powerful comparative framework to study the evolution and adaptation of Fagaceae trees. Mol. Ecol. Resour. 2022, 22, 1178–1189. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.J.; Pang, X.B.; Liu, Z.Y.; Li, J.Y.; Zhang, W.Y.; Yin, S.L.; Wang, L.B.; Li, Y.C. Diversity of phenotypic characters of Quercus mongolica seeds from different provenances. J. Zhejiang Univ. 2022, 39, 1221–1228. (In Chinese) [Google Scholar]
- Chen, K.; Zhang, H.; Lei, X. Spatial pattern of Quercus mongolica in natural secondary forest. Acta Ecol. Sin. 2018, 38, 3462–3470. [Google Scholar]
- He, F.; Yang, B.S.; Wang, H.; Yan, Q.L.; Cao, Y.N.; He, X.H. Changes in composition and diversity of fungal communities along Quercus mongolica forests developments in Northeast China. Appl. Soil Ecol. 2016, 100, 162–171. [Google Scholar] [CrossRef]
- Chen, S.; Xu, Z.Y.; Adil, M.F.; Zhang, G.P. Cultivar-, stress duration- and leaf age-specific hub genes and co-expression networks responding to waterlogging in barley. Environ. Exp. Bot. 2021, 191, 104599. [Google Scholar] [CrossRef]
- Yang, Y.F.; Shi, D.Y.; Wang, Y.N.; Zhang, L.; Chen, X.G.; Yang, X.P. Transcript profiling for regulation of sweet potato skin color in Sushu8 and its mutant Zhengshu20. Plant Physiol. Bioch. 2020, 148, 1–9. [Google Scholar] [CrossRef]
- Lu, C.F.; Pu, Y.; Liu, Y.T.; Li, Y.J.; Qu, J.P.; Huang, H.; Dai, S.L. Comparative transcriptomics and weighted gene co-expression correlation network analysis (WGCNA) reveal potential regulation mechanism of carotenoid accumulation in Chrysanthemum × morifolium. Plant Physiol. Bioch. 2019, 142, 415–428. [Google Scholar] [CrossRef]
- He, Y.J.; Wang, Z.W.; Ge, H.Y.; Liu, Y.; Chen, H.Y. Weighted gene co-expression network analysis identifies genes related to anthocyanin biosynthesis and functional verification of hub gene SmWRKY44. Plant Sci. 2021, 309, 110935. [Google Scholar] [CrossRef]
- Jiang, M.; Li, X.M.; Yuan, Y.C.; Zhang, G.W.; Pang, J.S.; Ren, J.J.; Wang, J.M.; Yang, M.S. Integrated physiological and transcriptomic analyses reveal the molecular mechanism behind the response to cultivation in Quercus mongolica. Front. Plant Sci. 2022, 13, 947696. [Google Scholar] [CrossRef]
- Jagtap, A.B.; Yadav, I.S.; Vikal, Y.; Praba, U.P.; Kaur, N.; Gill, A.S.; Johal, G.S. Transcriptional dynamics of maize leaves, pollens and ovules to gain insights into heat stress-related responses. Front. Plant Sci. 2023, 14, 1117136. [Google Scholar] [CrossRef]
- Liu, X.Q.; Chen, A.P.; Wang, Y.X.; Jin, G.L.; Zhang, Y.H.; Gu, L.L.; Li, C.J.; Shao, X.Q.; Wang, K. Physiological and transcriptomic insights into adaptive responses of Seriphidium transiliense seedlings to drought stress. Environ. Exp. Bot. 2022, 194, 104736. [Google Scholar] [CrossRef]
- Wu, W.Q.; Liao, T.; Du, K.; Wei, H.R.; Kang, X.Y. Transcriptome comparison of different ploidy reveals the mechanism of photosynthetic efficiency superiority of triploid poplar. Genomics 2021, 113, 2211–2220. [Google Scholar] [CrossRef]
- Holmes, J.J.; Weger, H.G.; Turpin, D.H. Chlorophyll a Fluorescence Predicts Total Photosynthetic Electron Flow to CO2 or NO3−/NO2− under Transient Conditions. J. Plant Physiol. 1989, 91, 331–337. [Google Scholar] [CrossRef] [Green Version]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of proteindye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Buysse, J.; Merckx, R. An Improved Colorimetric Method to Quantify Sugar Content of Plant Tissue. J. Exp. Bot. 1993, 44, 1627–1629. [Google Scholar] [CrossRef]
- Guinagui, N.B.; Sorho, F.; Souleymane, S.; Koné, B.; Koné, D. Effect of Rice yellow mottle virus, Sobemovirus on the Contents of N P K Ca and Mg in Leaves of Infected Rice. Annu. Res. Rev. Biol. 2019, 30, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.F.; Zhou, Y.Q.; Chen, Y.R.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Chatwin, W.B.; Carpenter, K.K.; Jimenez, F.R.; Elzinga, D.B.; Johnson, L.A.; Maughan, P.J. Microsatellite primer development for Post Oak, Quercus stellata (Fagaceae). Appl. Plant Sci. 2014, 2, 3. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Nie, M.X.; Huan, J.Y.; Huang, J.L.; Chen, Z.Y.; Lin, L.Z.; Luo, F.L.; Zhang, X.; Xiao, S. Differences in the components of Neolamarckia cadamba from different provenance and the drug resistance reversal activity of characteristic alkaloid. Ind. Crop. Prod. 2022, 186, 115145. [Google Scholar] [CrossRef]
- Vendramini, F.; Díaz, S.; Gurvich, D.E.; Wilson, P.J.; Thompson, K.; Hodgson, J.G. Leaf traits as indicators of resource-use strategy in floras with succulent species. New Phyto. 2002, 154, 147–157. [Google Scholar] [CrossRef]
- Westoby, M.A. Leaf-height-seed (LHS) plant ecology strategy scheme. Plant Soil 1998, 199, 213–227. [Google Scholar] [CrossRef]
- Xu, Z.Q.; Huang, X.R.; Xu, L.C.; Xu, Q.; Ji, X.L. The impacts of light conditions on the growth and morphology of Quercus mongolica seedlings. Acta Ecol. Sin. 2009, 29, 1121–1128. [Google Scholar]
- Wang, Y.; Han, X.; Ai, W.; Zhan, H.; Ma, S.; Lu, X. Non-Structural Carbohydrates and Growth Adaptation Strategies of Quercus mongolica Fisch. ex Ledeb. Seedlings under Drought Stress. Forests 2023, 14, 404. [Google Scholar] [CrossRef]
- Li, H.X.; Li, Z.H.; Bai, H.; Xing, Y.J. Effects of Different Phosphorus Treatments on the Growth and Physiological Characteristics of Mongolian Oak Seedlings. For. Sci. Technol. 2021, 46, 1–3. [Google Scholar]
- Wang, X.Y.; Zhang, Y.; He, H.J.; Fan, X.H. Response of photosynthetic and fluorescence characteristics of Quercus mongolica seedlings to simulated nitrogen deposition in Shulan, Jilin Province. Chin. J. Appl. Environ. Biol. 2023, 1–11. [Google Scholar] [CrossRef]
- Zhou, R.L.; Hou, Y.P.; Zuo, J.C.; Wang, Y.F. The physiological adaptation mechanisms of four common desert species in response to desert environments. Acta Ecol. Sin. 2015, 35, 340–349. [Google Scholar]
- Shi, W.J.; Wang, X.J.; Liu, H.; Cai, Z.A.; Lu, C.F.; Chen, Y.Z. A novel ABA-insensitive mutant in Arabidopsis reveals molecular network of ABA-induced anthocyanin accumulation and abiotic stress tolerance. J. Plant Physiol. 2022, 278, 153810. [Google Scholar] [CrossRef]
- Huang, X.; Shi, H.; Hu, Z.; Liu, A.; Amombo, E.; Chen, L.; Fu, J. ABA is involved in regulation of cold stress response in bermudagrass. Front. Plant Sci. 2017, 8, 1613. [Google Scholar] [CrossRef] [Green Version]
- Yue, D.; Wang, Y.K. Study on the Relationship between Endogenous Hormone Content and Cold Resistance of Apricot Trees. J. Anhui Agri. Sci. 2008, 10023, 9951–9952. [Google Scholar]
- Jędrzejuk, A.; Kuźma, N.; Nawrot, K.; Budzyński, R.; Orłowski, A. Mechanical stimulation affects growth dynamics, IAA content and activity of POD and IAA oxidase in Petunia × atkinsiana. Sci. Hortic. 2020, 274, 109661. [Google Scholar] [CrossRef]
- Xie, J.R.; Xiang, D.Y.; Mei, H.; Tan, F. The Relationship between Changes in Cold Resistance of Southern Taxus and Endogenous Hormones. J. South Chin. Norm. Univ. 2002, 2, 231–234. [Google Scholar]
- Theocharis, A.; Clement, C.; Barka, E.A. Physiological and molecular changes in plants grown at low temperatures. Planta 2012, 235, 1091–1105. [Google Scholar] [CrossRef]
- Yan, L.; Shah, T.; Cheng, Y.; Lü, Y.; Zhang, X.; Zou, X. Physiological and molecular responses to cold stress in rapeseed (Brassica napus L.). J. Integr. Agric. 2019, 18, 2742–2752. [Google Scholar] [CrossRef]
- Wu, S.W.; Li, M.; Zhang, C.M.; Tan, Q.L.; Yang, X.Z.; Sun, X.C.; Pan, Z.Y. Effects of phosphorus on fruit soluble sugar and citric acid accumulations in citrus. Plant Physiol. Biochem. 2021, 160, 73–81. [Google Scholar] [CrossRef]
- Adrià, L.C.; Paula, S.; Clara, G.; Anna, A.; Elena, G.F. Quality comparison of recombinant soluble proteins and proteins solubilized from bacterial inclusion bodies. New Biotechnol. 2022, 72, 58–63. [Google Scholar]
- Liang, S.X.; Meng, Q.X.; Shi, M.J.; Luo, G.T.; Wang, G.Z. tudy on the Relationship between the Content of Soluble Protein and Soluble Sugar in the Branches of Hazelnut and Its Cold Resistance. Chin. Agri. Sci. Bull. 2015, 31, 14–18. [Google Scholar]
- Zhang, H.; Jiang, C.J.; Lei, J.N.; Dong, J.L.; Ren, J.Y.; Shi, X.L.; Zhong, C.; Wang, X.G.; Zhao, X.H.; Yu, H.Q. Comparative physiological and transcriptomic analyses reveal key regulatory networks and potential hub genes controlling peanut chilling tolerance. Genomics 2022, 114, 110285. [Google Scholar] [CrossRef]
- Wang, P.; Liu, D.; Yang, F.H.; Ge, H.; Zhao, X.; Chen, H.G.; Du, T. Identification of key gene networks controlling vernalization development characteristics of Isatis indigotica by full-length transcriptomes and gene expression profiles. Physiol. Mol. Biol. Plants 2021, 27, 2679–2693. [Google Scholar] [CrossRef]
- Jia, Y.; Wong, D.C.J.; Sweetman, C.; Bruning, J.B.; Ford, C.M. New insights into the evolutionary history of plant sorbitol dehydrogenase. BMC Plant Biol. 2015, 15, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archbold, D.D.; Nosarzewski, M.; Wu, B.H.; Downie, A.B. The role of sorbitol dehydrogenase in Arabidopsis thaliana. Funct. Plant Biol. 2012, 39, 462–470. [Google Scholar]
- Wang, N.; Huang, H.J.; Ren, S.T.; Li, J.J.; Sun, Y.; Sun, D.Y.; Zhang, S.Q. The rice wall-associated receptor-like kinase gene OsDEES1 plays a role in female gametophyte development. Plant Physiol. 2012, 160, 696–707. [Google Scholar] [CrossRef] [Green Version]
- Wagner, T.A.; Kohorn, B.D. Wall-associated kinases are expressed throughout plant development and are required for cell expansion. Plant Cell 2001, 13, 303–318. [Google Scholar] [CrossRef] [Green Version]
- Kato, Y.; Murakami, S.; Yamamoto, Y.; Chatani, H.; Kondo, Y.; Nakano, T.; Yokota, A.; Sato, F. The DNA-binding protease, CND41, and the degradation of ribulose-1,5-bisphosphate carboxylase/oxygenase in senescent leaves of tobacco. Planta 2004, 220, 97–104. [Google Scholar] [CrossRef]
- Asakura, T.; Watanabe, H.; Abe, K.; Arai, S. Rice aspartic proteinase, oryzasin, expressed during seed ripening and germination, has a gene organization distinct from those of animal and microbial aspartic proteinases. Eur. J. Biochem. 1995, 232, 77–83. [Google Scholar] [CrossRef]
- Guevara, M.G.; Oliva, C.R.; Huarte, M.; Daleo, G.R. An aspartic protease with antimicrobial activity is induced after infection and wounding in intercellular fluids of potato tubers. Eur. J. Plant Pathol. 2002, 108, 131–137. [Google Scholar] [CrossRef]
- Ge, X.C.; Dietrich, C.; Matsuno, M.; Li, G.J.; Berg, H.; Xia, Y.J. An Arabidopsis aspartic protease functions as an anti-cell-death component in reproduction and embryogenesis. Embo Rep. 2005, 6, 282–288. [Google Scholar] [CrossRef] [Green Version]
- Sebastián, D.; Fernando, F.D.; Raúl, D.G.; Gabriela, G.M. Overexpression of Arabidopsis aspartic protease APA1 gene confers drought tolerance. Plant Sci. 2020, 292, 110406. [Google Scholar] [CrossRef]
- Zhang, P.; Zhang, Z.; Zhang, L.J.; Wang, J.J.; Wu, C.S. Glycosyltransferase GT1 family: Phylogenetic distribution, substrates cover-age, and representative structural features. Comput. Struct. Biotechnol. J. 2020, 18, 1383–1390. [Google Scholar] [CrossRef]
- Yin, Q.G.; Shen, G.A.; Di, S.K.; Fan, C.Y.; Chang, Z.Z.; Pang, Y.Z. Genome-wide identification and functional characterization of UDP-glucosyltransferase genes involved in flavonoid biosynthesis in Glycine max. Plant Cell Physiol. 2017, 58, 1558–1572. [Google Scholar] [CrossRef] [Green Version]
- Zheng, R.R.; Zhu, Z.Y.; Wang, Y.L.; Hu, S.Y.; Xi, W.; Xiao, W.; Qu, X.L.; Zhong, L.L.; Fu, Q.; Wang, C.Y. UGT85A84 catalyzes the glycosylation of aromatic monoterpenes in Osmanthus fragrans Lour. flowers. Front. Plant Sci. 2019, 10, 1376. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Huang, X.X.; Zhao, S.M.; Xiao, D.W.; Xiao, L.T.; Tong, J.H.; Wang, W.S.; Li, Y.J.; Ding, Z.J.; Hou, B.K. IPyA glucosylation mediates light and temperature signaling to regulate auxin -dependent hypocotyl elongation in Arabidopsis. Proc. Natl. Acad. Sci. USA 2020, 117, 6910. [Google Scholar] [CrossRef]
- Mohnike, L.; Rekhter, D.; Huang, W.J.; Feussner, K.; Tian, H.Y.; Herrfurth, C.; Zhang, Y.L.; Feussne, L. The glycosyltransferase UGT76B1 modulates N-hydroxy-pipecolicacid homeostasis and plant immunity. Plant Cell 2021, 33, 735–749. [Google Scholar] [CrossRef] [PubMed]
- Nayidu, N.K.; Wang, L.; Xie, W.B.; Zhang, C.J.; Fan, C.Z.; Zhang, Q.F.; Xiong, L.Z. Comprehensive sequence and expression profile analysis of PEX11 gene family in rice. Gene 2008, 412, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Mindthoff, S.; Grunau, S.; Steinfort, L.L.; Girzalsky, W.; Hiltunen, J.K.; Erdmann, R.; Antonenkov, V.D. Peroxisomal Pex11 is a pore-forming protein homologous to TRPM channels. BBA-Mol. Cell Res. 2016, 1863, 271–283. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Jiang, M.; Ren, J.; Liu, Z.; Zhang, W.; Li, G.; Wang, J.; Yang, M. Transcriptomic Determination of the Core Genes Regulating the Growth and Physiological Traits of Quercus mongolica Fisch. ex Ledeb. Forests 2023, 14, 1313. https://doi.org/10.3390/f14071313
Li X, Jiang M, Ren J, Liu Z, Zhang W, Li G, Wang J, Yang M. Transcriptomic Determination of the Core Genes Regulating the Growth and Physiological Traits of Quercus mongolica Fisch. ex Ledeb. Forests. 2023; 14(7):1313. https://doi.org/10.3390/f14071313
Chicago/Turabian StyleLi, Xinman, Min Jiang, Junjie Ren, Zhaohua Liu, Wanying Zhang, Guifen Li, Jinmao Wang, and Minsheng Yang. 2023. "Transcriptomic Determination of the Core Genes Regulating the Growth and Physiological Traits of Quercus mongolica Fisch. ex Ledeb" Forests 14, no. 7: 1313. https://doi.org/10.3390/f14071313
APA StyleLi, X., Jiang, M., Ren, J., Liu, Z., Zhang, W., Li, G., Wang, J., & Yang, M. (2023). Transcriptomic Determination of the Core Genes Regulating the Growth and Physiological Traits of Quercus mongolica Fisch. ex Ledeb. Forests, 14(7), 1313. https://doi.org/10.3390/f14071313