

Chemical Composition of Larch Oleoresin before and during Thermal Modification


Abstract
:1. Introduction
2. Materials and Methods
2.1. Thermal Modification
2.2. Gas Chromatography–Mass Spectrometry of Oleoresin
2.3. Fourier Transform Infrared (FTIR) Spectroscopy
2.4. Temperature Simulations Using TGA/DSC
3. Results
3.1. FTIR Spectra
3.2. GC-MS for Oleoresins from Untreated and Thermally Treated Wood

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Larch Oleoresin | Oleoresin from Mild Thermally Modified Wood |
|---|---|---|
| α-pinene | dominant | medium |
| β-pinene | large | small |
| camphene | minor | absent |
| Δ-3-carene | medium | absent |
| d-limonene | minor | absent |
| l-α-terpineol | minor | minor |
| α-terpineol acetate | minor | absent |
| longifolene | minor | absent |
| γ-elemene | minor | minor |
| one of the germacrenes | minor | absent |
| germacrene D | small | absent |
| β-cadinene (or δ-cadinene) | minor | absent |
| germacrene B | minor | absent |
| thunbergol | medium | absent |
| epi-manool | medium | present—strong |
| isopimara 7,15-dien-3-one | small | minor |
| isopimara 7,15-dienol? | medium | minor |
| a compound peak mixture | medium | one component still present |
| larixol | medium | absent |
| torulosol | small | strong |
| dehydroabietol or neoabietinol | small | minor |
| palustric acid | large | absent |
| torulosol acetate | small | medium—large |
| isopimaric acid (?) | small | minor |
| abietic acid | small | minor |
| neoabietic acid | small | absent |
| thunbergene | not present | present—medium |
| pimaradiene | not present | present—small |
| benzenedicarboxylic acid, ester | not present | (33.76) |
| unidentified | not present | (35.29) |
| squalene (C30H50) | not present | present—dominant peak (37.08) |

| α-Pinene | β-Pinene | Sum of Sesquiterpenes | Sum of Diterpenes | |
|---|---|---|---|---|
| Untreated CC | 20.67% | 6.66% | 2.37% | 70.30% |
| TM CC mild | 6.41% | 1.14% | 2.51% | 89.9% |
| TM CC moderate, liquid resin | 10.71% | 0.67% | 0.66% | 87.96% |
| TM CC moderate, dry resin | 9.48% | 0.60% | 0.98% | 88.94% |
| Untreated CM | 35.19% | 2.94% | 3.04% | 58.84% |
| TM high CMT | 31.7% | 3.64% | 4.52% | 60.14% |
3.3. TGA/DSC Temperature Simulations
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brasier, C.; Webber, J. Sudden larch death. Nature 2010, 466, 824–825. [Google Scholar] [CrossRef] [PubMed]
- Spear, M.; Binding, T.; Jenkins, D.; Nicholls, J.; Ormondroyd, G. Mild thermal modification to enhance the machinability of larch. In Proceedings of the 7th European Conference on Wood Modification, Lisbon, Portugal, 10–12 March 2014. [Google Scholar]
- Spear, M.J.; Binding, T.; Nicholls, J.; Jenkins, D.; Ormondroyd, G.A. Physical properties of UK grown larch subjected to mild and moderate thermal modification processes. In Proceedings of the 11th Meeting of the Nordic European Network for Wood Sciences and Engineering, Poznan, Poland, 14–15 September 2015; Perdoch, W., Broda, M., Eds.; pp. 98–104. [Google Scholar]
- Esteves, B.; Pereira, H. Wood modification by heat treatment: A review. Bioresources 2009, 4, 370–404. [Google Scholar] [CrossRef]
- Fritz, B.; Lamb, B.; Westberg, H.; Folk, R.; Knighton, B.; Grimsrud, E. Pilot- and full-scale measurements of VOC emissions from lumber drying of inland Northwest species. For. Prod. J. 2004, 54, 50–56. [Google Scholar]
- Dahlen, J.; Prewitt, L.; Schmulsky, R.; Jones, D. Hazardous air pollutants and volatile organic compounds emitted during kiln drying of Southern pine lumber to interior and export moisture specifications. For. Prod. J. 2011, 61, 229–234. [Google Scholar] [CrossRef]
- Pang, S. Emissions from kiln drying of Pinus radiata timber: Analysis, recovery and treatment. Dry. Technol. 2012, 30, 1099–1104. [Google Scholar] [CrossRef]
- Manninen, A.M.; Pasanen, P.; Holopainen, J.K. Comparing the VOC emissions between air-dried and heat-treated Scots pine wood. Atmos. Environ. 2002, 36, 1763–1768. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, Y.; Shen, Y.; Li, C.; Wang, Y.; Ma, Y.; Sun, W. New perspective on wood thermal modification: Relevance between the evolution of chemical structure and physical-mechanical properties, and online analysis of release of VOCs. Polymers 2019, 11, 1145. [Google Scholar] [CrossRef] [PubMed]
- Granström, K. Emissions of monoterpenes and VOCs during drying of sawdust in a spouted bed. For. Prod. J. 2003, 53, 43–55. [Google Scholar]
- Banerjee, S.; Su, W.; Wild, M.P.; Otwell, L.P.; Hittmeier, M.E.; Nichols, K.M. Wet line extension reduces VOCs from softwood drying. Environ. Sci. Technol. 1998, 32, 1303–1307. [Google Scholar] [CrossRef]
- Banerjee, S. Mechanisms of terpene release during sawdust and flake drying. Holzforschung 2001, 55, 413–416. [Google Scholar] [CrossRef]
- Peters, J.; Fischer, K.; Fischer, S. Characterisation of emissions from thermally modified wood and their reduction by chemical treatment. BioResources 2008, 3, 491–502. [Google Scholar] [CrossRef]
- Herrera, R.; da Silva, D.T.; Llano-Ponte, R.; Labidi, J. Characterization of pine wood liquid and solid residues generated during industrial hydrothermal treatment. Biomass Bioenergy 2016, 95, 174–181. [Google Scholar] [CrossRef]
- Hyttinen, M.; Masalinweijo, M.; Kalliokoski, P.; Pasanen, P. Comparison of VOC emissions between air-dried and heat-treated Norway spruce (Picea abies), Scots pine (Pinus sylvesteris) and European aspen (Populus tremula) wood. Atmos. Environ. 2010, 44, 5028–5033. [Google Scholar] [CrossRef]
- Kotilainen, R.A.; Toivanen, T.-J.; Alén, R.J. FTIR monitoring of chemical changes in softwood during heating. J. Wood Chem. Technol. 2000, 20, 307–320. [Google Scholar] [CrossRef]
- Sjöström, E. Wood Chemistry: Fundamentals and Applications; Academic Press: San Diego, CA, USA, 1993; 293p. [Google Scholar]
- Mills, J.S. Diterpenes of Larix oleoresins. Phytochemistry 1973, 12, 2407–2412. [Google Scholar] [CrossRef]
- Derrick, M. Fourier Transform Infrared spectral analysis of natural resins used in furniture finishes. J. Am. Inst. Conserv. 1988, 28, 43–56. [Google Scholar] [CrossRef]
- Banthorpe, D.V. Terpenoids. In Natural Products: Their Chemistry and Biological Significance; Longman Scientific and Technical: Harlow, UK, 1994; pp. 289–359. [Google Scholar]
- Holmbom, T.; Reunanen, M.; Fardim, P. Composition of callus resin of Norway spruce, Scots pine, European larch and Douglas fir. Holzforschung 2008, 62, 417–422. [Google Scholar] [CrossRef]
- Holmbom, B.; Avela, E.; Pekkala, S. Capillary gas-chromatography-mass spectrometry of resin acids in tall oil rosin. J. Am. Oil Chem. Soc. 1974, 51, 397–399. [Google Scholar] [CrossRef]
- Sato, M.; Seki, K.; Kita, K.; Moriguchi, Y.; Hashimoto, M.; Yunoki, K.; Ohnishi, M. Comparative analysis of diterpene composition in the bark of the hybrid larch F1, Larix gmelinii var. japonica × L. kaempferi and their parent trees. J. Wood Sci. 2009, 55, 32–40. [Google Scholar] [CrossRef]
- Spear, M.; Binding, T.; Dimitriou, A.; Cowley, C.; Ormondroyd, G. Chemical changes in mild thermal treatment of larch: To what extent to they differ from full thermal modification? In Proceedings of the 8th Eighth European Conference on Wood Modification, Helsinki, Finland, 26–27 October 2015. [Google Scholar]
- Babushok, V.I.; Linstrom, P.J.; Zenkevich, I.G. Retention indices for frequently reported compounds of plant essential oils. J. Phys. Chem. Ref. Data 2011, 40, 043101. [Google Scholar] [CrossRef]
- Silverstein, R.M.; Bassler, G.C.; Morrill, T.C. Spectrometric Identification of Organic Compounds, 4th ed.; Wiley: Hoboken, NJ, USA, 1982. [Google Scholar]
- Chernenko, G.F.; Kobzar, E.A.; Salakhutdinov, N.F.; Schmidt, E.N.; Bagrayanskaya, I.Y.; Galitov, Y.V. Transformations of terpinoids on synthetic zeolites. Khimiya Prir. Soedin. 1991, 5, 657–667, Translated Plenum Publishing Corp, 1992. [Google Scholar]
- Kosyukova, L.V.; Khorguani, T.V. Retention indices of diterpenes isolated from resins of coniferous trees. Zhurnal Anal. Khimii (J. Anal. Chem. USSR) 1989, 44, 1309–1313. [Google Scholar]
- Granström, K.M. Emissions of sesquiterpenes from spruce sawdust during drying. Eur. J. Wood Wood Prod. 2009, 67, 343–350. [Google Scholar] [CrossRef]
- Russo, M.V.; Avino, P. Characterization and identification of natural terpenic resins employed in “Madonna con bambino e angeli” by Antonello da Messina using Gas Chromatography-Mass Spectrometry. Chem. Cent. J. 2012, 6, 59. [Google Scholar] [CrossRef]
- Grüll, G.; Forsthuber, B.; Truskaller, M.; Illy, A. Methods to reduce resin exudation from Larch wood. In Advances in Modified and Functional Bio-Based Surfaces, Proceedings of the COST Action FP1006, Thessaloniki, Greece, 8–9 April 2015; Bangor University: Bangor, UK, 2015. [Google Scholar]
- Kačik, F.; Velkova, V.; Šmira, P.; Nasswettrova, D.; Reinprecht, L. Release of terpenes from fir wood during its long-term use and in thermal treatment. Molecules 2012, 17, 9990–9999. [Google Scholar] [CrossRef]




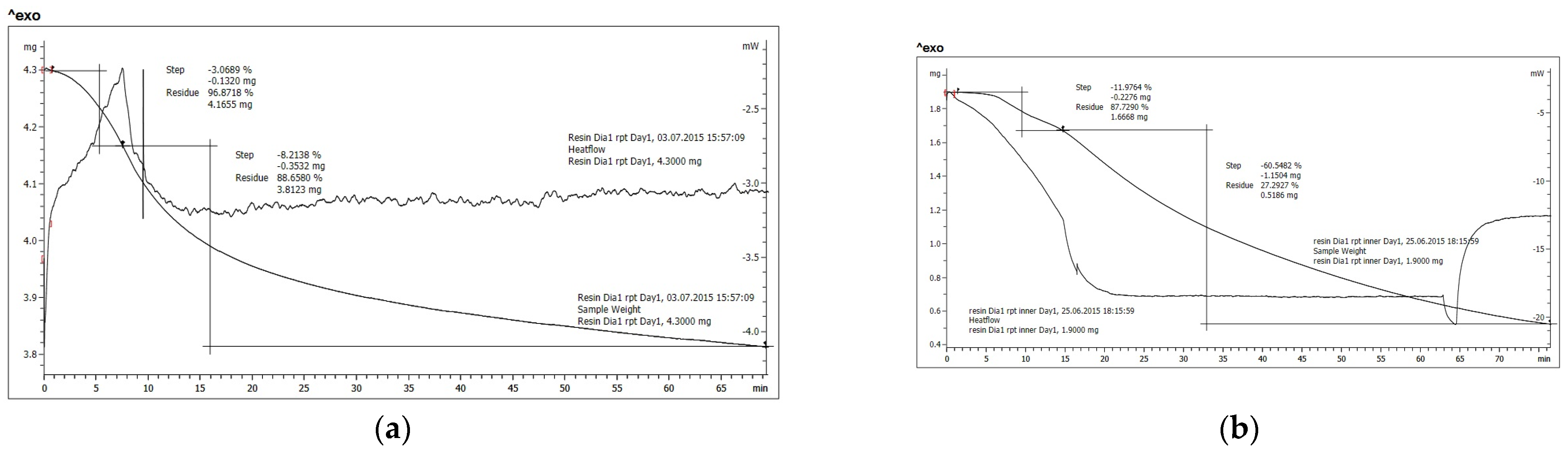


| 120 °C Isotherm | 150 °C Isotherm | 170 °C Isotherm | 190 °C Isotherm | ||
|---|---|---|---|---|---|
| Untreated Resin A | ramp | - | −2.3270% | −3.6629% | - |
| isotherm | - | −12.5246% | −23.1080% | - | |
| Untreated resin CC | ramp | −1.0444% | −2.0191% | −7.1936% | −10.5407% |
| isotherm | −9.5621% | −14.8054% | −31.0611% | −48.5842% | |
| Untreated resin D1 | ramp | −3.0689% | −3.1069% | −7.3155% | −11.9764% |
| isotherm | −8.2138% | −15.5338% | −27.6076% | −60.5482% | |
| Untreated resin D2 | ramp | −2.6315% | −4.8903% | −6.4693% | - |
| isotherm | −8.1963% | −11.3445% | −28.0898% | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spear, M.J.; Dimitriou, A.; Marriott, R. Chemical Composition of Larch Oleoresin before and during Thermal Modification. Forests 2024, 15, 904. https://doi.org/10.3390/f15060904
Spear MJ, Dimitriou A, Marriott R. Chemical Composition of Larch Oleoresin before and during Thermal Modification. Forests. 2024; 15(6):904. https://doi.org/10.3390/f15060904
Chicago/Turabian StyleSpear, Morwenna J., Athanasios Dimitriou, and Ray Marriott. 2024. "Chemical Composition of Larch Oleoresin before and during Thermal Modification" Forests 15, no. 6: 904. https://doi.org/10.3390/f15060904