Role of Viral Ribonucleoproteins in Human Papillomavirus Type 16 Gene Expression


Abstract
:1. Introduction
2. HPV Life Cycle
3. HPV16 Gene Regulation
3.1. The Switch from HPV16 Early to Late Gene Expression
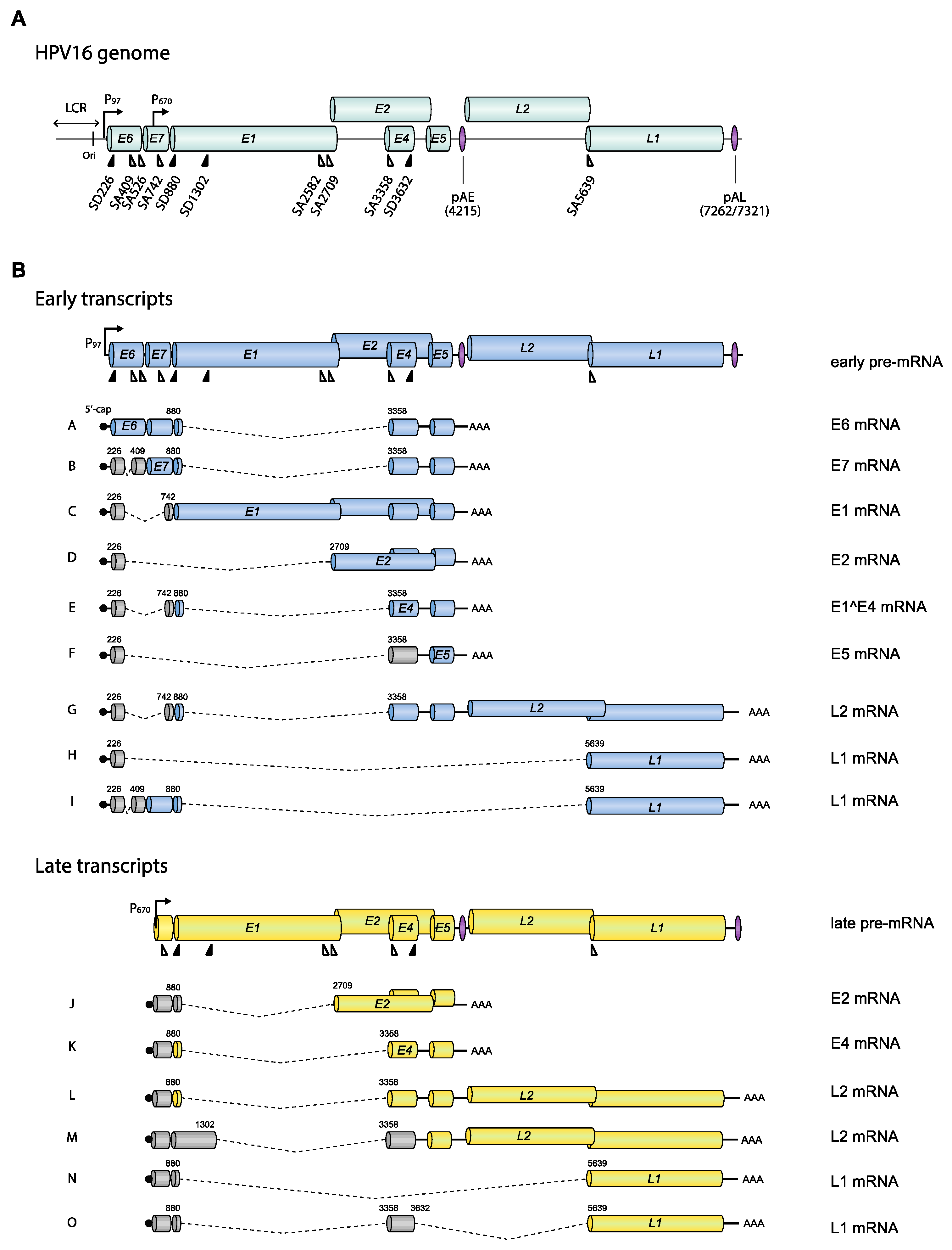
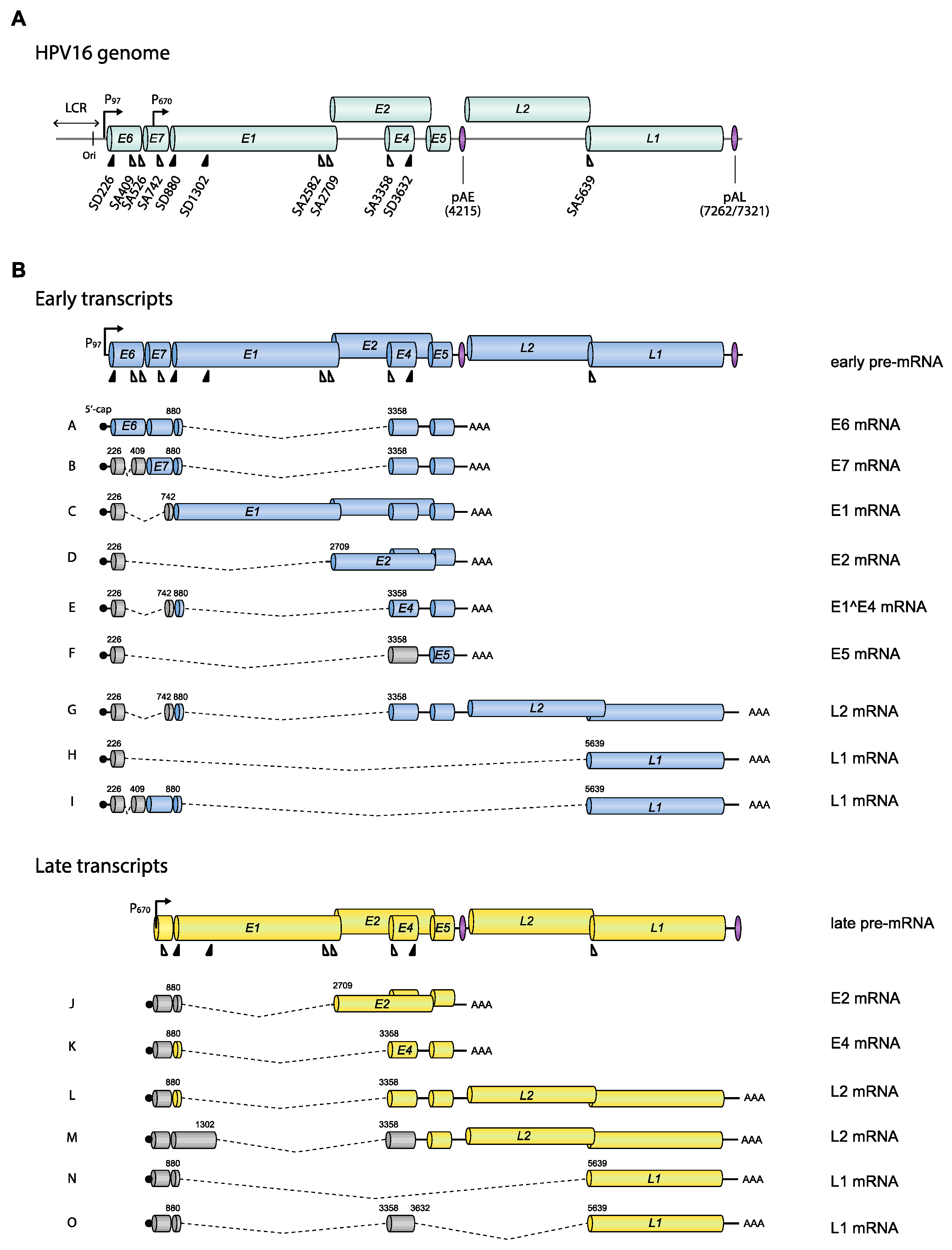
3.2. Exons and Introns on HPV16 mRNAs
4. Regulation of HPV16 Gene Expression Mediated by Interactions between HPV16 RNA and Cellular Proteins
4.1. Alternative Splicing and Polyadenylation
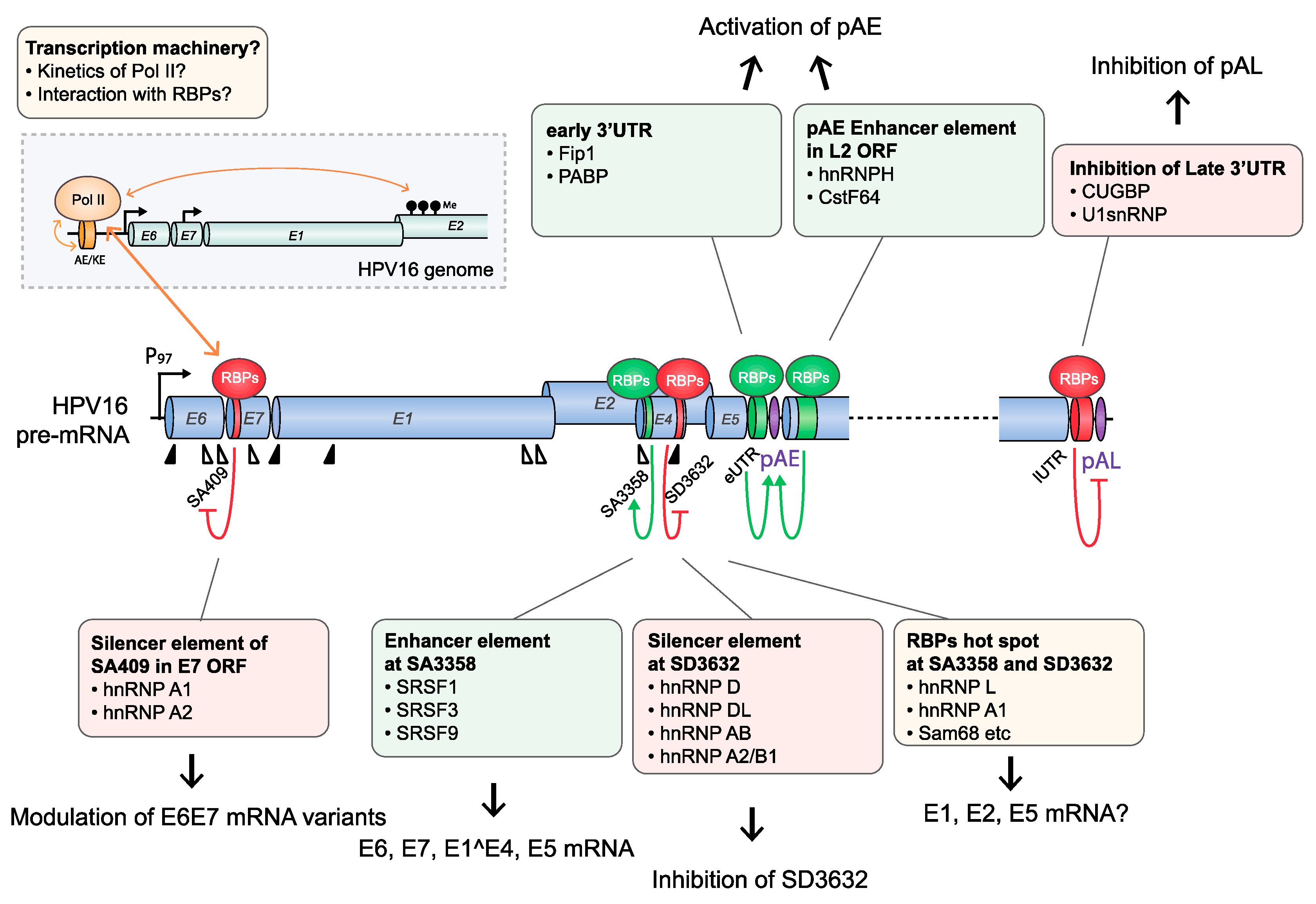
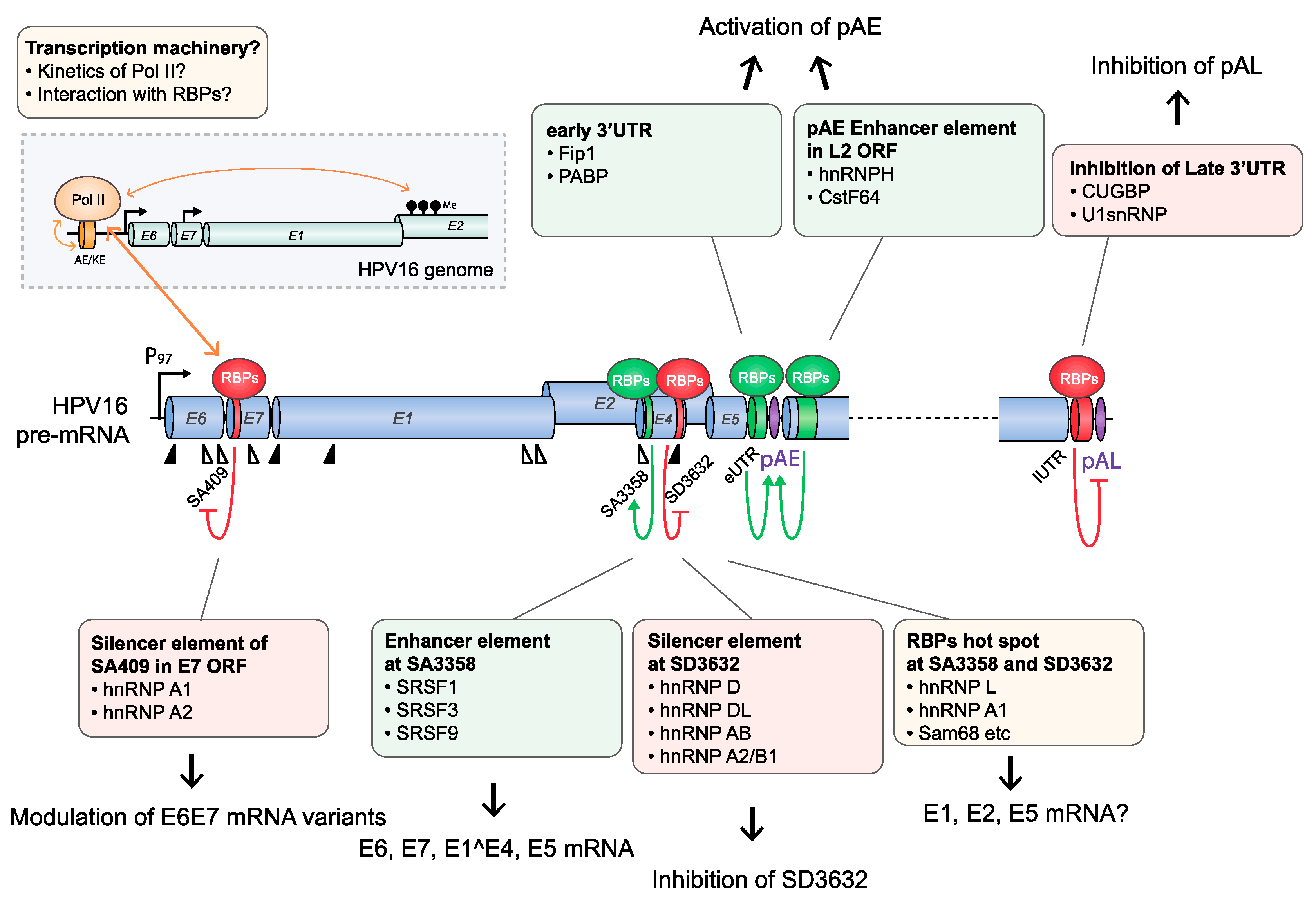
4.1.1. HPV16 Splice Sites
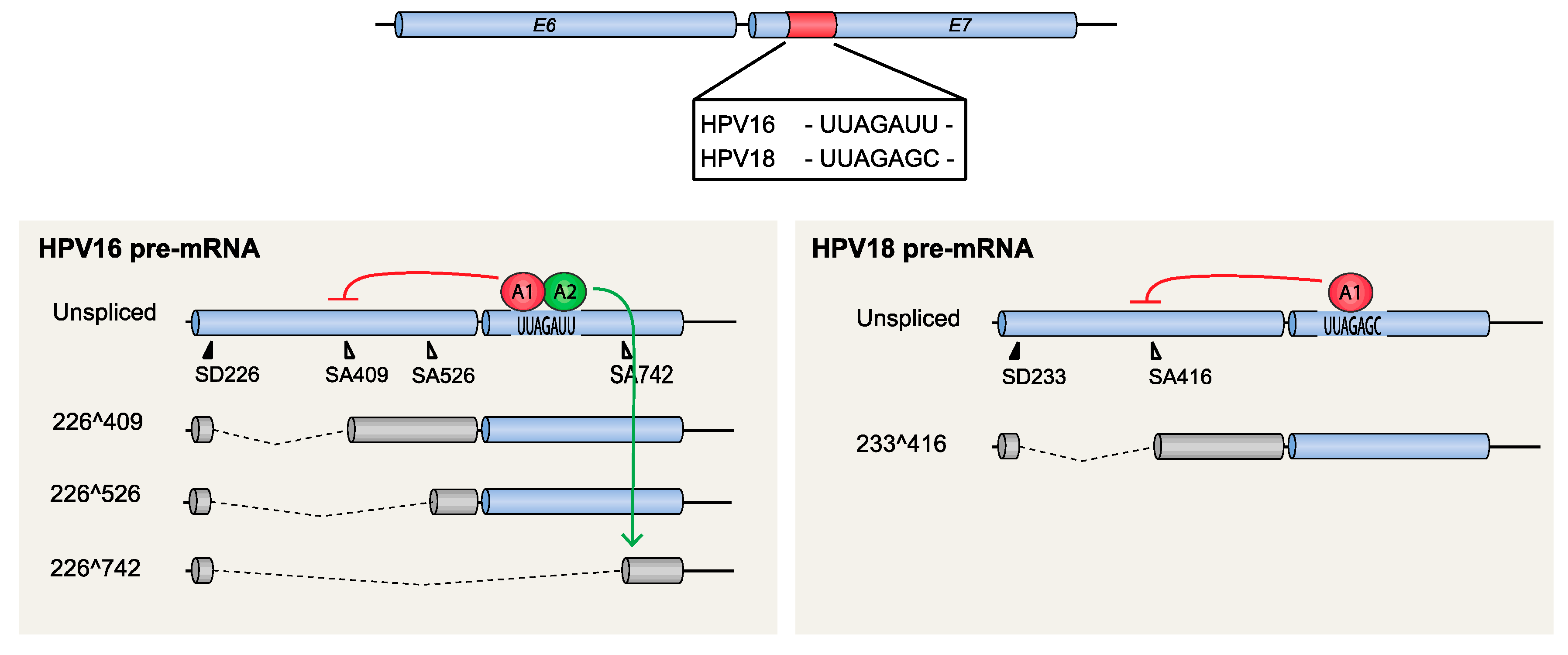
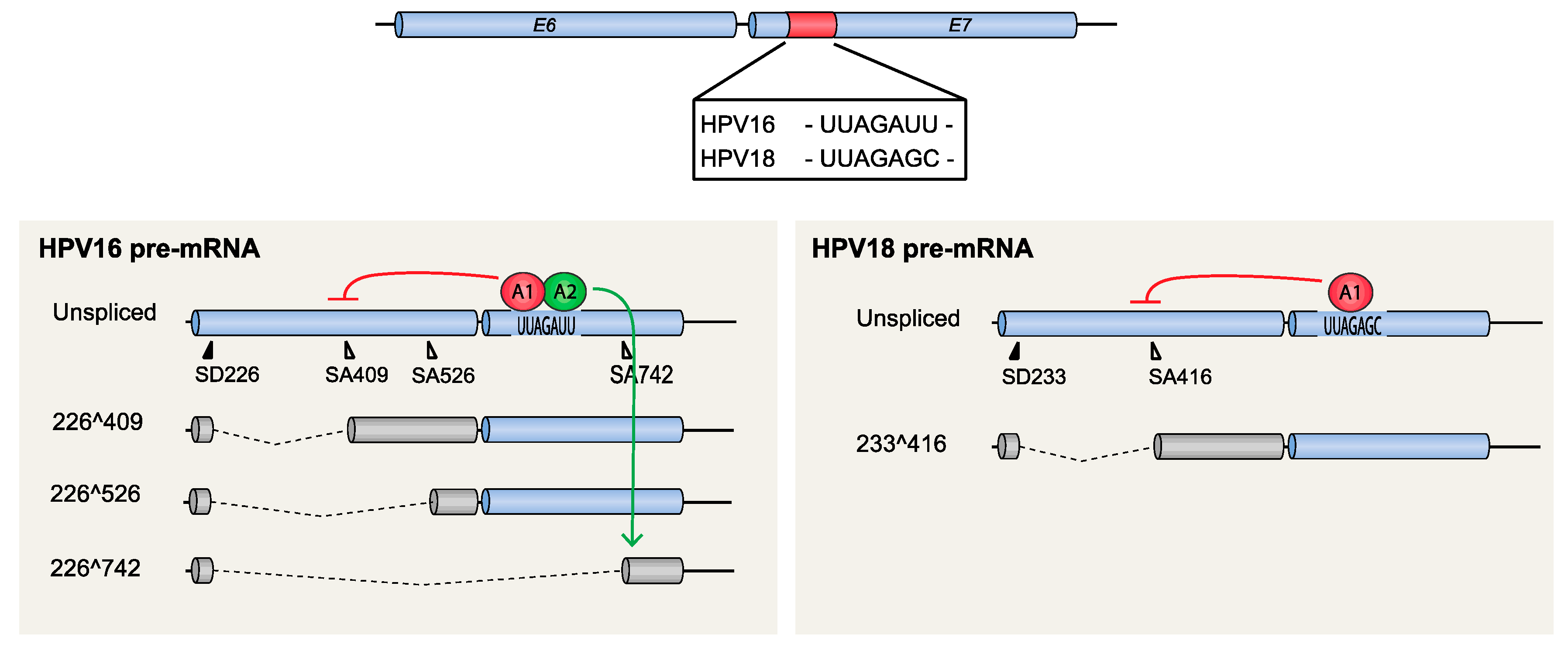
4.1.2. HPV16 Splice Sites SD226, SA409, SA526, and SA742
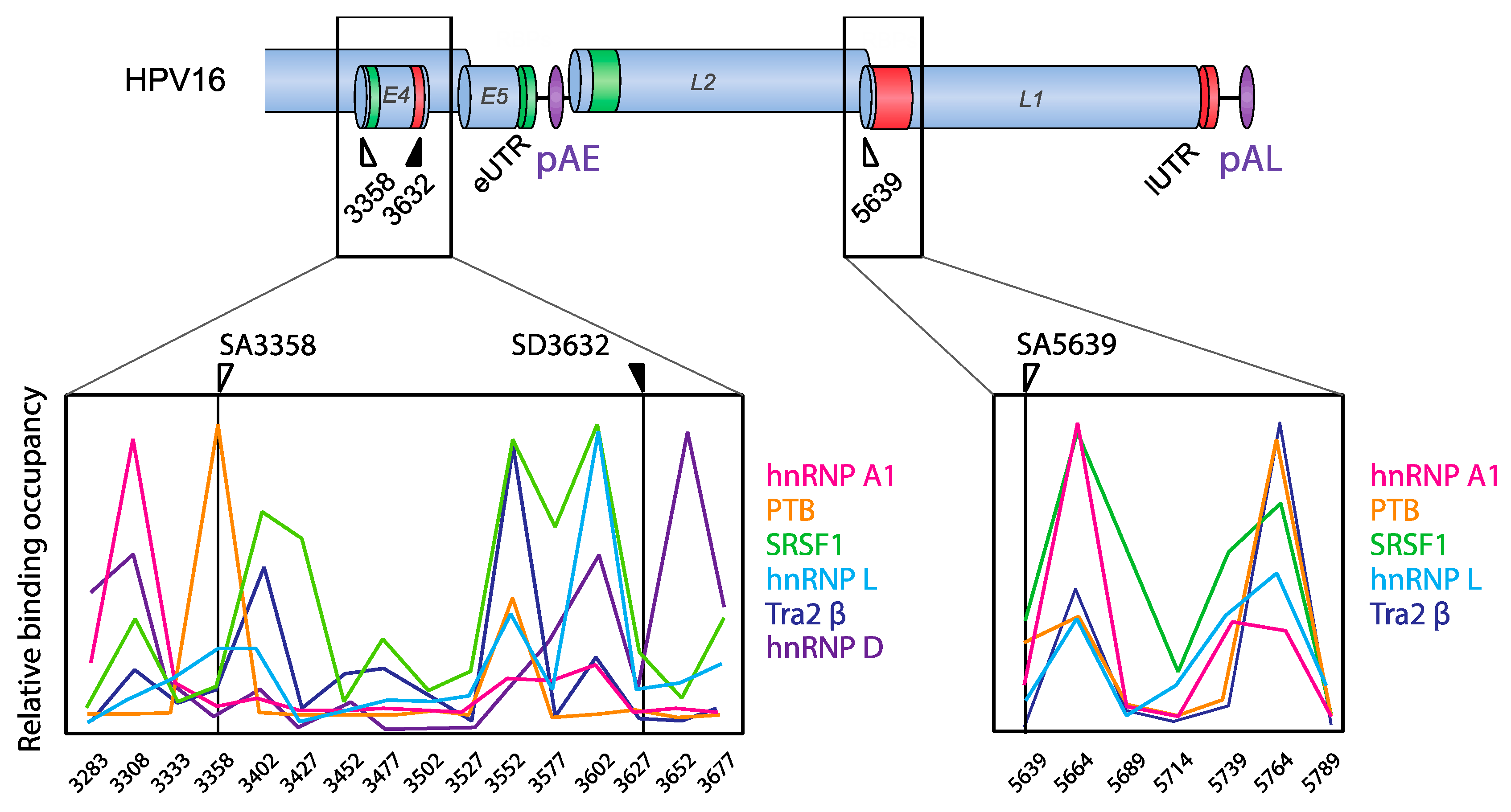
4.1.3. HPV16 Splice Site SA3358
4.1.4. HPV16 Splice Sites SD3632 and SA5639
4.1.5. HPV16 Early and Late 3′-UTR Sequences and Polyadenylation Signals
4.1.6. The 5′-Untranslated Region of HPV16
4.1.7. What Do the Various HPV16 mRNAs Produce?
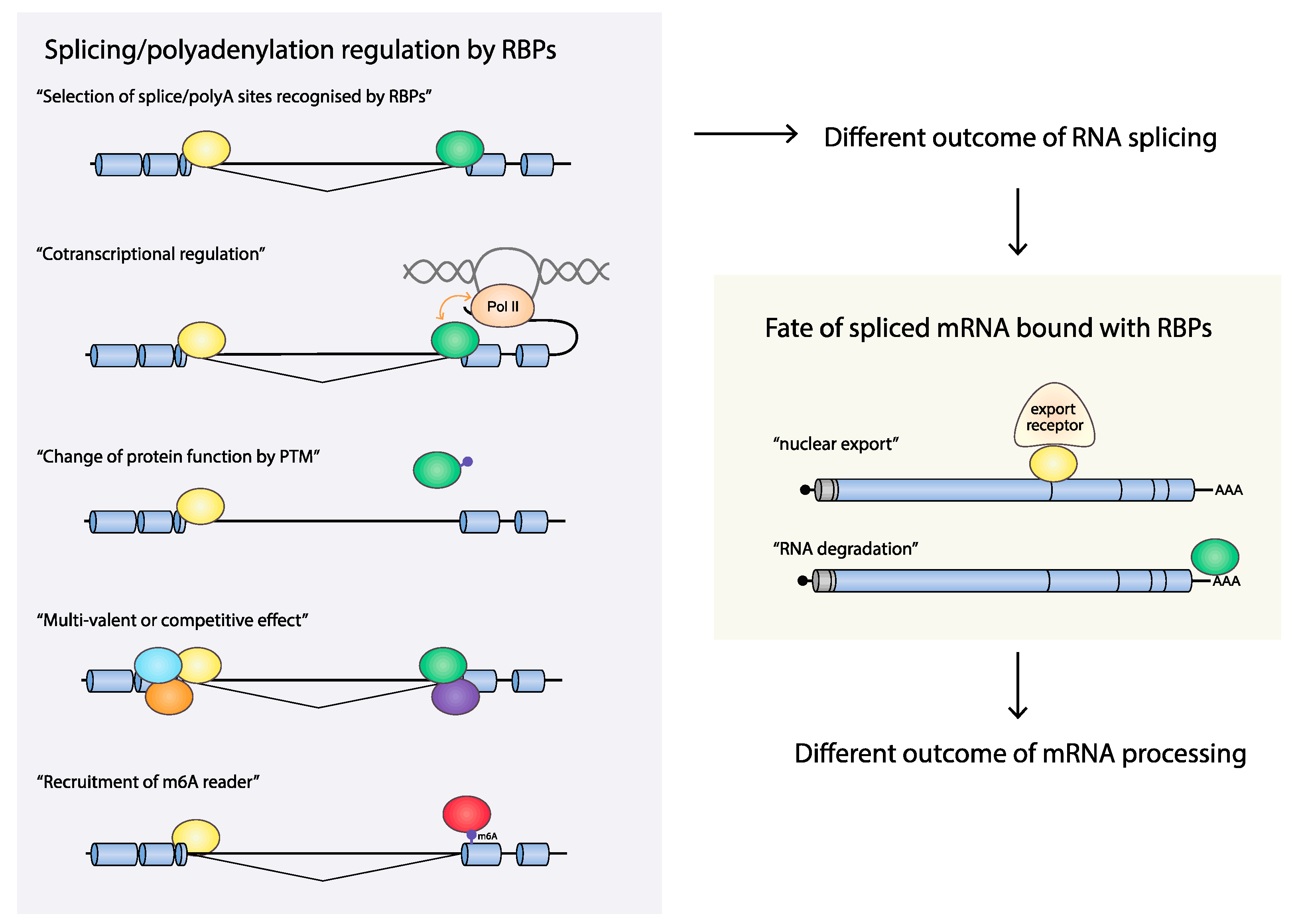
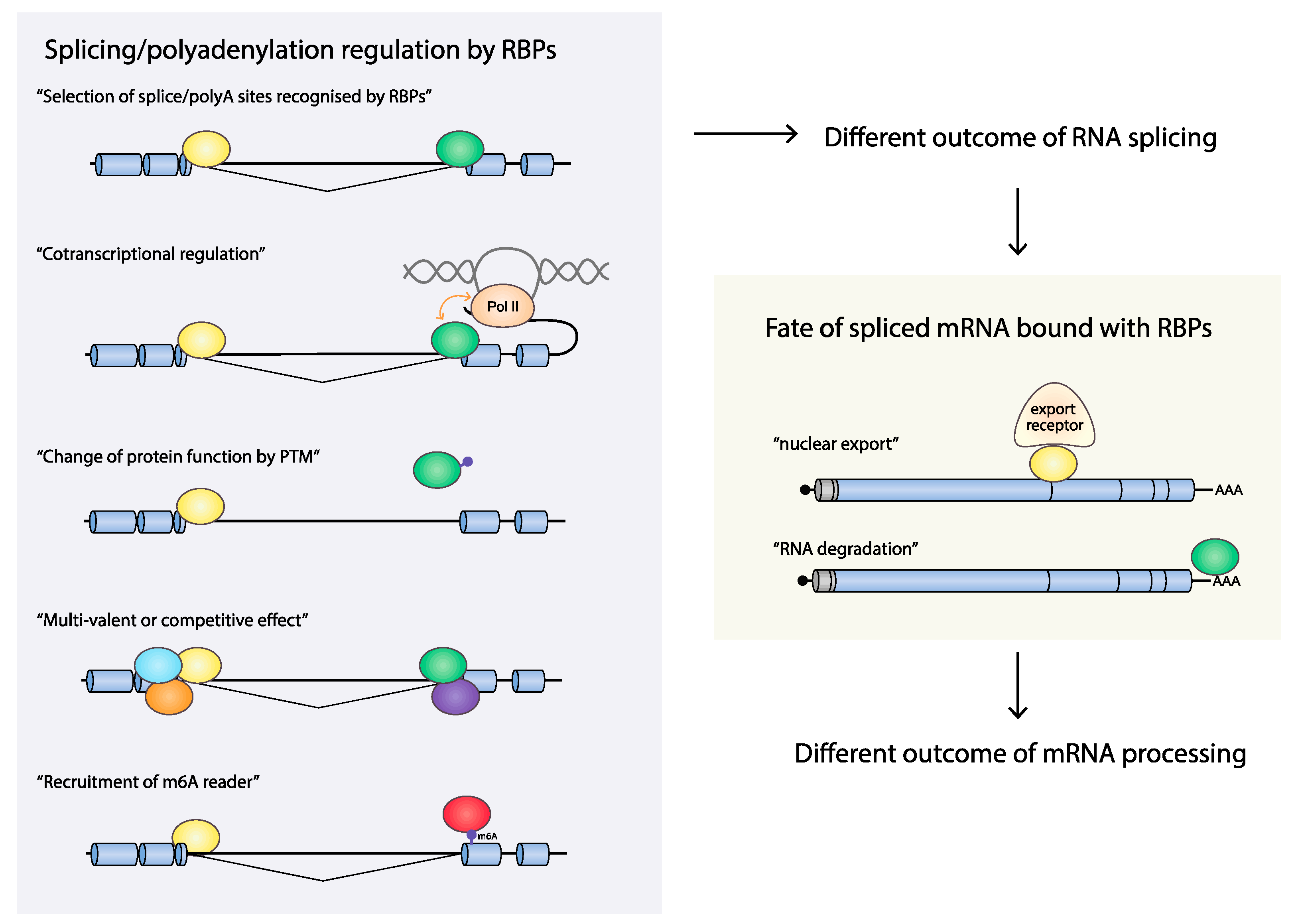
4.2. Are Splicing and Polyadenylation of HPV mRNAs Cotranscriptionally Controlled?
4.2.1. Cotranscriptional Control of RNA Processing
4.2.2. HPV16 RNA Processing May Be Cotranscriptionally Regulated
4.3. mRNA Export
4.4. RNA Stability
4.5. m6A Modification on HPV mRNAs
5. The HPV Life Cycle and Ribonucleoproteins
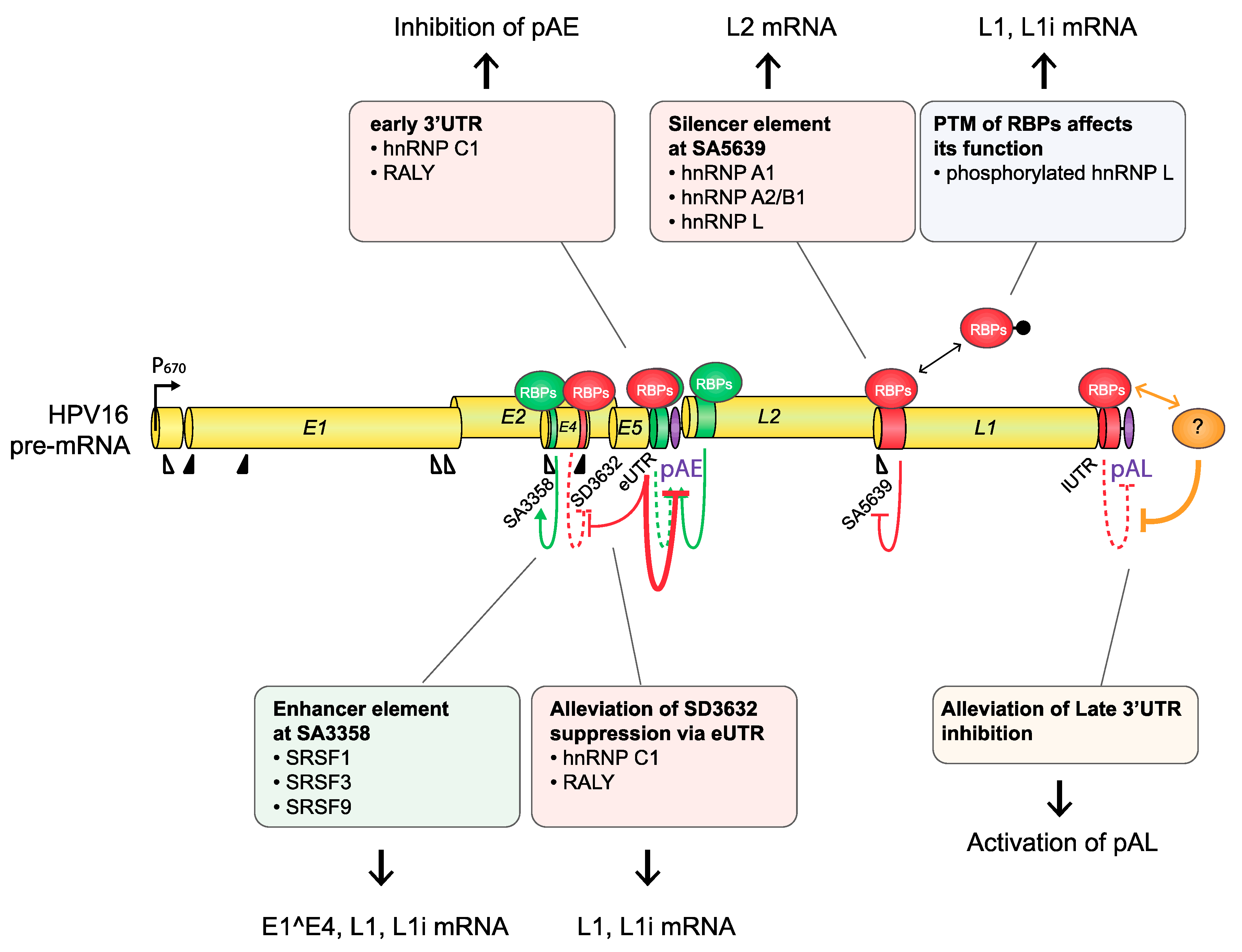
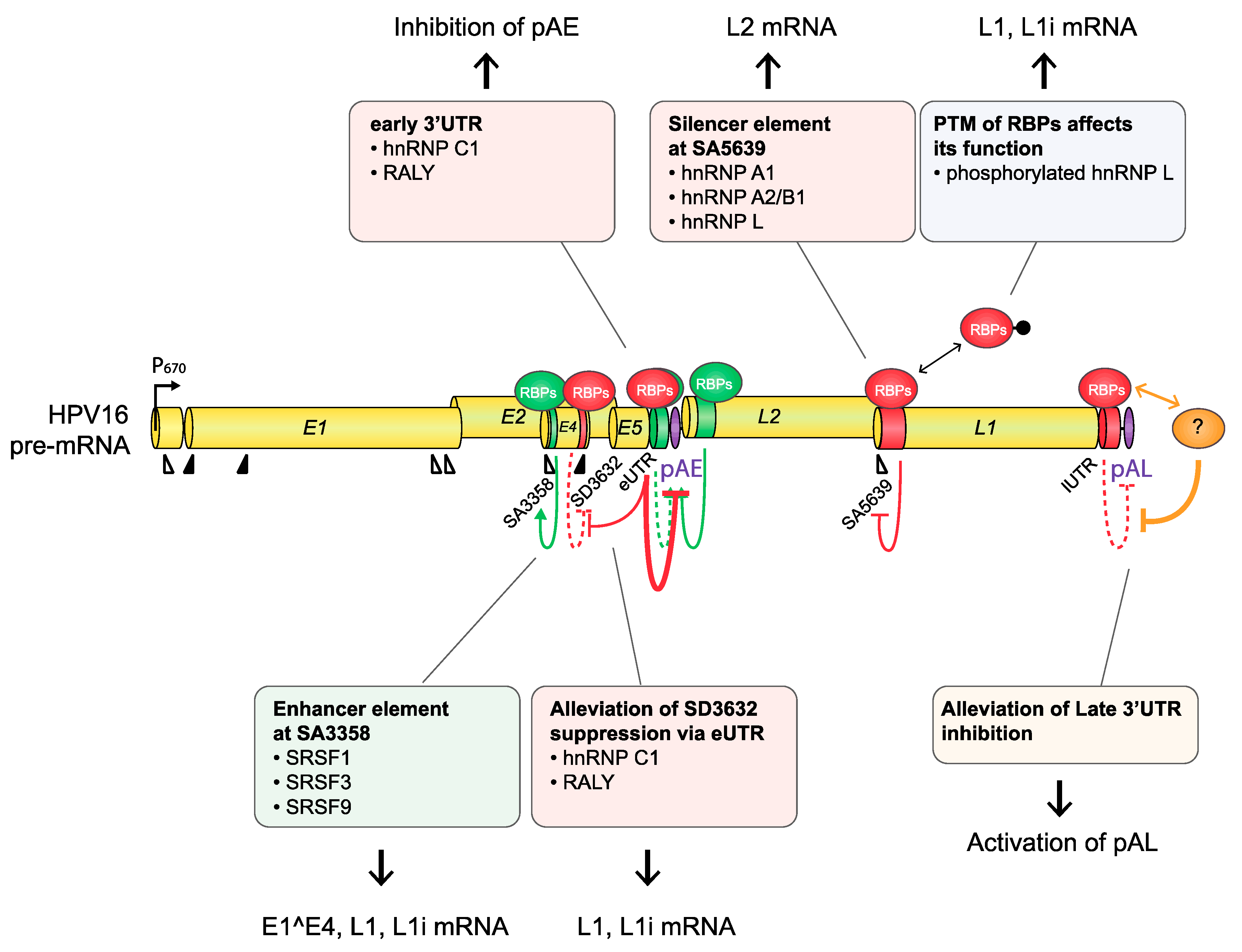
5.1. Expression Levels of RNA-Binding Proteins Contribute to the Control of HPV mRNA Processing
5.2. Posttranslational Modifications of RNA-Binding Proteins Contribute to the Control of HPV mRNA Processing

5.3. Differentiation and HPV RNA Processing
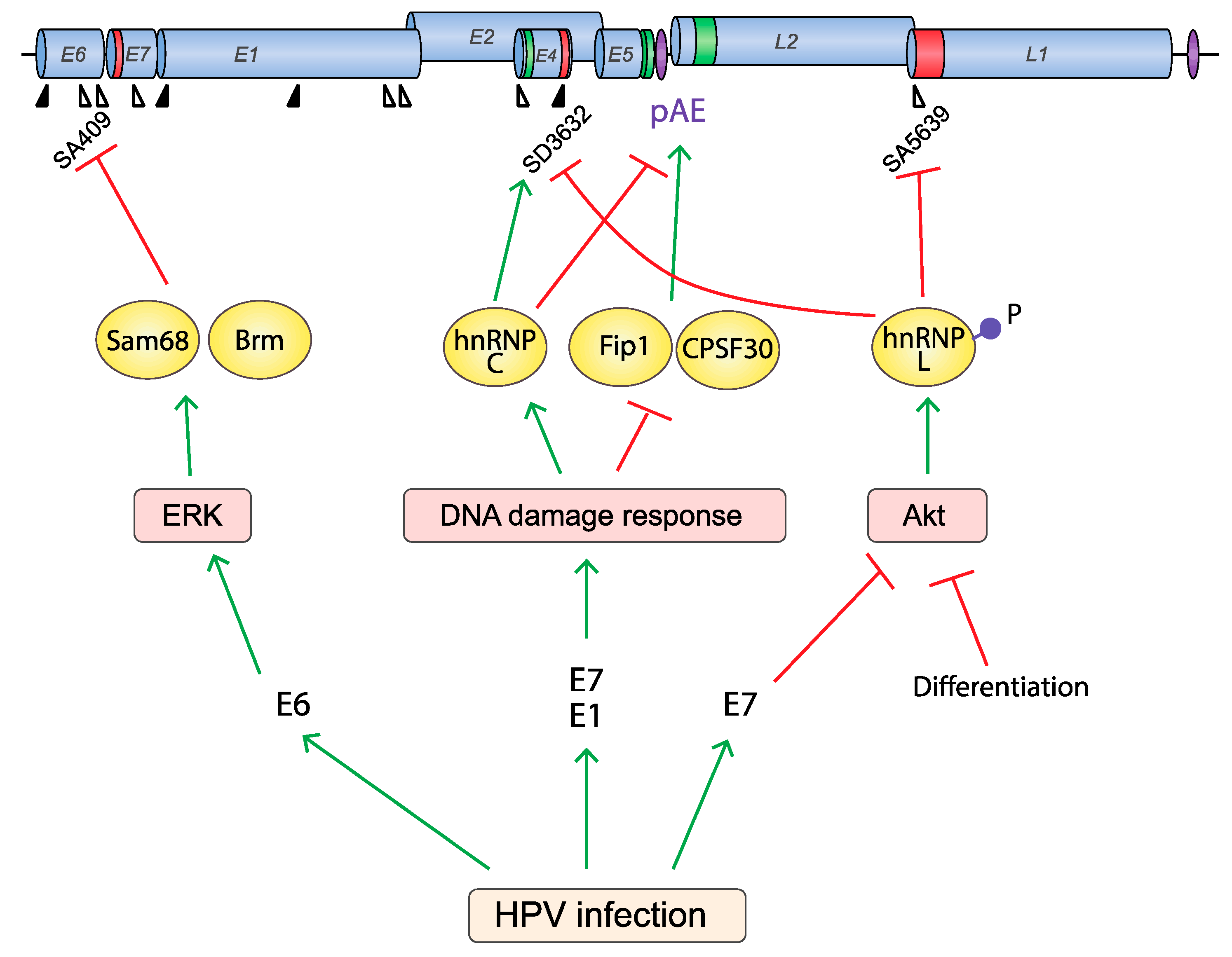
5.4. Do HPV Proteins Affect HPV RNA Processing?
6. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| DDR | DNA damage response |
| CDS | coding sequence |
| hnRNP | heterogeneous nuclear ribonucleoprotein |
| HPV | human papillomavirus |
| ORF | open reading frame |
| Ori | origin of replication |
| pAE | early polyadenylation signal |
| pAL | late polyadenylation signal |
| Pol II | RNA polymerase II |
| RBP | RNA binding proteins |
| SA | splice acceptor (3′-splice site) |
| SD | splice donor (5′-splice site) |
| SR | serine-arginine rich protein |
| UTR | untranslated region |
References
- Zur Hausen, H. Papillomaviruses and cancer: From basic studies to clinical application. Nat. Rev. Cancer 2002, 2, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Howley, P.M.; Lowy, D.R. Papillomaviridae. In Virology, 5th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott/The Williams & Wilkins Co.: Philadelphia, PA, USA, 2006; Volume 2, pp. 2299–2354. [Google Scholar]
- Moscicki, A.B.; Schiffman, M.; Burchell, A.; Albero, G.; Giuliano, A.R.; Goodman, M.T.; Kjaer, S.K.; Palefsky, J. Updating the natural history of human papillomavirus and anogenital cancers. Vaccine 2012, 30, F24–F33. [Google Scholar] [CrossRef] [Green Version]
- Woodman, C.B.; Collins, S.; Winter, H.; Bailey, A.; Ellis, J.; Prior, P.; Yates, M.; Rollason, T.P.; Young, L.S. Natural history of cervical human papillomavirus infection in young women: A longitudinal cohort study. Lancet 2001, 357, 1831–1836. [Google Scholar] [CrossRef]
- Chow, L.T.; Broker, T.R.; Steinberg, B.M. The natural history of human papillomavirus infections of the mucosal epithelia. APMIS 2010, 118, 422–449. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; Banks, L. Molecular mechanisms underlying human papillomavirus e6 and e7 oncoprotein-induced cell transformation. Mutat. Res. Rev. Mutat. Res. 2017, 772, 23–35. [Google Scholar] [CrossRef]
- Moody, C.A.; Laimins, L.A. Human papillomavirus oncoproteins: Pathways to transformation. Nat. Rev. Cancer 2010, 10, 550–560. [Google Scholar] [CrossRef]
- Spurgeon, M.E.; Lambert, P.F. Human papillomavirus and the stroma: Bidirectional crosstalk during the virus life cycle and carcinogenesis. Viruses 2017, 9, 219. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, A.L.; Caodaglio, A.S.; Sichero, L. Regulation of hpv transcription. Clinics 2018, 73, e486s. [Google Scholar] [CrossRef]
- Bernard, H.U. Regulatory elements in the viral genome. Virology 2013, 445, 197–204. [Google Scholar] [CrossRef] [Green Version]
- Graham, S.V.; Faizo, A.A.A. Control of human papillomavirus gene expression by alternative splicing. Virus Res. 2017, 231, 83–95. [Google Scholar] [CrossRef] [Green Version]
- Ajiro, M.; Zheng, Z.M. Oncogenes and rna splicing of human tumor viruses. Emerg. Microbes Infect. 2014, 3, e63. [Google Scholar] [CrossRef] [PubMed]
- Johansson, C.; Schwartz, S. Regulation of human papillomavirus gene expression by splicing and polyadenylation. Nat. Rev. Microbiol. 2013, 11, 239–251. [Google Scholar] [CrossRef]
- Kajitani, N.; Schwartz, S. Rna binding proteins that control human papillomavirus gene expression. Biomolecules 2015, 5, 758–774. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S. Papillomavirus transcripts and posttranscriptional regulation. Virology 2013, 445, 187–196. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Kajitani, N.; Schwartz, S. Splicing and polyadenylation of human papillomavirus type 16 mrnas. Int. J. Mol. Sci. 2017, 18, 366. [Google Scholar] [CrossRef] [PubMed]
- Cerasuolo, A.; Buonaguro, L.; Buonaguro, F.M.; Tornesello, M.L. The role of rna splicing factors in cancer: Regulation of viral and human gene expression in human papillomavirus-related cervical cancer. Front. Cell Dev. Biol. 2020, 8, 474. [Google Scholar] [CrossRef] [PubMed]
- Kadaja, M.; Silla, T.; Ustav, E.; Ustav, M. Papillomavirus DNA replication from initiation to genomic instability. Virology 2009, 384, 360–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McBride, A.A. Mechanisms and strategies of papillomavirus replication. Biol. Chem. 2017, 398, 919–927. [Google Scholar] [CrossRef]
- Doorbar, J.; Parton, A.; Hartley, K.; Banks, L.; Crook, T.; Stanley, M.; Crawford, L. Detection of novel splicing patterns in a hpv16-containing keratinocyte cell line. Virology 1990, 178, 254–262. [Google Scholar] [CrossRef]
- Milligan, S.G.; Veerapraditsin, T.; Ahamet, B.; Mole, S.; Graham, S.V. Analysis of novel human papillomavirus type 16 late mrnas in differentiated w12 cervical epithelial cells. Virology 2007, 360, 172–181. [Google Scholar] [CrossRef] [Green Version]
- Doorbar, J.; Quint, W.; Banks, L.; Bravo, I.G.; Stoler, M.; Broker, T.R.; Stanley, M.A. The biology and life-cycle of human papillomaviruses. Vaccine 2012, 30, F55–F70. [Google Scholar] [CrossRef] [PubMed]
- Archambault, J.; Melendy, T. Targeting human papillomavirus genome replication for antiviral drug discovery. Antivir. Ther. 2013, 18, 271–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horvath, C.A.; Boulet, G.A.; Renoux, V.M.; Delvenne, P.O.; Bogers, J.P. Mechanisms of cell entry by human papillomaviruses: An overview. Virol. J. 2010, 7, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longworth, M.S.; Laimins, L.A. Pathogenesis of human papillomaviruses in differentiating epithelia. Microbiol. Mol. Biol. Rev. 2004, 68, 362–372. [Google Scholar] [CrossRef] [Green Version]
- Roman, A.; Munger, K. The papillomavirus e7 proteins. Virology 2013, 445, 138–168. [Google Scholar] [CrossRef] [Green Version]
- Vande Pol, S.B.; Klingelhutz, A.J. Papillomavirus e6 oncoproteins. Virology 2013, 445, 115–137. [Google Scholar] [CrossRef] [Green Version]
- Anacker, D.C.; Moody, C.A. Modulation of the DNA damage response during the life cycle of human papillomaviruses. Virus Res. 2017, 231, 41–49. [Google Scholar] [CrossRef] [Green Version]
- McBride, A.A. The papillomavirus e2 proteins. Virology 2013, 445, 57–79. [Google Scholar] [CrossRef] [Green Version]
- Bergvall, M.; Melendy, T.; Archambault, J. The e1 proteins. Virology 2013, 445, 35–56. [Google Scholar] [CrossRef] [Green Version]
- Doorbar, J. The e4 protein; structure, function and patterns of expression. Virology 2013, 445, 80–98. [Google Scholar] [CrossRef] [Green Version]
- DiMaio, D.; Petti, L.M. The e5 proteins. Virology 2013, 445, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Siddiqa, A.; Broniarczyk, J.; Banks, L. Papillomaviruses and endocytic trafficking. Int. J. Mol. Sci. 2018, 19, 2619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, S.V. Keratinocyte differentiation-dependent human papillomavirus gene regulation. Viruses 2017, 9, 245. [Google Scholar] [CrossRef]
- Hubert, W.G.; Laimins, L.A. Human papillomavirus type 31 replication modes during the early phases of the viral life cycle depend on transcriptional and posttranscriptional regulation of e1 and e2 expression. J. Virol. 2002, 76, 2263–2273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spink, K.M.; Laimins, L.A. Induction of the human papillomavirus type 31 late promoter requires differentiation but not DNA amplification. J. Virol. 2005, 79, 4918–4926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, C.; Somberg, M.; Li, X.; Backström Winquist, E.; Fay, J.; Ryan, F.; Pim, D.; Banks, L.; Schwartz, S. Hpv-16 e2 contributes to induction of hpv-16 late gene expression by inhibiting early polyadenylation. EMBO J. 2012, 31, 3212–3227. [Google Scholar] [CrossRef]
- Terhune, S.S.; Hubert, W.G.; Thomas, J.T.; Laimins, L.A. Early polyadenylation signals of human papillomavirus type 31 negatively regulate capsid gene expression. J. Virol. 2001, 75, 8147–8157. [Google Scholar] [CrossRef] [Green Version]
- Oberg, D.; Collier, B.; Zhao, X.; Schwartz, S. Mutational inactivation of two distinct negative rna elements in the human papillomavirus type 16 l2 coding region induces production of high levels of l2 in human cells. J. Virol. 2003, 77, 11674–11684. [Google Scholar] [CrossRef] [Green Version]
- Ozbun, M.A.; Meyers, C. Characterization of late gene transcripts expressed during vegetative replication of human papillomavirus type 31b. J. Virol. 1997, 71, 5161–5172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ule, J.; Blencowe, B.J. Alternative splicing regulatory networks: Functions, mechanisms, and evolution. Mol. Cell 2019, 76, 329–345. [Google Scholar] [CrossRef] [PubMed]
- Sibley, C.R.; Blazquez, L.; Ule, J. Lessons from non-canonical splicing. Nat. Rev. Genet. 2016, 17, 407–421. [Google Scholar] [CrossRef] [PubMed]
- Busch, A.; Hertel, K.J. Evolution of sr protein and hnrnp splicing regulatory factors. Wiley Interdiscip. Rev. RNA 2012, 3, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piovesan, A.; Antonaros, F.; Vitale, L.; Strippoli, P.; Pelleri, M.C.; Caracausi, M. Human protein-coding genes and gene feature statistics in 2019. BMC Res. Notes 2019, 12, 315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baralle, F.E.; Giudice, J. Alternative splicing as a regulator of development and tissue identity. Nat. Rev. Mol. Cell Biol. 2017, 18, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Pan, Z.; Zhang, Z.; Lin, L.; Xing, Y. The expanding landscape of alternative splicing variation in human populations. Am. J. Hum. Genet. 2018, 102, 11–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, B.; Manley, J.L. Alternative polyadenylation of mrna precursors. Nat. Rev. Mol. Cell Biol. 2017, 18, 18–30. [Google Scholar] [CrossRef]
- Kajitani, N.; Glahder, J.; Wu, C.; Yu, H.; Nilsson, K.; Schwartz, S. Hnrnp l controls hpv16 rna polyadenylation and splicing in an akt kinase-dependent manner. Nucleic Acids Res. 2017, 45, 9654–9678. [Google Scholar] [CrossRef]
- Ajiro, M.; Jia, R.; Zhang, L.; Liu, X.; Zheng, Z.M. Intron definition and a branch site adenosine at nt 385 control rna splicing of hpv16 e6*i and e7 expression. PLoS ONE 2012, 7, e46412. [Google Scholar] [CrossRef] [Green Version]
- Bernard, H.U.; Oltersdorf, T.; Seedorf, K. Expression of the human papillomavirus type 18 e7 gene by a cassette-vector system for the transcription and translation of open reading frames in eukaryotic cells. EMBO J. 1987, 6, 133–138. [Google Scholar] [CrossRef]
- Guccione, E.; Pim, D.; Banks, L. Hpv-18 e6*i modulates hpv-18 full-length e6 functions in a cell cycle dependent manner. Int. J. Cancer 2004, 110, 928–933. [Google Scholar] [CrossRef]
- Pim, D.; Massimi, P.; Banks, L. Alternatively spliced hpv-18 e6* protein inhibits e6 mediated degradation of p53 and suppresses transformed cell growth. Oncogene 1997, 15, 257–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olmedo-Nieva, L.; Muñoz-Bello, J.O.; Contreras-Paredes, A.; Lizano, M. The role of e6 spliced isoforms (e6*) in human papillomavirus-induced carcinogenesis. Viruses 2018, 10, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filippova, M.; Evans, W.; Aragon, R.; Filippov, V.; Williams, V.M.; Hong, L.; Reeves, M.E.; Duerksen-Hughes, P. The small splice variant of hpv16 e6, e6, reduces tumor formation in cervical carcinoma xenografts. Virology 2014, 450, 153–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mesplède, T.; Gagnon, D.; Bergeron-Labrecque, F.; Azar, I.; Sénéchal, H.; Coutlée, F.; Archambault, J. P53 degradation activity, expression, and subcellular localization of e6 proteins from 29 human papillomavirus genotypes. J. Virol. 2012, 86, 94–107. [Google Scholar] [CrossRef] [Green Version]
- Ajiro, M.; Tang, S.; Doorbar, J.; Zheng, Z.M. Serine/arginine-rich splicing factor 3 and heterogeneous nuclear ribonucleoprotein a1 regulate alternative rna splicing and gene expression of human papillomavirus 18 through two functionally distinguishable cis elements. J. Virol. 2016, 90, 9138–9152. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Jönsson, J.; Hao, C.; Chaghervand, S.S.; Cui, X.; Kajitani, N.; Gong, L.; Wu, C.; Schwartz, S. Hnrnp a1 and hnrnp a2 inhibit splicing to hpv16 splice site sa409 through a uag-containing sequence in the e7 coding region. J. Virol. 2020. [Google Scholar] [CrossRef]
- Jia, R.; Zheng, Z.M. Regulation of bovine papillomavirus type 1 gene expression by rna processing. Front. Biosci. 2009, 14, 1270–1282. [Google Scholar] [CrossRef] [Green Version]
- Somberg, M.; Schwartz, S. Multiple asf/sf2 sites in the human papillomavirus type 16 (hpv-16) e4-coding region promote splicing to the most commonly used 3′-splice site on the hpv-16 genome. J. Virol. 2010, 84, 8219–8230. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Johansson, C.; Cardoso Palacios, C.; Mossberg, A.; Dhanjal, S.; Bergvall, M.; Schwartz, S. Eight nucleotide substitutions inhibit splicing to hpv-16 3′-splice site sa3358 and reduce the efficiency by which hpv-16 increases the life span of primary human keratinocytes. PLoS ONE 2013, 8, e72776. [Google Scholar] [CrossRef]
- Somberg, M.; Li, X.; Johansson, C.; Orru, B.; Chang, R.; Rush, M.; Fay, J.; Ryan, F.; Schwartz, S. Serine/arginine-rich protein 30c activates human papillomavirus type 16 l1 mrna expression via a bimodal mechanism. J. Gen. Virol. 2011, 92, 2411–2421. [Google Scholar] [CrossRef]
- Jia, R.; Liu, X.; Tao, M.; Kruhlak, M.; Guo, M.; Meyers, C.; Baker, C.C.; Zheng, Z.M. Control of the papillomavirus early-to-late switch by differentially expressed srp20. J. Virol. 2009, 83, 167–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rush, M.; Zhao, X.; Schwartz, S. A splicing enhancer in the e4 coding region of human papillomavirus type 16 is required for early mrna splicing and polyadenylation as well as inhibition of premature late gene expression. J. Virol. 2005, 79, 12002–12015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Johansson, C.; Glahder, J.; Mossberg, A.K.; Schwartz, S. Suppression of hpv-16 late l1 5′-splice site sd3632 by binding of hnrnp d proteins and hnrnp a2/b1 to upstream auagua rna motifs. Nucleic Acids Res. 2013, 41, 10488–10508. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Rush, M.; Carlsson, A.; Schwartz, S. The presence of inhibitory rna elements in the late 3′-untranslated region is a conserved property of human papillomaviruses. Virus Res. 2007, 125, 135–144. [Google Scholar] [CrossRef]
- Zhao, X.; Rush, M.; Schwartz, S. Identification of an hnrnp a1-dependent splicing silencer in the human papillomavirus type 16 l1 coding region that prevents premature expression of the late l1 gene. J. Virol. 2004, 78, 10888–10905. [Google Scholar] [CrossRef] [Green Version]
- Oberg, D.; Fay, J.; Lambkin, H.; Schwartz, S. A downstream polyadenylation element in human papillomavirus type 16 l2 encodes multiple ggg motifs and interacts with hnrnp h. J. Virol. 2005, 79, 9254–9269. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Oberg, D.; Rush, M.; Fay, J.; Lambkin, H.; Schwartz, S. A 57-nucleotide upstream early polyadenylation element in human papillomavirus type 16 interacts with hfip1, cstf-64, hnrnp c1/c2, and polypyrimidine tract binding protein. J. Virol. 2005, 79, 4270–4288. [Google Scholar] [CrossRef] [Green Version]
- Terhune, S.S.; Milcarek, C.; Laimins, L.A. Regulation of human papillomavirus type 31 polyadenylation during the differentiation-dependent life cycle. J. Virol. 1999, 73, 7185–7192. [Google Scholar] [CrossRef] [Green Version]
- Geuens, T.; Bouhy, D.; Timmerman, V. The hnrnp family: Insights into their role in health and disease. Hum. Genet. 2016, 135, 851–867. [Google Scholar] [CrossRef] [Green Version]
- Somberg, M.; Zhao, X.; Fröhlich, M.; Evander, M.; Schwartz, S. Polypyrimidine tract binding protein induces human papillomavirus type 16 late gene expression by interfering with splicing inhibitory elements at the major late 5′ splice site, sd3632. J. Virol. 2008, 82, 3665–3678. [Google Scholar] [CrossRef] [Green Version]
- Dhanjal, S.; Kajitani, N.; Glahder, J.; Mossberg, A.K.; Johansson, C.; Schwartz, S. Heterogeneous nuclear ribonucleoprotein c proteins interact with the human papillomavirus type 16 (hpv16) early 3′-untranslated region and alleviate suppression of hpv16 late l1 mrna splicing. J. Biol. Chem. 2015, 290, 13354–13371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, K.; Wu, C.; Kajitani, N.; Yu, H.; Tsimtsirakis, E.; Gong, L.; Winquist, E.B.; Glahder, J.; Ekblad, L.; Wennerberg, J.; et al. The DNA damage response activates hpv16 late gene expression at the level of rna processing. Nucleic Acids Res. 2018, 46, 5029–5049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goraczniak, R.; Gunderson, S.I. The regulatory element in the 3′-untranslated region of human papillomavirus 16 inhibits expression by binding cug-binding protein 1. J. Biol. Chem. 2008, 283, 2286–2296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunderson, S.I.; Polycarpou-Schwarz, M.; Mattaj, I.W. U1 snrnp inhibits pre-mrna polyadenylation through a direct interaction between u1 70k and poly(a) polymerase. Mol. Cell 1998, 1, 255–264. [Google Scholar] [CrossRef]
- Furth, P.A.; Choe, W.T.; Rex, J.H.; Byrne, J.C.; Baker, C.C. Sequences homologous to 5′ splice sites are required for the inhibitory activity of papillomavirus late 3′ untranslated regions. Mol. Cell. Biol. 1994, 14, 5278–5289. [Google Scholar] [CrossRef] [Green Version]
- Graham, S.V. Papillomavirus 3′ utr regulatory elements. Front. Biosci. 2008, 13, 5646–5663. [Google Scholar] [CrossRef]
- Desaintes, C.; Demeret, C. Control of papillomavirus DNA replication and transcription. Semin. Cancer Biol. 1996, 7, 339–347. [Google Scholar] [CrossRef]
- Thierry, F. Transcriptional regulation of the papillomavirus oncogenes by cellular and viral transcription factors in cervical carcinoma. Virology 2009, 384, 375–379. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.H.; Baker, C.C.; Stünkel, W.; Bernard, H.U. A transcriptional initiator overlaps with a conserved yy1 binding site in the long control region of human papillomavirus type 16. Virology 2003, 305, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.; Tao, M.; McCoy, J.P., Jr.; Zheng, Z.M. The e7 oncoprotein is translated from spliced e6*i transcripts in high-risk human papillomavirus type 16- or type 18-positive cervical cancer cell lines via translation reinitiation. J. Virol. 2006, 80, 4249–4263. [Google Scholar] [CrossRef] [Green Version]
- Stacey, S.N.; Jordan, D.; Williamson, A.J.; Brown, M.; Coote, J.H.; Arrand, J.R. Leaky scanning is the predominant mechanism for translation of human papillomavirus type 16 e7 oncoprotein from e6/e7 bicistronic mrna. J. Virol. 2000, 74, 7284–7297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, K.; Norberg, C.; Mossberg, A.K.; Schwartz, S. Hpv16 e5 is produced from an hpv16 early mrna spliced from sd226 to sa3358. Virus Res. 2018, 244, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Cui, X.; Nilsson, K.; Yu, H.; Gong, L.; Wu, C.; Schwartz, S. Efficient production of hpv16 e2 protein from hpv16 late mrnas spliced from sd880 to sa2709. Virus Res. 2020, 285, 198004. [Google Scholar] [CrossRef] [PubMed]
- Stacey, S.N.; Jordan, D.; Snijders, P.J.; Mackett, M.; Walboomers, J.M.; Arrand, J.R. Translation of the human papillomavirus type 16 e7 oncoprotein from bicistronic mrna is independent of splicing events within the e6 open reading frame. J. Virol. 1995, 69, 7023–7031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozak, M. Regulation of translation via mrna structure in prokaryotes and eukaryotes. Gene 2005, 361, 13–37. [Google Scholar] [CrossRef] [PubMed]
- Luukkonen, B.G.; Tan, W.; Schwartz, S. Efficiency of reinitiation of translation on human immunodeficiency virus type 1 mrnas is determined by the length of the upstream open reading frame and by intercistronic distance. J. Virol. 1995, 69, 4086–4094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merkhofer, E.C.; Hu, P.; Johnson, T.L. Introduction to cotranscriptional rna splicing. Methods Mol. Biol. 2014, 1126, 83–96. [Google Scholar] [PubMed] [Green Version]
- Dujardin, G.; Lafaille, C.; Petrillo, E.; Buggiano, V.; Gómez Acuña, L.I.; Fiszbein, A.; Godoy Herz, M.A.; Nieto Moreno, N.; Muñoz, M.J.; Alló, M.; et al. Transcriptional elongation and alternative splicing. Biochim. Biophys. Acta 2013, 1829, 134–140. [Google Scholar] [CrossRef]
- de la Mata, M.; Alonso, C.R.; Kadener, S.; Fededa, J.P.; Blaustein, M.; Pelisch, F.; Cramer, P.; Bentley, D.; Kornblihtt, A.R. A slow rna polymerase ii affects alternative splicing in vivo. Mol. Cell 2003, 12, 525–532. [Google Scholar] [CrossRef]
- Kornblihtt, A.R.; de la Mata, M.; Fededa, J.P.; Munoz, M.J.; Nogues, G. Multiple links between transcription and splicing. RNA 2004, 10, 1489–1498. [Google Scholar] [CrossRef] [Green Version]
- Howe, K.J.; Kane, C.M.; Ares, M., Jr. Perturbation of transcription elongation influences the fidelity of internal exon inclusion in saccharomyces cerevisiae. RNA 2003, 9, 993–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, S.; Meshorer, E.; Ast, G. Chromatin organization marks exon-intron structure. Nat. Struct. Mol. Biol. 2009, 16, 990–995. [Google Scholar] [CrossRef] [PubMed]
- Tilgner, H.; Nikolaou, C.; Althammer, S.; Sammeth, M.; Beato, M.; Valcárcel, J.; Guigó, R. Nucleosome positioning as a determinant of exon recognition. Nat. Struct. Mol. Biol. 2009, 16, 996–1001. [Google Scholar] [CrossRef]
- Kornblihtt, A.R.; Schor, I.E.; Alló, M.; Dujardin, G.; Petrillo, E.; Muñoz, M.J. Alternative splicing: A pivotal step between eukaryotic transcription and translation. Nat. Rev. Mol. Cell Biol. 2013, 14, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Ip, J.Y.; Schmidt, D.; Pan, Q.; Ramani, A.K.; Fraser, A.G.; Odom, D.T.; Blencowe, B.J. Global impact of rna polymerase ii elongation inhibition on alternative splicing regulation. Genome Res. 2011, 21, 390–401. [Google Scholar] [CrossRef] [Green Version]
- Corden, J.L. Tails of rna polymerase ii. Trends Biochem. Sci. 1990, 15, 383–387. [Google Scholar] [CrossRef]
- West, M.L.; Corden, J.L. Construction and analysis of yeast rna polymerase ii ctd deletion and substitution mutations. Genetics 1995, 140, 1223–1233. [Google Scholar]
- Buratowski, S. Progression through the rna polymerase ii ctd cycle. Mol. Cell 2009, 36, 541–546. [Google Scholar] [CrossRef] [Green Version]
- Barboric, M.; Lenasi, T.; Chen, H.; Johansen, E.B.; Guo, S.; Peterlin, B.M. 7sk snrnp/p-tefb couples transcription elongation with alternative splicing and is essential for vertebrate development. Proc. Natl. Acad. Sci. USA 2009, 106, 7798–7803. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.; Coutinho-Mansfield, G.; Wang, D.; Pandit, S.; Fu, X.D. The splicing factor sc35 has an active role in transcriptional elongation. Nat. Struct. Mol. Biol. 2008, 15, 819–826. [Google Scholar] [CrossRef] [Green Version]
- Nojima, T.; Rebelo, K.; Gomes, T.; Grosso, A.R.; Proudfoot, N.J.; Carmo-Fonseca, M. Rna polymerase ii phosphorylated on ctd serine 5 interacts with the spliceosome during co-transcriptional splicing. Mol. Cell 2018, 72, 369–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortillaro, M.J.; Blencowe, B.J.; Wei, X.; Nakayasu, H.; Du, L.; Warren, S.L.; Sharp, P.A.; Berezney, R. A hyperphosphorylated form of the large subunit of rna polymerase ii is associated with splicing complexes and the nuclear matrix. Proc. Natl. Acad. Sci. USA 1996, 93, 8253–8257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, C.J.; Boyne, A.R.; Millhouse, S.R.; Manley, J.L. The rna polymerase ii c-terminal domain promotes splicing activation through recruitment of a u2af65-prp19 complex. Genes Dev. 2011, 25, 972–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, R.; Yu, J.; Zhang, Z.; Gygi, M.P.; Krainer, A.R.; Gygi, S.P.; Reed, R. Sr proteins function in coupling rnap ii transcription to pre-mrna splicing. Mol. Cell 2007, 26, 867–881. [Google Scholar] [CrossRef]
- Cramer, P.; Pesce, C.G.; Baralle, F.E.; Kornblihtt, A.R. Functional association between promoter structure and transcript alternative splicing. Proc. Natl. Acad. Sci. USA 1997, 94, 11456–11460. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Li, W.; Yao, X.; Lin, Q.J.; Yin, J.W.; Liang, Y.; Heiner, M.; Tian, B.; Hui, J.; Wang, G. Mediator complex regulates alternative mrna processing via the med23 subunit. Mol. Cell 2012, 45, 459–469. [Google Scholar] [CrossRef] [Green Version]
- Lemieux, B.; Blanchette, M.; Monette, A.; Mouland, A.J.; Wellinger, R.J.; Chabot, B. A function for the hnrnp a1/a2 proteins in transcription elongation. PLoS ONE 2015, 10, e0126654. [Google Scholar] [CrossRef]
- Li, J.; Chen, Y.; Xu, X.; Jones, J.; Tiwari, M.; Ling, J.; Wang, Y.; Harismendy, O.; Sen, G.L. Hnrnpk maintains epidermal progenitor function through transcription of proliferation genes and degrading differentiation promoting mrnas. Nat. Commun. 2019, 10, 4198. [Google Scholar] [CrossRef] [Green Version]
- Jeong, S. Sr proteins: Binders, regulators, and connectors of rna. Mol. Cells 2017, 40, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Durzynska, J.; Lesniewicz, K.; Poreba, E. Human papillomaviruses in epigenetic regulations. Mutat. Res. Rev. Mutat. Res. 2017, 772, 36–50. [Google Scholar] [CrossRef]
- Clarke, M.A.; Wentzensen, N.; Mirabello, L.; Ghosh, A.; Wacholder, S.; Harari, A.; Lorincz, A.; Schiffman, M.; Burk, R.D. Human papillomavirus DNA methylation as a potential biomarker for cervical cancer. Cancer Epidemiol. Biomark. Prev. 2012, 21, 2125–2137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.; Garner-Hamrick, P.A.; Fisher, C.; Lee, D.; Lambert, P.F. Methylation patterns of papillomavirus DNA, its influence on e2 function, and implications in viral infection. J. Virol. 2003, 77, 12450–12459. [Google Scholar] [CrossRef] [Green Version]
- Johansson, C.; Jamal Fattah, T.; Yu, H.; Nygren, J.; Mossberg, A.K.; Schwartz, S. Acetylation of intragenic histones on hpv16 correlates with enhanced hpv16 gene expression. Virology 2015, 482, 244–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johannsen, E.; Lambert, P.F. Epigenetics of human papillomaviruses. Virology 2013, 445, 205–212. [Google Scholar] [CrossRef] [Green Version]
- Paris, C.; Pentland, I.; Groves, I.; Roberts, D.C.; Powis, S.J.; Coleman, N.; Roberts, S.; Parish, J.L. Ccctc-binding factor recruitment to the early region of the human papillomavirus 18 genome regulates viral oncogene expression. J. Virol. 2015, 89, 4770–4785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pentland, I.; Campos-León, K.; Cotic, M.; Davies, K.J.; Wood, C.D.; Groves, I.J.; Burley, M.; Coleman, N.; Stockton, J.D.; Noyvert, B.; et al. Disruption of ctcf-yy1-dependent looping of the human papillomavirus genome activates differentiation-induced viral oncogene transcription. PLoS Biol. 2018, 16, e2005752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lace, M.J.; Anson, J.R.; Turek, L.P.; Haugen, T.H. Functional mapping of the human papillomavirus type 16 e1 cistron. J. Virol. 2008, 82, 10724–10734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedorova, M.; Vinokurova, S.; Pavlova, L.; Komel’kov, A.; Korolenkova, L.; Kisseljov, F.; Kisseljova, N. Human papillomavirus types 16 e1 mrna is transcribed from p14 early promoter in cervical neoplasms. Virology 2016, 488, 196–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, C.C.; Phelps, W.C.; Lindgren, V.; Braun, M.J.; Gonda, M.A.; Howley, P.M. Structural and transcriptional analysis of human papillomavirus type 16 sequences in cervical carcinoma cell lines. J. Virol. 1987, 61, 962–971. [Google Scholar] [CrossRef] [Green Version]
- Haller, K.; Stubenrauch, F.; Pfister, H. Differentiation-dependent transcription of the epidermodysplasia verruciformis-associated human papillomavirus type 5 in benign lesions. Virology 1995, 214, 245–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stubenrauch, F.; Leigh, I.M.; Pfister, H. E2 represses the late gene promoter of human papillomavirus type 8 at high concentrations by interfering with cellular factors. J. Virol. 1996, 70, 119–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wettstein, F.O.; Barbosa, M.S.; Nasseri, M. Identification of the major cottontail rabbit papillomavirus late rna cap site and mapping and quantitation of an e2 and minor e6 coding mrna in papillomas and carcinomas. Virology 1987, 159, 321–328. [Google Scholar] [CrossRef]
- Björk, P.; Wieslander, L. Mechanisms of mrna export. Semin. Cell Dev. Biol. 2014, 32, 47–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strässer, K.; Masuda, S.; Mason, P.; Pfannstiel, J.; Oppizzi, M.; Rodriguez-Navarro, S.; Rondón, A.G.; Aguilera, A.; Struhl, K.; Reed, R.; et al. Trex is a conserved complex coupling transcription with messenger rna export. Nature 2002, 417, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Masuda, S.; Das, R.; Cheng, H.; Hurt, E.; Dorman, N.; Reed, R. Recruitment of the human trex complex to mrna during splicing. Genes Dev. 2005, 19, 1512–1517. [Google Scholar] [CrossRef] [Green Version]
- Reed, R.; Cheng, H. Trex, sr proteins and export of mrna. Curr. Opin. Cell Biol. 2005, 17, 269–273. [Google Scholar] [CrossRef]
- Huang, Y.; Yario, T.A.; Steitz, J.A. A molecular link between sr protein dephosphorylation and mrna export. Proc. Natl. Acad. Sci. USA 2004, 101, 9666–9670. [Google Scholar] [CrossRef] [Green Version]
- Lévesque, L.; Guzik, B.; Guan, T.; Coyle, J.; Black, B.E.; Rekosh, D.; Hammarskjöld, M.L.; Paschal, B.M. Rna export mediated by tap involves nxt1-dependent interactions with the nuclear pore complex. J. Biol. Chem. 2001, 276, 44953–44962. [Google Scholar] [CrossRef] [Green Version]
- Cullen, B.R. Nuclear mrna export: Insights from virology. Trends Biochem. Sci. 2003, 28, 419–424. [Google Scholar] [CrossRef]
- Felber, B.K.; Valentin, A.; Rosati, M.; Bergamaschi, C.; Pavlakis, G.N. Hiv DNA vaccine: Stepwise improvements make a difference. Vaccines 2014, 2, 354–379. [Google Scholar] [CrossRef]
- Brennan, C.M.; Gallouzi, I.E.; Steitz, J.A. Protein ligands to hur modulate its interaction with target mrnas in vivo. J. Cell Biol. 2000, 151, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topisirovic, I.; Siddiqui, N.; Lapointe, V.L.; Trost, M.; Thibault, P.; Bangeranye, C.; Piñol-Roma, S.; Borden, K.L. Molecular dissection of the eukaryotic initiation factor 4e (eif4e) export-competent rnp. EMBO J. 2009, 28, 1087–1098. [Google Scholar] [CrossRef]
- Natalizio, B.J.; Wente, S.R. Postage for the messenger: Designating routes for nuclear mrna export. Trends Cell Biol. 2013, 23, 365–373. [Google Scholar] [CrossRef] [Green Version]
- Sokolowski, M.; Tan, W.; Jellne, M.; Schwartz, S. Mrna instability elements in the human papillomavirus type 16 l2 coding region. J. Virol. 1998, 72, 1504–1515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlsson, A.; Schwartz, S. Inhibitory activity of the human papillomavirus type 1 au-rich element correlates inversely with the levels of the elav-like hur protein in the cell cytoplasm. Arch. Virol. 2000, 145, 491–503. [Google Scholar] [CrossRef] [PubMed]
- Cumming, S.A.; Chuen-Im, T.; Zhang, J.; Graham, S.V. The rna stability regulator hur regulates l1 protein expression in vivo in differentiating cervical epithelial cells. Virology 2009, 383, 142–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barksdale, S.K.; Baker, C.C. The human immunodeficiency virus type 1 rev protein and the rev-responsive element counteract the effect of an inhibitory 5′ splice site in a 3′ untranslated region. Mol. Cell. Biol. 1995, 15, 2962–2971. [Google Scholar] [CrossRef] [Green Version]
- Tan, W.; Schwartz, S. The rev protein of human immunodeficiency virus type 1 counteracts the effect of an au-rich negative element in the human papillomavirus type 1 late 3′ untranslated region. J. Virol. 1995, 69, 2932–2945. [Google Scholar] [CrossRef] [Green Version]
- Tan, W.; Felber, B.K.; Zolotukhin, A.S.; Pavlakis, G.N.; Schwartz, S. Efficient expression of the human papillomavirus type 16 l1 protein in epithelial cells by using rev and the rev-responsive element of human immunodeficiency virus or the cis-acting transactivation element of simian retrovirus type 1. J. Virol. 1995, 69, 5607–5620. [Google Scholar] [CrossRef] [Green Version]
- Piñol-Roma, S.; Dreyfuss, G. Shuttling of pre-mrna binding proteins between nucleus and cytoplasm. Nature 1992, 355, 730–732. [Google Scholar] [CrossRef]
- Dreyfuss, G.; Matunis, M.J.; Piñol-Roma, S.; Burd, C.G. Hnrnp proteins and the biogenesis of mrna. Annu. Rev. Biochem. 1993, 62, 289–321. [Google Scholar] [CrossRef] [PubMed]
- Michael, W.M.; Choi, M.; Dreyfuss, G. A nuclear export signal in hnrnp a1: A signal-mediated, temperature-dependent nuclear protein export pathway. Cell 1995, 83, 415–422. [Google Scholar] [CrossRef] [Green Version]
- Herrick, D.; Parker, R.; Jacobson, A. Identification and comparison of stable and unstable mrnas in saccharomyces cerevisiae. Mol. Cell. Biol. 1990, 10, 2269–2284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sachs, A.B. Messenger rna degradation in eukaryotes. Cell 1993, 74, 413–421. [Google Scholar] [CrossRef]
- Pérez-Ortín, J.E.; Alepuz, P.; Chávez, S.; Choder, M. Eukaryotic mrna decay: Methodologies, pathways, and links to other stages of gene expression. J. Mol. Biol. 2013, 425, 3750–3775. [Google Scholar] [CrossRef]
- Friedel, C.C.; Dölken, L.; Ruzsics, Z.; Koszinowski, U.H.; Zimmer, R. Conserved principles of mammalian transcriptional regulation revealed by rna half-life. Nucleic Acids Res. 2009, 37, e115. [Google Scholar] [CrossRef] [Green Version]
- Neff, A.T.; Lee, J.Y.; Wilusz, J.; Tian, B.; Wilusz, C.J. Global analysis reveals multiple pathways for unique regulation of mrna decay in induced pluripotent stem cells. Genome Res. 2012, 22, 1457–1467. [Google Scholar] [CrossRef] [Green Version]
- Rabani, M.; Levin, J.Z.; Fan, L.; Adiconis, X.; Raychowdhury, R.; Garber, M.; Gnirke, A.; Nusbaum, C.; Hacohen, N.; Friedman, N.; et al. Metabolic labeling of rna uncovers principles of rna production and degradation dynamics in mammalian cells. Nat. Biotechnol. 2011, 29, 436–442. [Google Scholar] [CrossRef] [Green Version]
- Yang, E.; van Nimwegen, E.; Zavolan, M.; Rajewsky, N.; Schroeder, M.; Magnasco, M.; Darnell, J.E., Jr. Decay rates of human mrnas: Correlation with functional characteristics and sequence attributes. Genome Res. 2003, 13, 1863–1872. [Google Scholar]
- Barreau, C.; Paillard, L.; Osborne, H.B. Au-rich elements and associated factors: Are there unifying principles? Nucleic Acids Res. 2005, 33, 7138–7150. [Google Scholar] [CrossRef]
- Zhang, T.; Kruys, V.; Huez, G.; Gueydan, C. Au-rich element-mediated translational control: Complexity and multiple activities of trans-activating factors. Biochem. Soc. Trans. 2002, 30, 952–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, B.; Hu, Y.; Brewer, G. Competitive binding of auf1 and tiar to myc mrna controls its translation. Nat. Struct. Mol. Biol. 2007, 14, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Pullmann, R., Jr.; Kim, H.H.; Abdelmohsen, K.; Lal, A.; Martindale, J.L.; Yang, X.; Gorospe, M. Analysis of turnover and translation regulatory rna-binding protein expression through binding to cognate mrnas. Mol. Cell. Biol. 2007, 27, 6265–6278. [Google Scholar] [CrossRef] [Green Version]
- Raineri, I.; Wegmueller, D.; Gross, B.; Certa, U.; Moroni, C. Roles of auf1 isoforms, hur and brf1 in are-dependent mrna turnover studied by rna interference. Nucleic Acids Res. 2004, 32, 1279–1288. [Google Scholar] [CrossRef] [PubMed]
- Ruggiero, T.; Trabucchi, M.; Ponassi, M.; Corte, G.; Chen, C.Y.; al-Haj, L.; Khabar, K.S.; Briata, P.; Gherzi, R. Identification of a set of ksrp target transcripts upregulated by pi3k-akt signaling. BMC Mol. Biol. 2007, 8, 28. [Google Scholar] [CrossRef] [Green Version]
- Myer, V.E.; Fan, X.C.; Steitz, J.A. Identification of hur as a protein implicated in auuua-mediated mrna decay. EMBO J. 1997, 16, 2130–2139. [Google Scholar] [CrossRef]
- Peng, S.S.; Chen, C.Y.; Xu, N.; Shyu, A.B. Rna stabilization by the au-rich element binding protein, hur, an elav protein. EMBO J. 1998, 17, 3461–3470. [Google Scholar] [CrossRef]
- Jeon, S.; Lambert, P.F. Integration of human papillomavirus type 16 DNA into the human genome leads to increased stability of e6 and e7 mrnas: Implications for cervical carcinogenesis. Proc. Natl. Acad. Sci. USA 1995, 92, 1654–1658. [Google Scholar] [CrossRef] [Green Version]
- Sokolowski, M.; Zhao, C.; Tan, W.; Schwartz, S. Au-rich mrna instability elements on human papillomavirus type 1 late mrnas and c-fos mrnas interact with the same cellular factors. Oncogene 1997, 15, 2303–2319. [Google Scholar] [CrossRef]
- Adhikari, S.; Xiao, W.; Zhao, Y.L.; Yang, Y.G. M(6)a: Signaling for mrna splicing. RNA Biol. 2016, 13, 756–759. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Fang, X.; Zhong, P.; Song, Z.; Hu, X. N6-methyladenosine modifications: Interactions with novel rna-binding proteins and roles in signal transduction. RNA Biol. 2019, 16, 991–1000. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Gong, L.; Wu, C.; Nilsson, K.; Li-Wang, X.; Schwartz, S. Hnrnp g prevents inclusion on the hpv16 l1 mrnas of the central exon between splice sites sa3358 and sd3632. J. Gen. Virol. 2018, 99, 328–343. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.I.; Parisien, M.; Dai, Q.; Liu, N.; Diatchenko, L.; Sachleben, J.R.; Pan, T. N(6)-methyladenosine modification in a long noncoding rna hairpin predisposes its conformation to protein binding. J. Mol. Biol. 2016, 428, 822–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, W.; Adhikari, S.; Dahal, U.; Chen, Y.S.; Hao, Y.J.; Sun, B.F.; Sun, H.Y.; Li, A.; Ping, X.L.; Lai, W.Y.; et al. Nuclear m(6)a reader ythdc1 regulates mrna splicing. Mol. Cell 2016, 61, 507–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alarcón, C.R.; Goodarzi, H.; Lee, H.; Liu, X.; Tavazoie, S.; Tavazoie, S.F. Hnrnpa2b1 is a mediator of m(6)a-dependent nuclear rna processing events. Cell 2015, 162, 1299–1308. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Dai, Q.; Zheng, G.; He, C.; Parisien, M.; Pan, T. N(6)-methyladenosine-dependent rna structural switches regulate rna-protein interactions. Nature 2015, 518, 560–564. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Zhou, K.I.; Parisien, M.; Dai, Q.; Diatchenko, L.; Pan, T. N6-methyladenosine alters rna structure to regulate binding of a low-complexity protein. Nucleic Acids Res. 2017, 45, 6051–6063. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Lee, E.E.; Kim, J.; Yang, R.; Chamseddin, B.; Ni, C.; Gusho, E.; Xie, Y.; Chiang, C.M.; Buszczak, M.; et al. Transforming activity of an oncoprotein-encoding circular rna from human papillomavirus. Nat. Commun. 2019, 10, 2300. [Google Scholar] [CrossRef] [Green Version]
- Cooper, T.A.; Wan, L.; Dreyfuss, G. Rna and disease. Cell 2009, 136, 777–793. [Google Scholar] [CrossRef] [Green Version]
- Kechavarzi, B.; Janga, S.C. Dissecting the expression landscape of rna-binding proteins in human cancers. Genome Biol. 2014, 15, R14. [Google Scholar] [CrossRef] [Green Version]
- Pereira, B.; Billaud, M.; Almeida, R. Rna-binding proteins in cancer: Old players and new actors. Trends Cancer 2017, 3, 506–528. [Google Scholar] [CrossRef] [PubMed]
- Fay, J.; Kelehan, P.; Lambkin, H.; Schwartz, S. Increased expression of cellular rna-binding proteins in hpv-induced neoplasia and cervical cancer. J. Med. Virol. 2009, 81, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Liu, H.; Yang, Y.; Wang, X.; Liu, P.; Li, Y.; Meyers, C.; Banerjee, N.S.; Wang, H.K.; Cam, M.; et al. Genome-wide profiling of cervical rna-binding proteins identifies human papillomavirus regulation of rnaseh2a expression by viral e7 and e2f1. mBio 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Mole, S.; Milligan, S.G.; Graham, S.V. Human papillomavirus type 16 e2 protein transcriptionally activates the promoter of a key cellular splicing factor, sf2/asf. J. Virol. 2009, 83, 357–367. [Google Scholar] [CrossRef] [Green Version]
- Sunthamala, N.; Thierry, F.; Teissier, S.; Pientong, C.; Kongyingyoes, B.; Tangsiriwatthana, T.; Sangkomkamhang, U.; Ekalaksananan, T. E2 proteins of high risk human papillomaviruses down-modulate sting and ifn-κ transcription in keratinocytes. PLoS ONE 2014, 9, e91473. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.R.; James, C.D.; Bristol, M.L.; Nulton, T.J.; Wang, X.; Kaur, N.; White, E.A.; Windle, B.; Morgan, I.M. Human papillomavirus 16 e2 regulates keratinocyte gene expression relevant to cancer and the viral life cycle. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Gauson, E.J.; Windle, B.; Donaldson, M.M.; Caffarel, M.M.; Dornan, E.S.; Coleman, N.; Herzyk, P.; Henderson, S.C.; Wang, X.; Morgan, I.M. Regulation of human genome expression and rna splicing by human papillomavirus 16 e2 protein. Virology 2014, 468, 10–18. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.; Kim, H.Z.; Jeong, K.W.; Shim, Y.S.; Horikawa, I.; Barrett, J.C.; Choe, J. Human papillomavirus e2 down-regulates the human telomerase reverse transcriptase promoter. J. Biol. Chem. 2002, 277, 27748–27756. [Google Scholar] [CrossRef] [Green Version]
- Steger, G.; Schnabel, C.; Schmidt, H.M. The hinge region of the human papillomavirus type 8 e2 protein activates the human p21(waf1/cip1) promoter via interaction with sp1. J. Gen. Virol. 2002, 83, 503–510. [Google Scholar] [CrossRef] [Green Version]
- Hadaschik, D.; Hinterkeuser, K.; Oldak, M.; Pfister, H.J.; Smola-Hess, S. The papillomavirus e2 protein binds to and synergizes with c/ebp factors involved in keratinocyte differentiation. J. Virol. 2003, 77, 5253–5265. [Google Scholar] [CrossRef] [Green Version]
- Lechner, M.S.; Mack, D.H.; Finicle, A.B.; Crook, T.; Vousden, K.H.; Laimins, L.A. Human papillomavirus e6 proteins bind p53 in vivo and abrogate p53-mediated repression of transcription. EMBO J. 1992, 11, 3045–3052. [Google Scholar] [CrossRef] [PubMed]
- Crook, T.; Tidy, J.A.; Vousden, K.H. Degradation of p53 can be targeted by hpv e6 sequences distinct from those required for p53 binding and trans-activation. Cell 1991, 67, 547–556. [Google Scholar] [CrossRef]
- Patel, D.; Huang, S.M.; Baglia, L.A.; McCance, D.J. The e6 protein of human papillomavirus type 16 binds to and inhibits co-activation by cbp and p300. EMBO J. 1999, 18, 5061–5072. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, H.; Degenkolbe, R.; Bernard, H.U.; O’Connor, M.J. The human papillomavirus type 16 e6 oncoprotein can down-regulate p53 activity by targeting the transcriptional coactivator cbp/p300. J. Virol. 1999, 73, 6209–6219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronco, L.V.; Karpova, A.Y.; Vidal, M.; Howley, P.M. Human papillomavirus 16 e6 oncoprotein binds to interferon regulatory factor-3 and inhibits its transcriptional activity. Genes Dev. 1998, 12, 2061–2072. [Google Scholar] [CrossRef] [Green Version]
- Shino, Y.; Shirasawa, H.; Kinoshita, T.; Simizu, B. Human papillomavirus type 16 e6 protein transcriptionally modulates fibronectin gene expression by induction of protein complexes binding to the cyclic amp response element. J. Virol. 1997, 71, 4310–4318. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, T.; Shirasawa, H.; Shino, Y.; Moriya, H.; Desbarats, L.; Eilers, M.; Simizu, B. Transactivation of prothymosin alpha and c-myc promoters by human papillomavirus type 16 e6 protein. Virology 1997, 232, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Veldman, T.; Horikawa, I.; Barrett, J.C.; Schlegel, R. Transcriptional activation of the telomerase htert gene by human papillomavirus type 16 e6 oncoprotein. J. Virol. 2001, 75, 4467–4472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desaintes, C.; Hallez, S.; Van Alphen, P.; Burny, A. Transcriptional activation of several heterologous promoters by the e6 protein of human papillomavirus type 16. J. Virol. 1992, 66, 325–333. [Google Scholar] [CrossRef] [Green Version]
- Etscheid, B.G.; Foster, S.A.; Galloway, D.A. The e6 protein of human papillomavirus type 16 functions as a transcriptional repressor in a mechanism independent of the tumor suppressor protein, p53. Virology 1994, 205, 583–585. [Google Scholar] [CrossRef] [PubMed]
- McPhillips, M.G.; Veerapraditsin, T.; Cumming, S.A.; Karali, D.; Milligan, S.G.; Boner, W.; Morgan, I.M.; Graham, S.V. Sf2/asf binds the human papillomavirus type 16 late rna control element and is regulated during differentiation of virus-infected epithelial cells. J. Virol. 2004, 78, 10598–10605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roundtree, I.A.; Evans, M.E.; Pan, T.; He, C. Dynamic rna modifications in gene expression regulation. Cell 2017, 169, 1187–1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Wu, W.; Han, Q.; Wang, Y.; Li, C.; Zhang, P.; Xu, H. Post-translational modification control of rna-binding protein hnrnpk function. Open Biol. 2019, 9, 180239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo Piccolo, L.; Mochizuki, H.; Nagai, Y. The lncrna hsrω regulates arginine dimethylation of human fus to cause its proteasomal degradation in Drosophila. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef] [Green Version]
- Bell, I.; Martin, A.; Roberts, S. The e1circumflexe4 protein of human papillomavirus interacts with the serine-arginine-specific protein kinase srpk1. J. Virol. 2007, 81, 5437–5448. [Google Scholar] [CrossRef] [Green Version]
- Prescott, E.L.; Brimacombe, C.L.; Hartley, M.; Bell, I.; Graham, S.; Roberts, S. Human papillomavirus type 1 e1^e4 protein is a potent inhibitor of the serine-arginine (sr) protein kinase srpk1 and inhibits phosphorylation of host sr proteins and of the viral transcription and replication regulator e2. J. Virol. 2014, 88, 12599–12611. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, K.; Wu, C.; Schwartz, S. Role of the DNA damage response in human papillomavirus rna splicing and polyadenylation. Int. J. Mol. Sci. 2018, 19, 1735. [Google Scholar] [CrossRef] [Green Version]
- Alam, H.; Sehgal, L.; Kundu, S.T.; Dalal, S.N.; Vaidya, M.M. Novel function of keratins 5 and 14 in proliferation and differentiation of stratified epithelial cells. Mol. Biol. Cell 2011, 22, 4068–4078. [Google Scholar] [CrossRef]
- Bossler, F.; Hoppe-Seyler, K.; Hoppe-Seyler, F. Pi3k/akt/mtor signaling regulates the virus/host cell crosstalk in hpv-positive cervical cancer cells. Int. J. Mol. Sci. 2019, 20, 2188. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, J.K.; Payton, J.E.; Rashmi, R.; Xiang, T.; Jia, Y.; Huettner, P.; Rogers, B.E.; Yang, Q.; Watson, M.; Rader, J.S.; et al. Pathway-specific analysis of gene expression data identifies the pi3k/akt pathway as a novel therapeutic target in cervical cancer. Clin. Cancer Res. 2012, 18, 1464–1471. [Google Scholar] [CrossRef] [Green Version]
- Strickland, S.W.; Vande Pol, S. The human papillomavirus 16 e7 oncoprotein attenuates akt signaling to promote internal ribosome entry site-dependent translation and expression of c-myc. J. Virol. 2016, 90, 5611–5621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Lin, B.; Liu, X.; Zhang, W.; Zhang, E.; Hu, L.; Ma, Y.; Li, X.; Tang, X. Erk signaling pathway is involved in hpv-16 e6 but not e7 oncoprotein-induced hif-1α protein accumulation in nsclc cells. Oncol. Res. 2016, 23, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Rosenberger, S.; De-Castro Arce, J.; Langbein, L.; Steenbergen, R.D.; Rösl, F. Alternative splicing of human papillomavirus type-16 e6/e6* early mrna is coupled to egf signaling via erk1/2 activation. Proc. Natl. Acad. Sci. USA 2010, 107, 7006–7011. [Google Scholar] [CrossRef] [Green Version]
- Hummel, M.; Hudson, J.B.; Laimins, L.A. Differentiation-induced and constitutive transcription of human papillomavirus type 31b in cell lines containing viral episomes. J. Virol. 1992, 66, 6070–6080. [Google Scholar] [CrossRef] [Green Version]
- Ozbun, M.A.; Meyers, C. Temporal usage of multiple promoters during the life cycle of human papillomavirus type 31b. J. Virol. 1998, 72, 2715–2722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grassmann, K.; Rapp, B.; Maschek, H.; Petry, K.U.; Iftner, T. Identification of a differentiation-inducible promoter in the e7 open reading frame of human papillomavirus type 16 (hpv-16) in raft cultures of a new cell line containing high copy numbers of episomal hpv-16 DNA. J. Virol. 1996, 70, 2339–2349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Meyers, C.; Wang, H.K.; Chow, L.T.; Zheng, Z.M. Construction of a full transcription map of human papillomavirus type 18 during productive viral infection. J. Virol. 2011, 85, 8080–8092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Xue, Y.; Poidinger, M.; Lim, T.; Chew, S.H.; Pang, C.L.; Abastado, J.P.; Thierry, F. Mapping of hpv transcripts in four human cervical lesions using rnaseq suggests quantitative rearrangements during carcinogenic progression. Virology 2014, 462, 14–24. [Google Scholar] [CrossRef] [Green Version]
- Ruesch, M.N.; Stubenrauch, F.; Laimins, L.A. Activation of papillomavirus late gene transcription and genome amplification upon differentiation in semisolid medium is coincident with expression of involucrin and transglutaminase but not keratin-10. J. Virol. 1998, 72, 5016–5024. [Google Scholar] [CrossRef] [Green Version]
- Mole, S.; Faizo, A.A.A.; Hernandez-Lopez, H.; Griffiths, M.; Stevenson, A.; Roberts, S.; Graham, S.V. Human papillomavirus type 16 infection activates the host serine arginine protein kinase 1 (srpk1) splicing factor axis. J. Gen. Virol. 2020, 101, 523–532. [Google Scholar] [CrossRef]
- Bodaghi, S.; Jia, R.; Zheng, Z.M. Human papillomavirus type 16 e2 and e6 are rna-binding proteins and inhibit in vitro splicing of pre-mrnas with suboptimal splice sites. Virology 2009, 386, 32–43. [Google Scholar] [CrossRef] [Green Version]
- Lai, M.C.; Teh, B.H.; Tarn, W.Y. A human papillomavirus e2 transcriptional activator. The interactions with cellular splicing factors and potential function in pre-mrna processing. J. Biol. Chem. 1999, 274, 11832–11841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, Y.; Bellanger, S.; Zhang, W.; Lim, D.; Low, J.; Lunny, D.; Thierry, F. Hpv16 e2 is an immediate early marker of viral infection, preceding e7 expression in precursor structures of cervical carcinoma. Cancer Res. 2010, 70, 5316–5325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.J.; Lee, A.; Kim, T.J.; Jin, H.T.; Seo, Y.B.; Park, J.S.; Lee, S.J. E2/e6 ratio and l1 immunoreactivity as biomarkers to determine hpv16-positive high-grade squamous intraepithelial lesions (cin2 and 3) and cervical squamous cell carcinoma. J. Gynecol. Oncol. 2018, 29, e38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zummeren, M.V.; Kremer, W.W.; Leeman, A.; Bleeker, M.C.G.; Jenkins, D.; Sandt, M.V.; Doorbar, J.; Heideman, D.A.M.; Steenbergen, R.D.M.; Snijders, P.J.F.; et al. Hpv e4 expression and DNA hypermethylation of cadm1, mal, and mir124-2 genes in cervical cancer and precursor lesions. Mod. Pathol. 2018, 31, 1842–1850. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HPV16 Splice Donors | |
| Consensus Sequence | (C/A)AG_GU(A/G)AGU |
| SD226 | GAG_GUAUAU |
| SD880 | CAG_GUACCA |
| SD1302 | CAG_GUAGAA |
| SD3632 | AAG_GUGAUG |
| HPV16 Splice Acceptors | |
| Consensus Sequence | (C/U)nX(C/U)AG_(A/G) |
| SA409 | GAUUUGUUAAUUAG_G |
| SA526 | AUGUCUUGUUGCAG_A |
| SA742 | CCUUUUGUUGCAAG_U |
| SA2582 | UAAUGCUGGUACAG_A |
| SA2709 | UCCUUUUUCUCAAG_G |
| SA3358 | ACAUCUGUGUUUAG_C |
| SA5639 | AUAUUUUUUUUCAG_A |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kajitani, N.; Schwartz, S. Role of Viral Ribonucleoproteins in Human Papillomavirus Type 16 Gene Expression. Viruses 2020, 12, 1110. https://doi.org/10.3390/v12101110
Kajitani N, Schwartz S. Role of Viral Ribonucleoproteins in Human Papillomavirus Type 16 Gene Expression. Viruses. 2020; 12(10):1110. https://doi.org/10.3390/v12101110
Chicago/Turabian StyleKajitani, Naoko, and Stefan Schwartz. 2020. "Role of Viral Ribonucleoproteins in Human Papillomavirus Type 16 Gene Expression" Viruses 12, no. 10: 1110. https://doi.org/10.3390/v12101110
APA StyleKajitani, N., & Schwartz, S. (2020). Role of Viral Ribonucleoproteins in Human Papillomavirus Type 16 Gene Expression. Viruses, 12(10), 1110. https://doi.org/10.3390/v12101110