Selective Blockade of TNFR1 Improves Clinical Disease and Bronchoconstriction in Experimental RSV Infection

 , ,
, , 

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
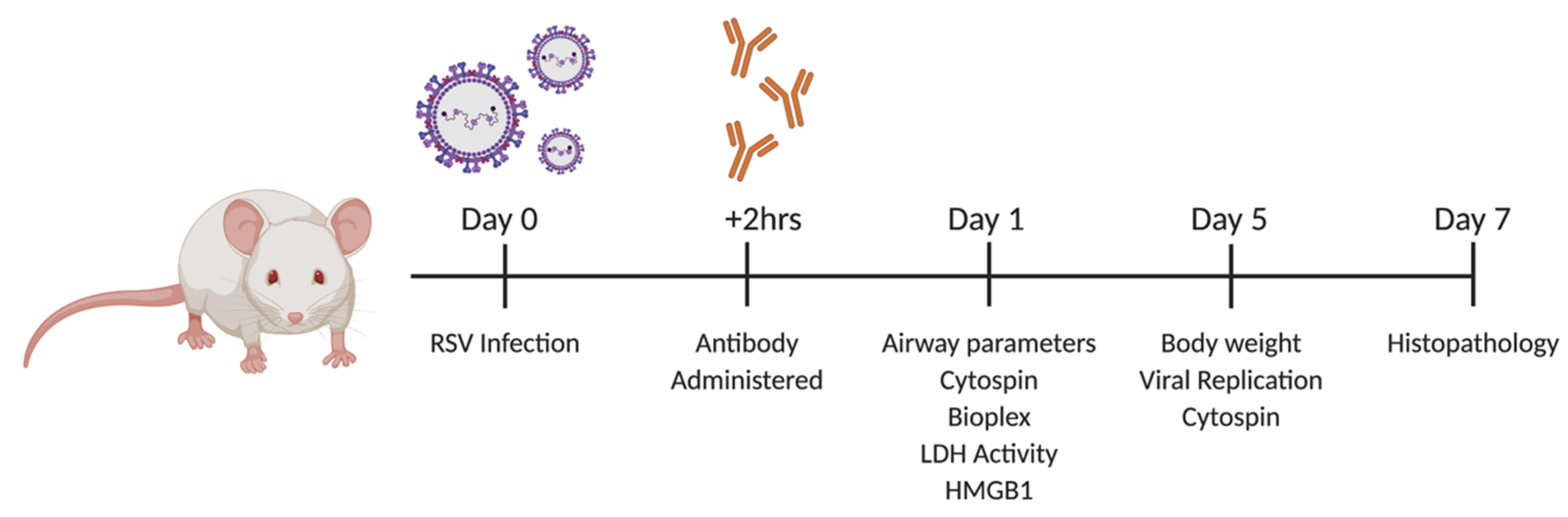
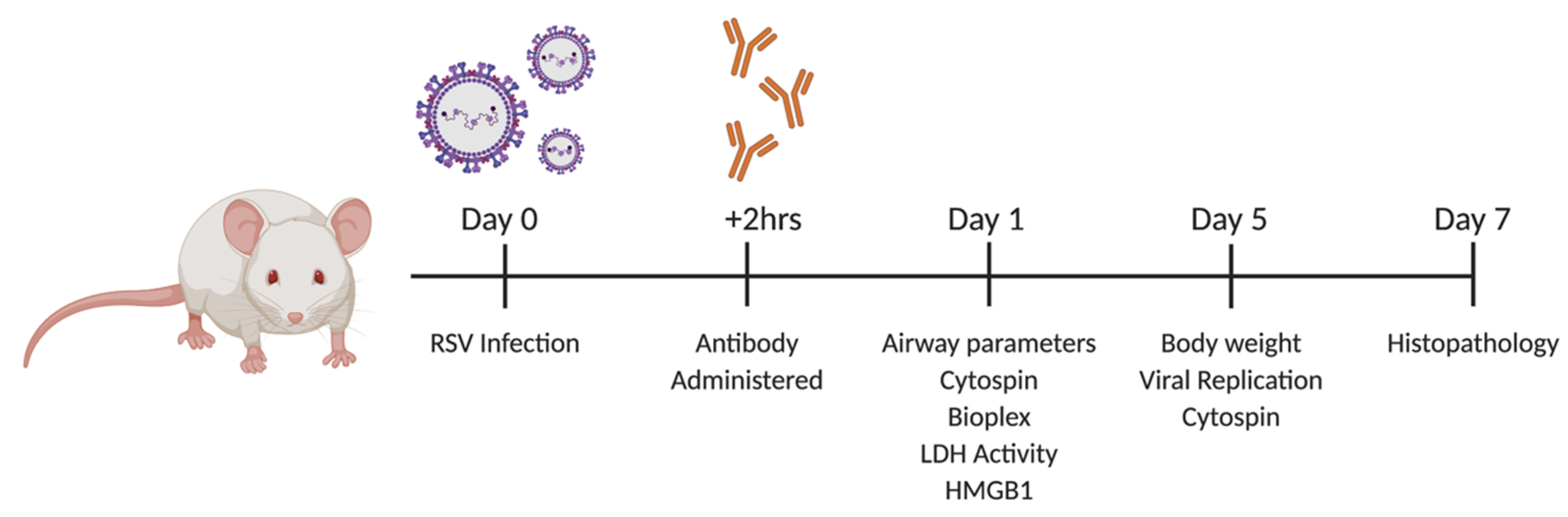
2.1. Viral Infection and TNFR Blockade in Balb/c Mice
2.2. Assessment of Airway Function
2.3. Bronchoalveolar Lavage
2.4. Measurement of Cytokines, Chemokines, Elastase, HMGB1, and LDH Activity
2.5. Histopathology
2.6. Statistical Analysis
3. Results
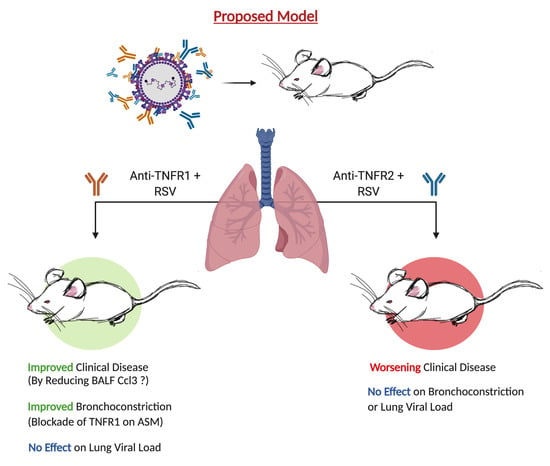
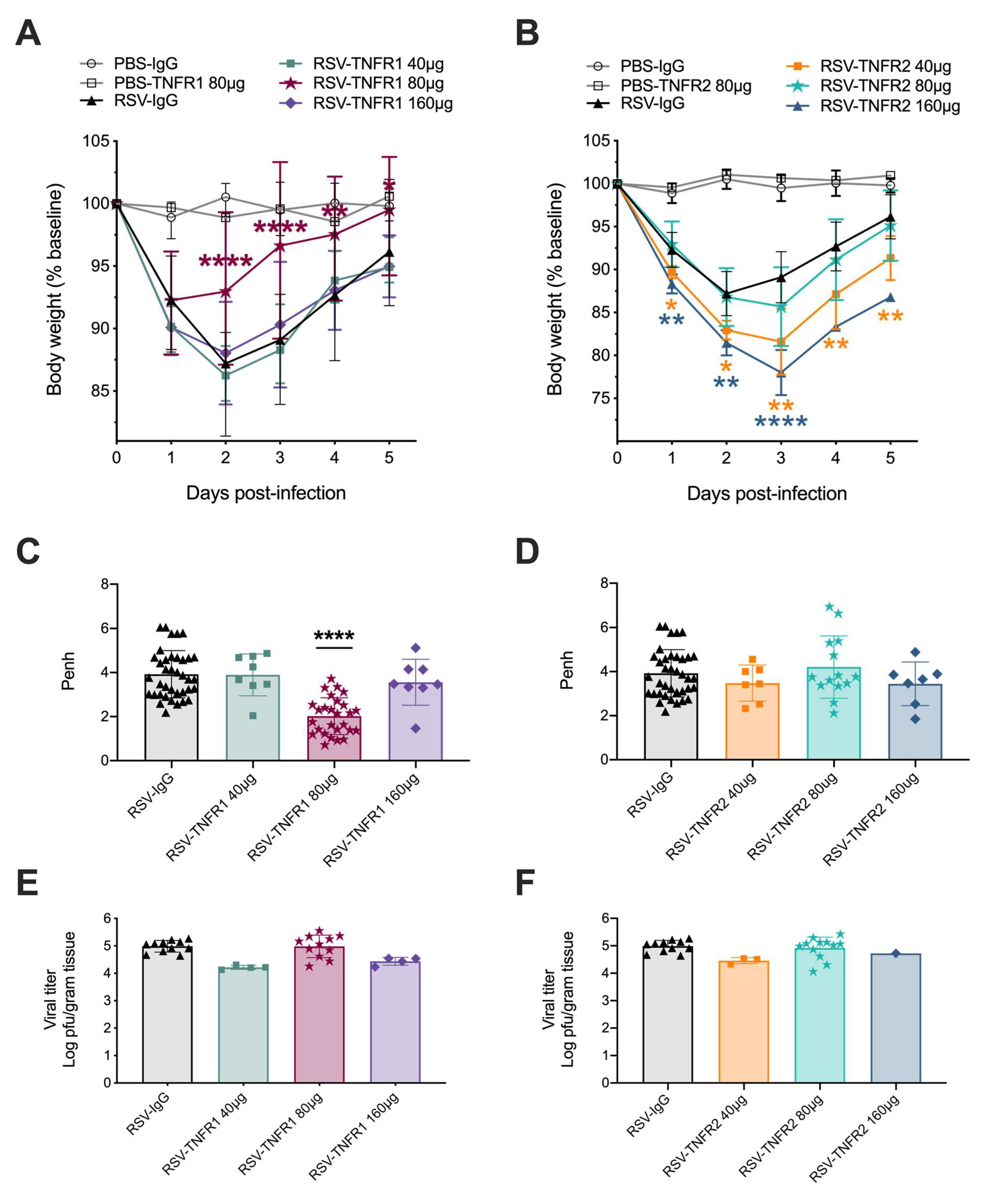
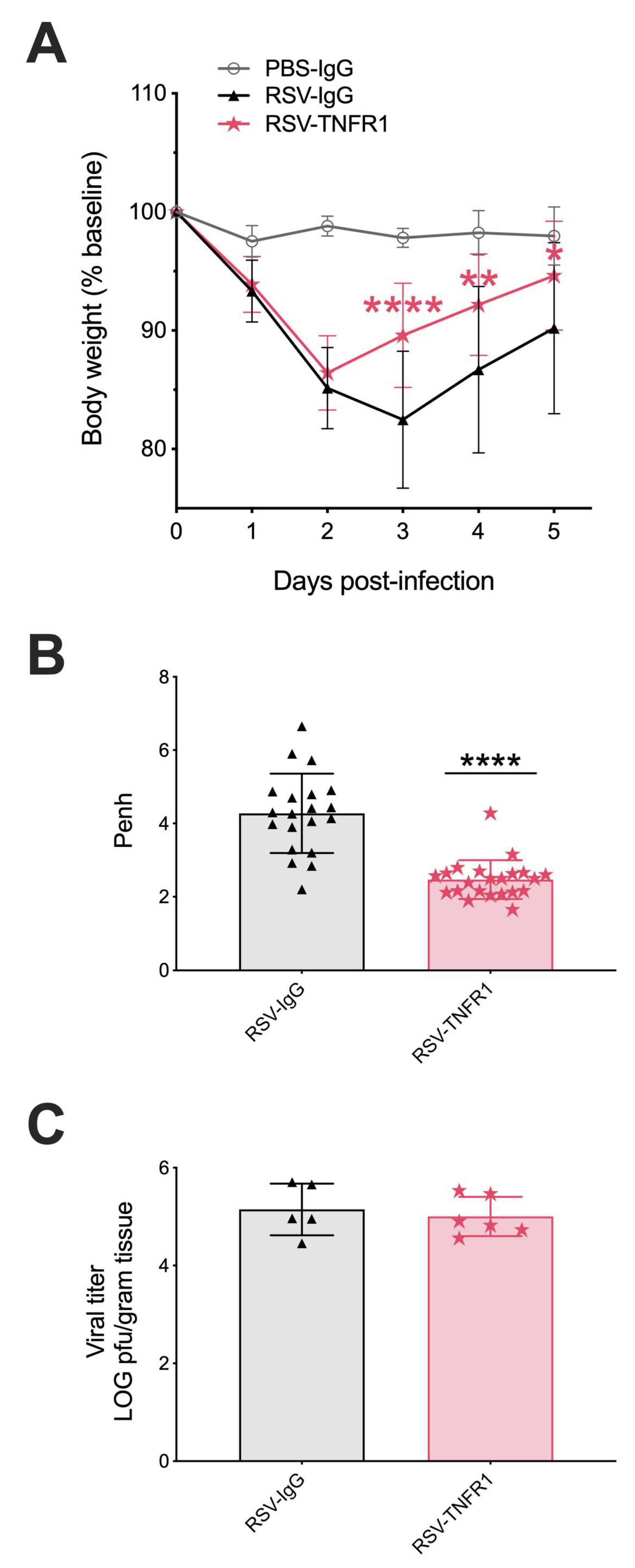
3.1. Blockade of TNFR1, but Not TNFR2, Improves Body Weight and Bronchoconstriction
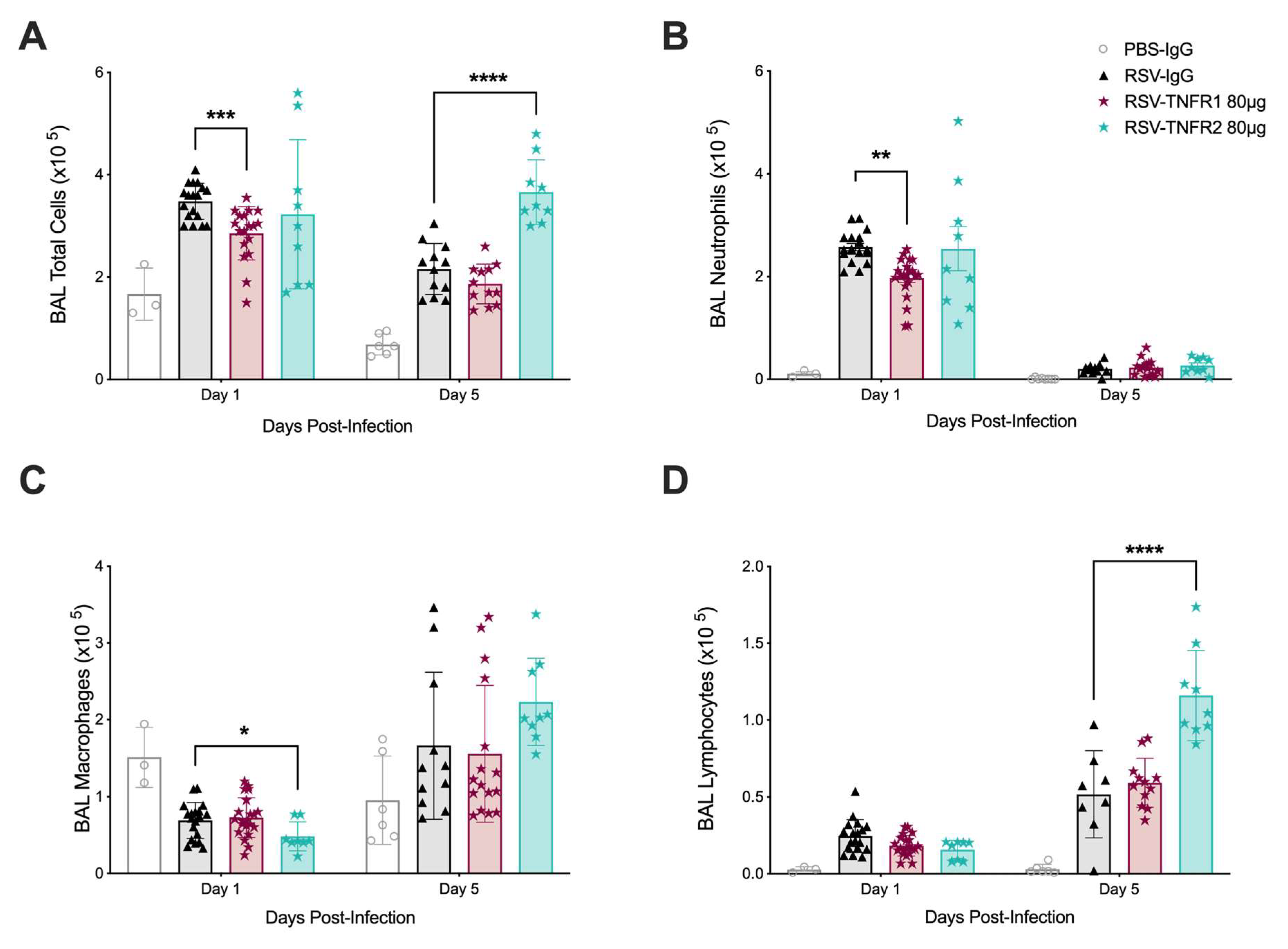
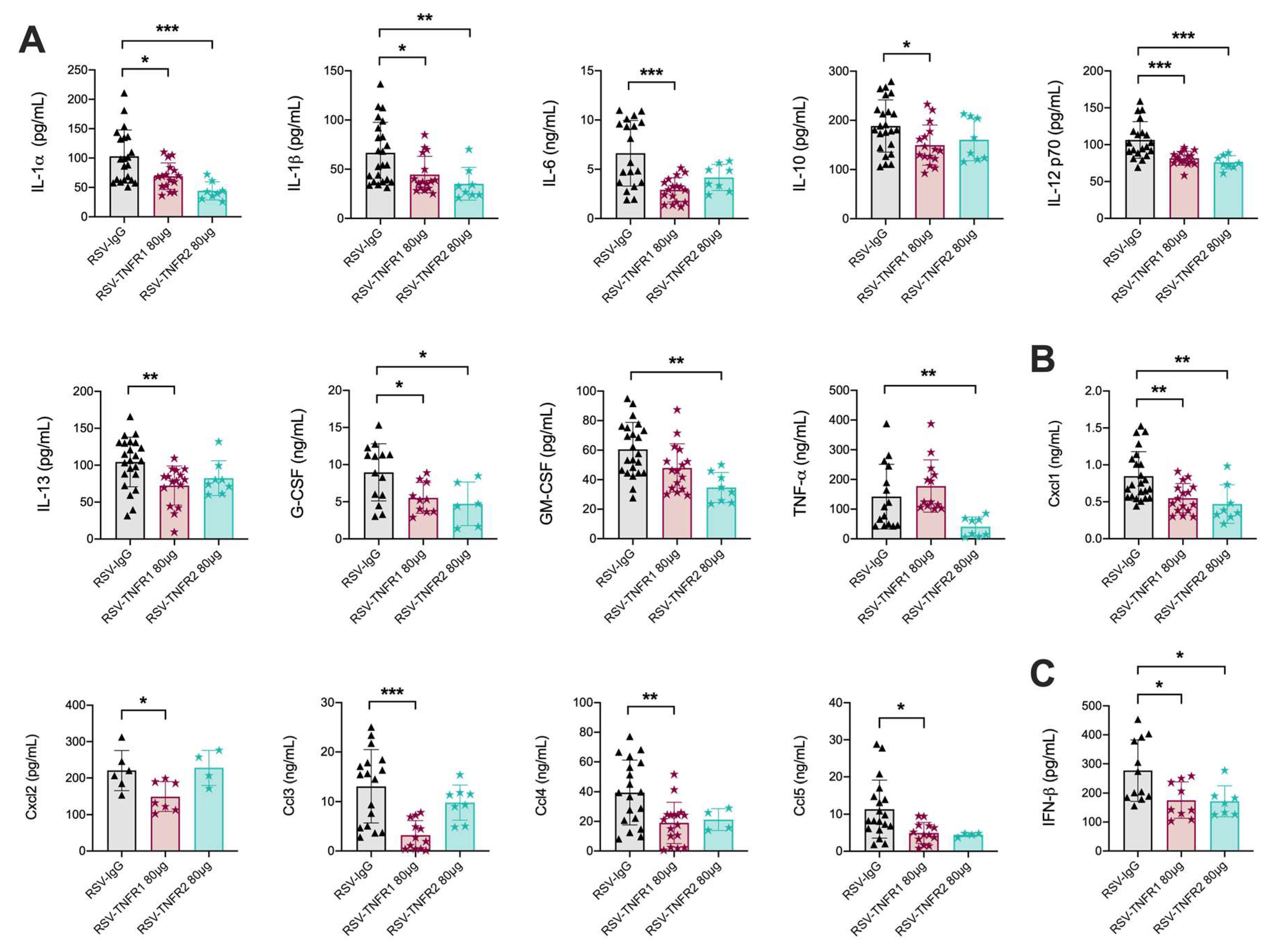
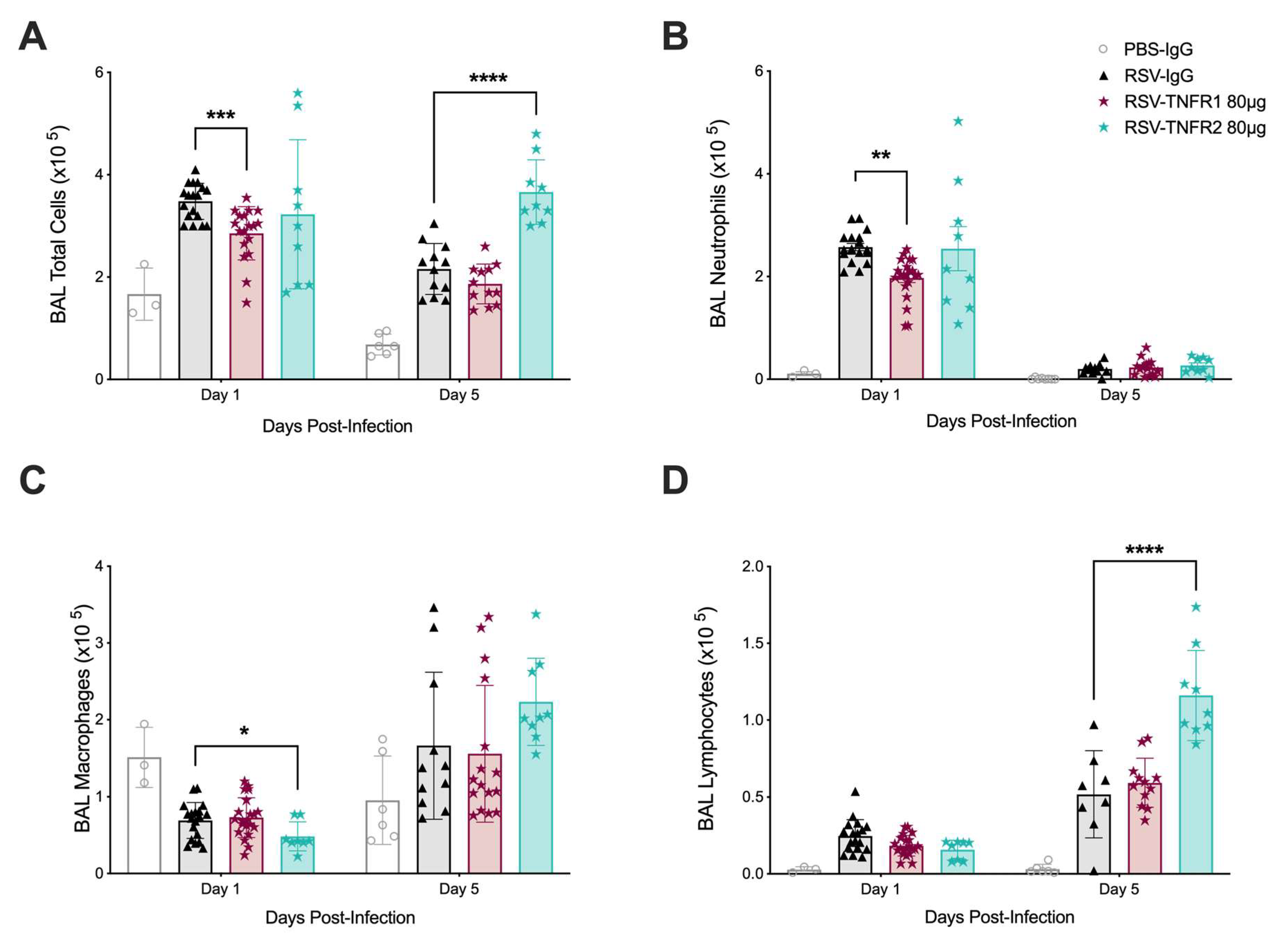
3.2. TNFR Blockade Reduces Cellular Inflammation and Cytokine Production
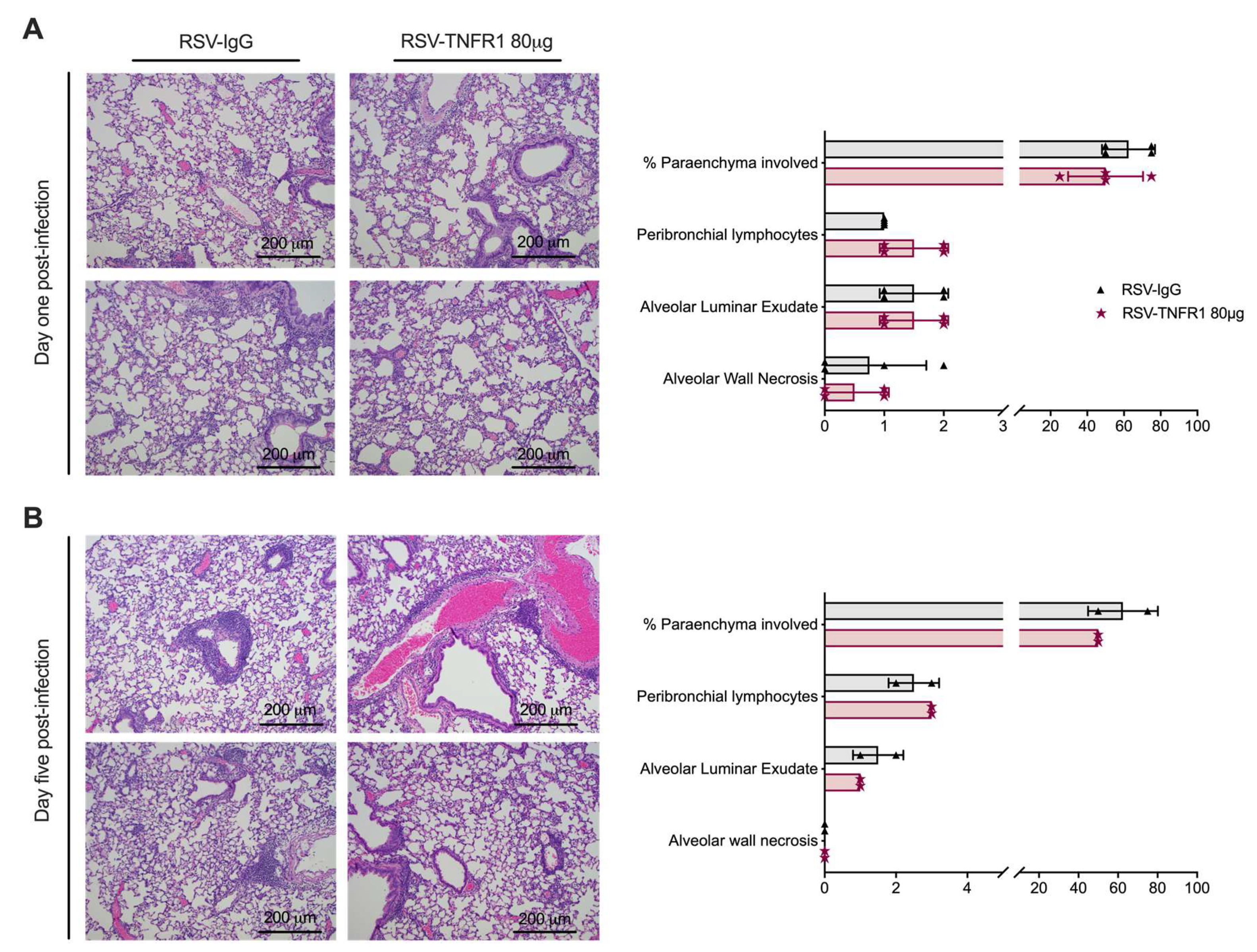
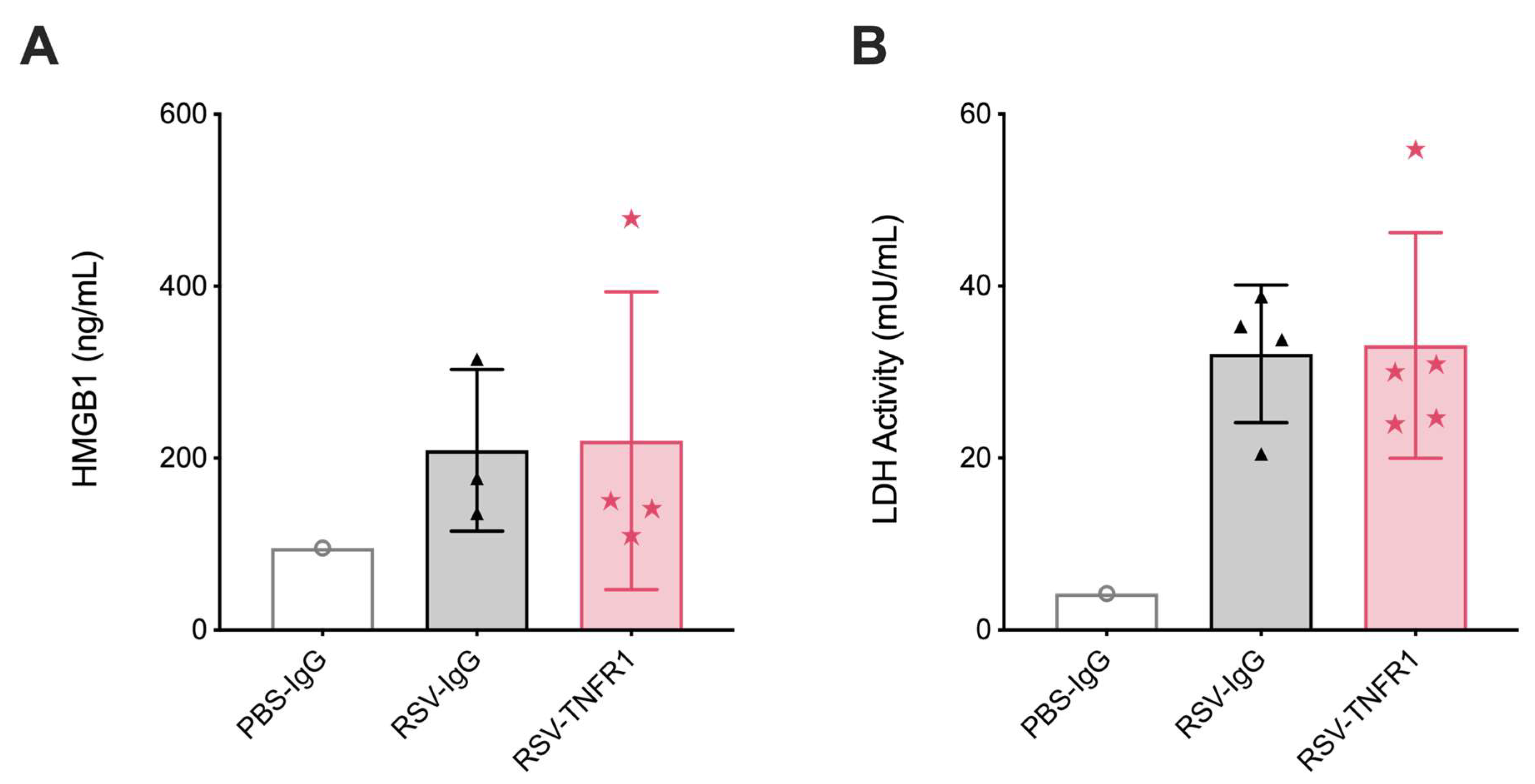
3.3. Cell Death or Lung Pathology in RSV Infections Are Not Affected by TNFR1 Blockade
3.4. Blockade of TNFR1 Improves Clinical Disease and Bronchoconstriction in a Model of Post-Infection Treatment
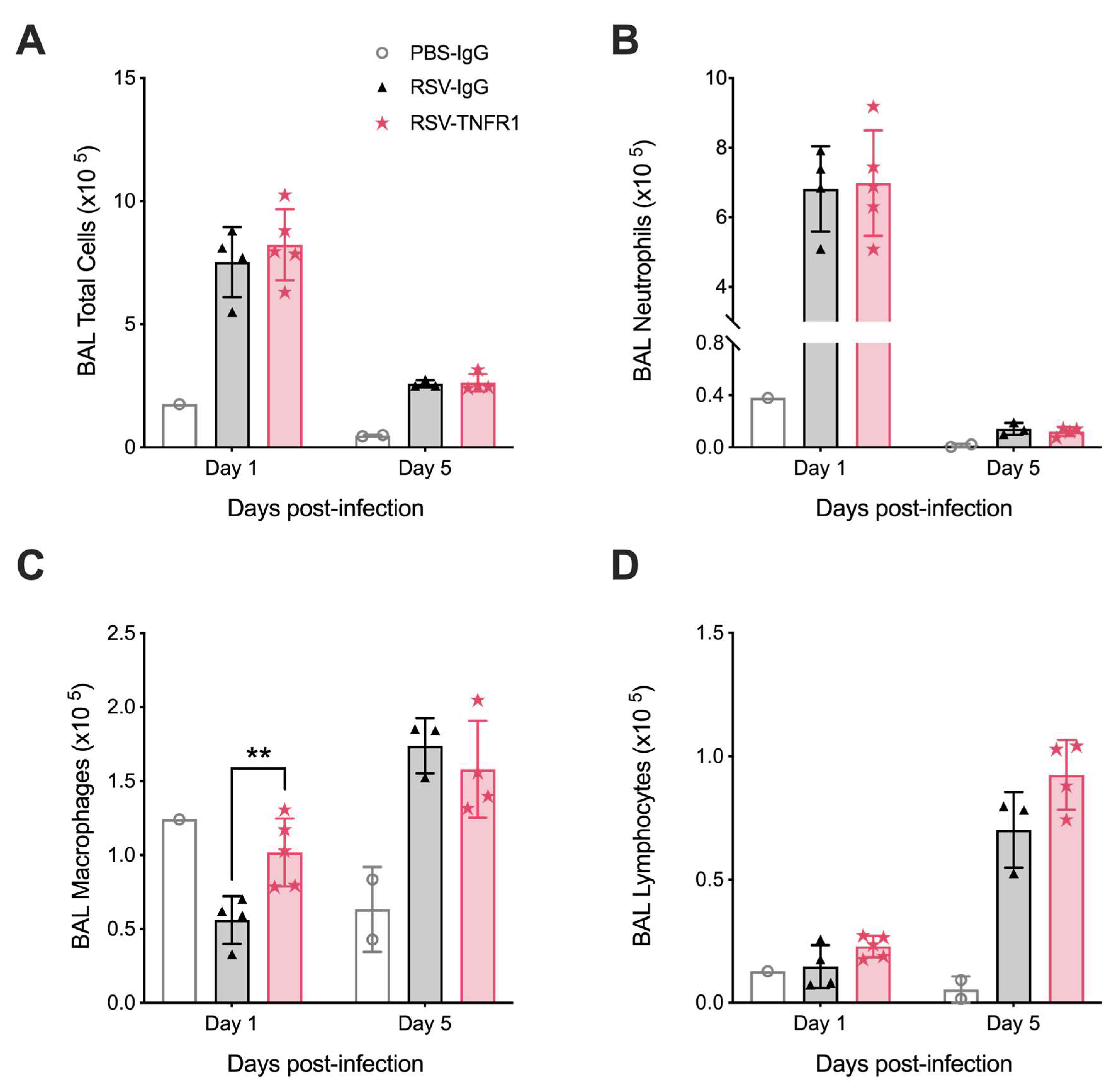
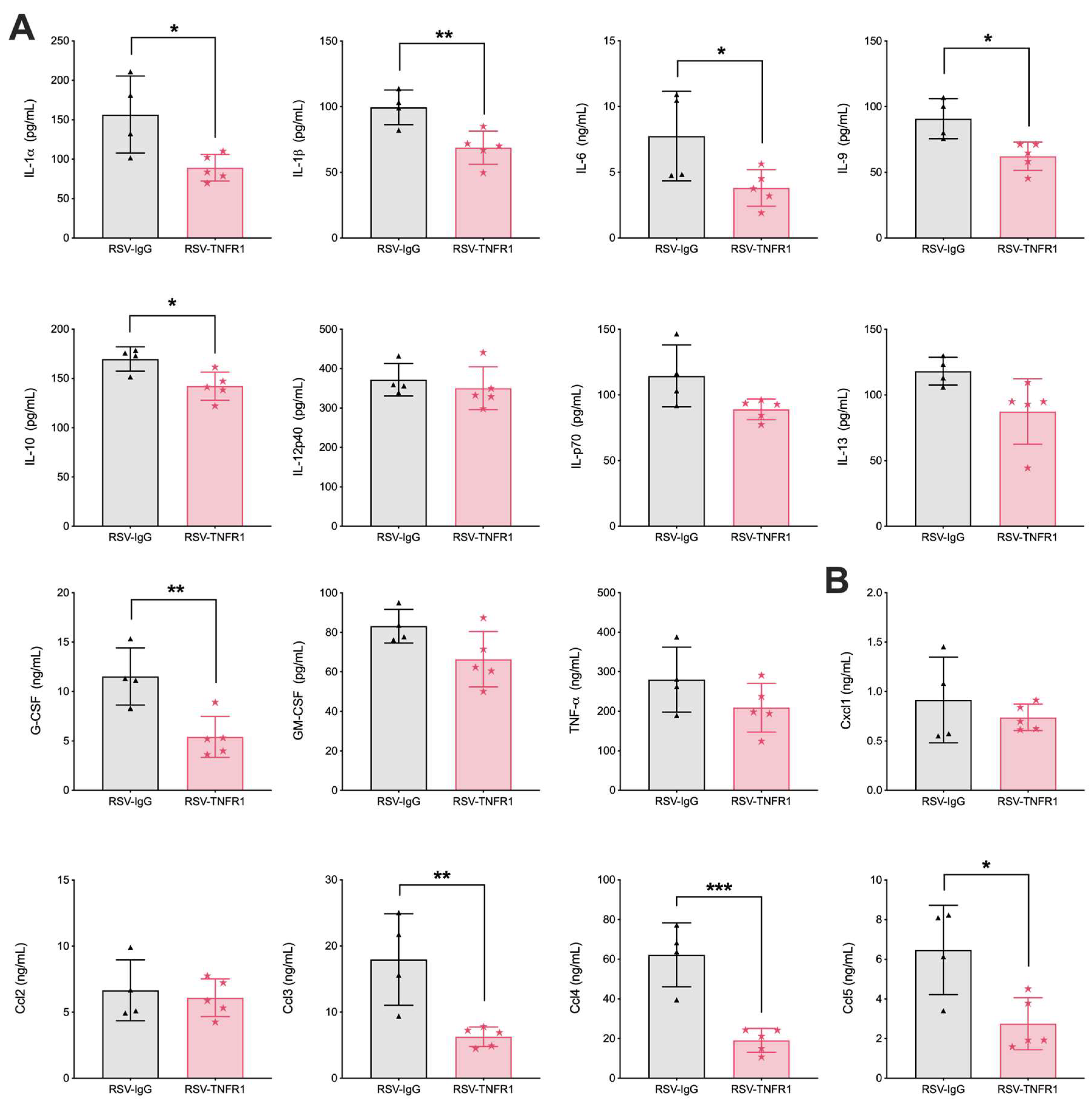
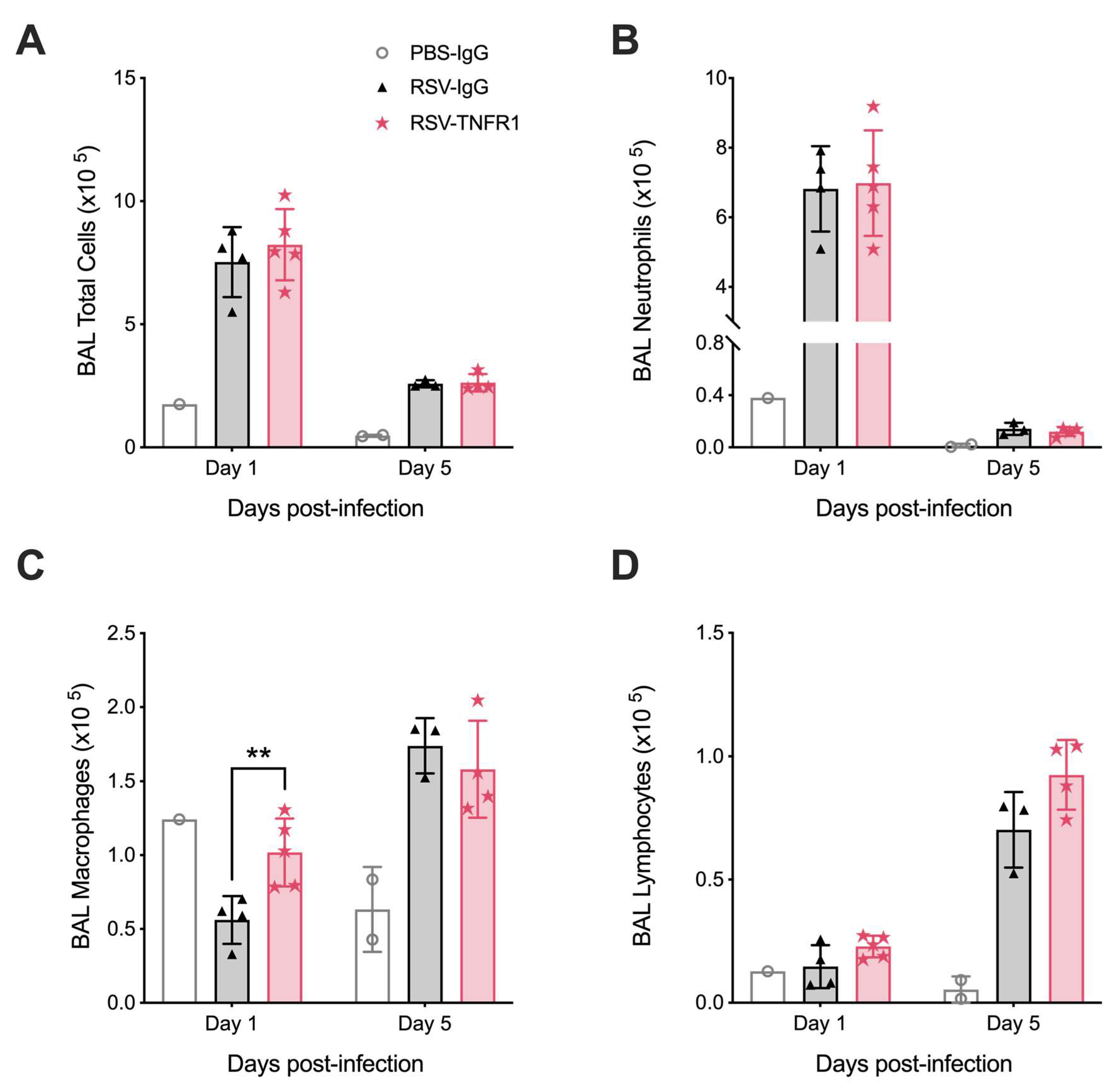
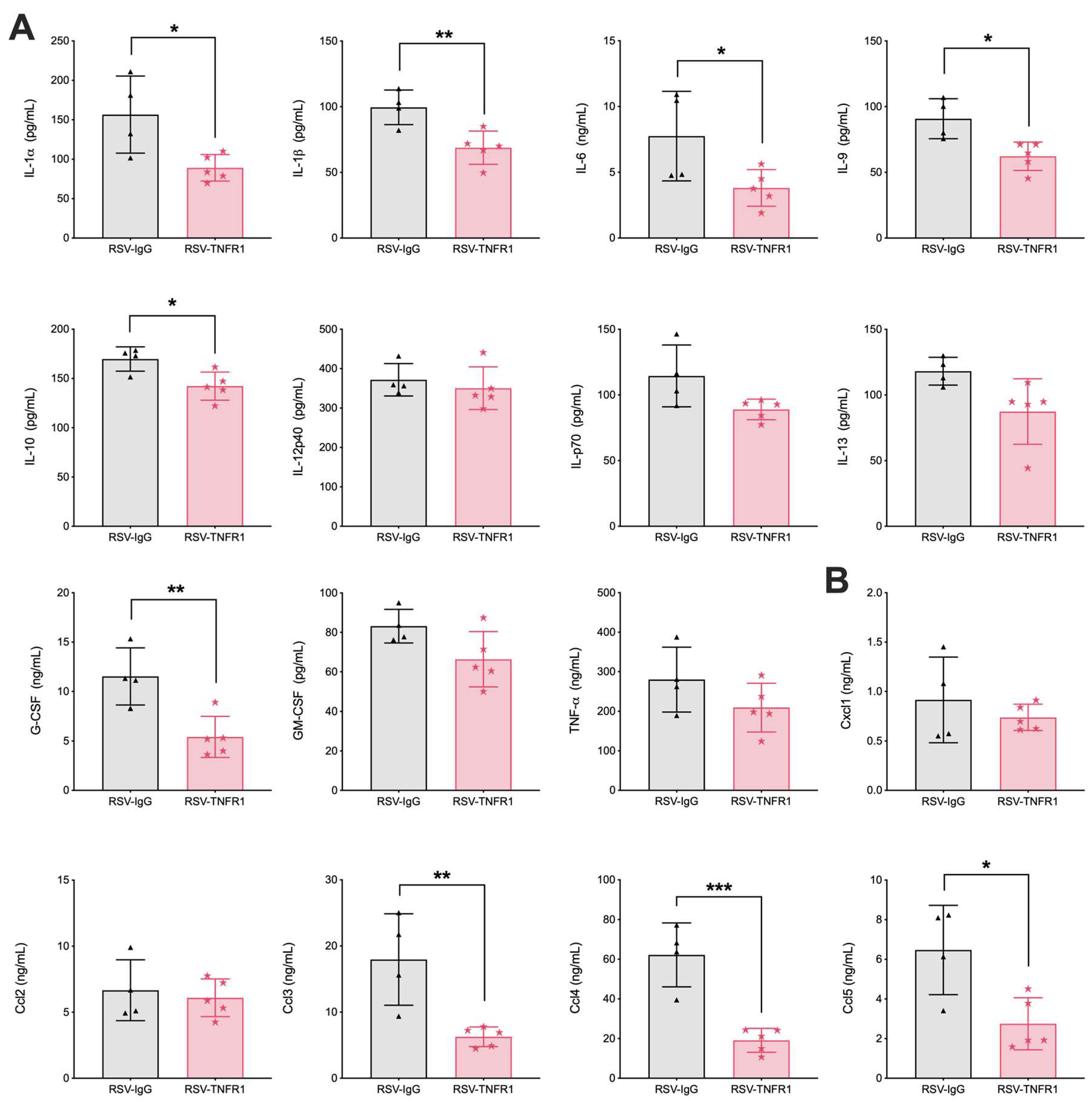
3.5. Blockade of TNFR1 Improves Macrophage Cell Counts and Reduces Pro-Inflammatory Cytokines in RSV Infection
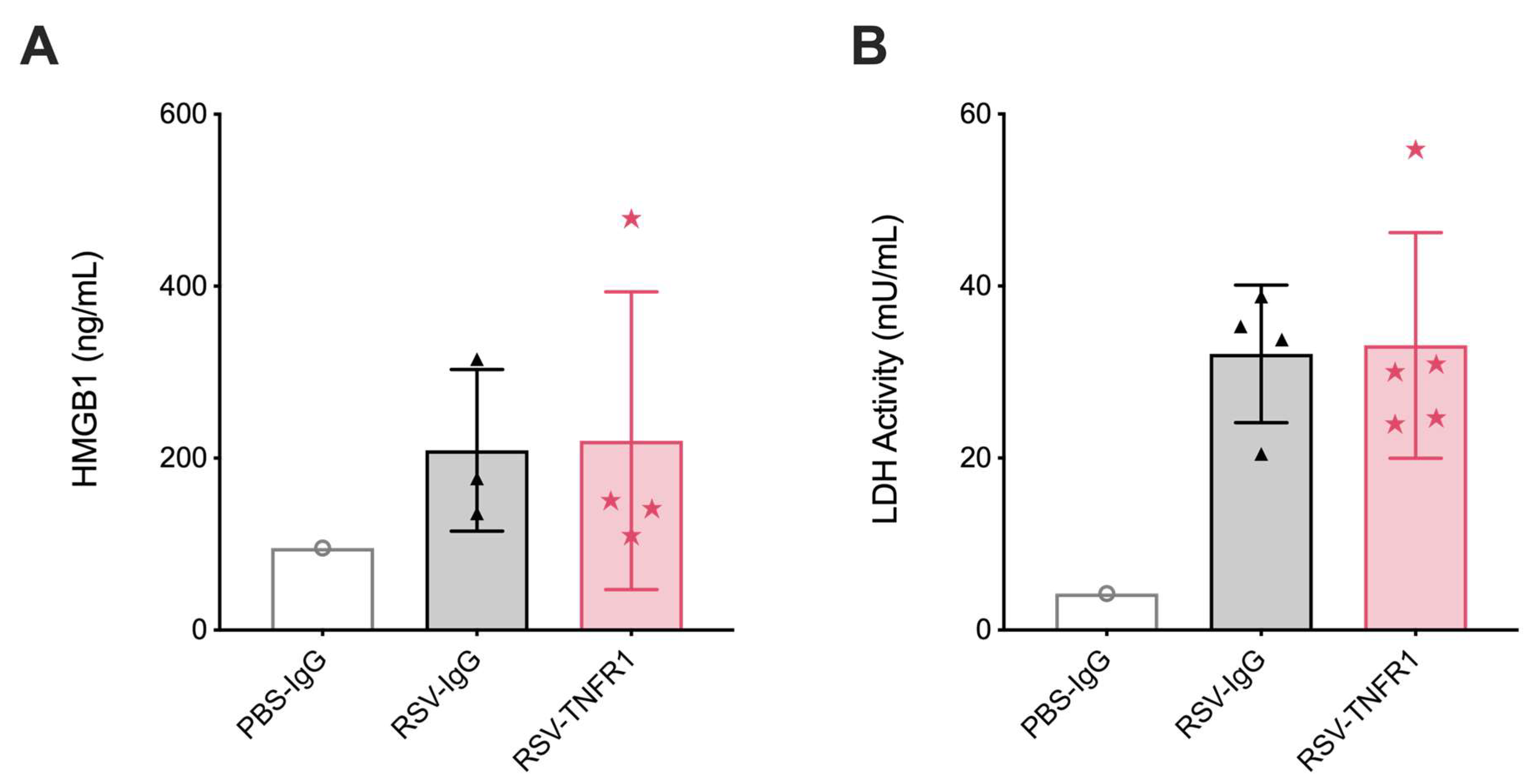
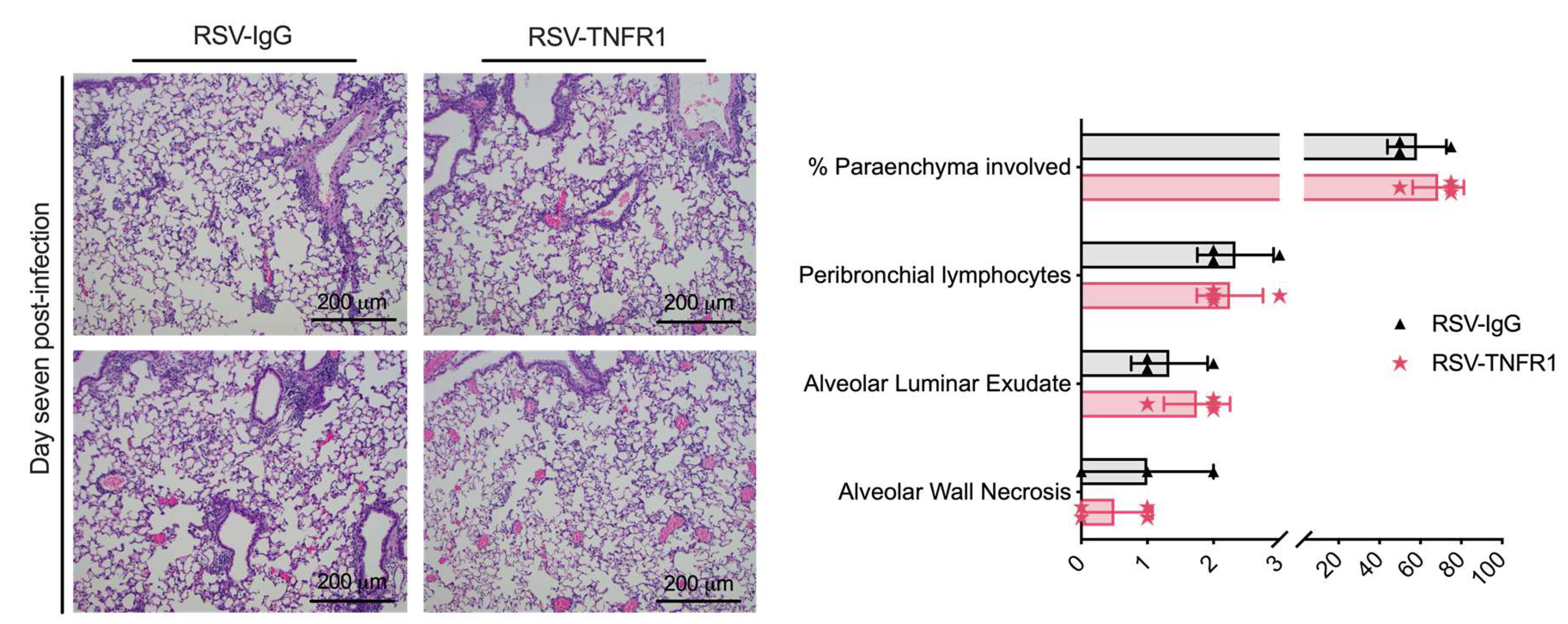
3.6. Blockade of TNFR1 after RSV Infection Does Not Affect Cell Death or Lung Pathology
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rima, B.; Collins, P.; Easton, A.; Fouchier, R.; Kurath, G.; Lamb, R.A.; Lee, B.; Maisner, A.; Rota, P.; Wang, L.; et al. ICTV Virus Taxonomy Profile: Pneumoviridae. J. Gen. Virol. 2017, 98, 2912–2913. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; McAllister, D.A.; O’Brien, K.L.; Simoes, E.A.F.; Madhi, S.A.; Gessner, B.D.; Polack, F.P.; Balsells, E.; Acacio, S.; Aguayo, C.; et al. Global, regional, and national disease burden estimates of acute lower respiratory infections due to respiratory syncytial virus in young children in 2015: A systematic review and modelling study. Lancet 2017, 390, 946–958. [Google Scholar] [CrossRef] [Green Version]
- Villenave, R.; Shields, M.D.; Power, U.F. Respiratory syncytial virus interaction with human airway epithelium. Trends Microbiol. 2013, 21, 238–244. [Google Scholar] [CrossRef]
- Touzelet, O.; Power, U. Cellular and Molecular Characteristics of RSV-Induced Disease in Humans. In Human Respiratory Syncytial Virus Infection; InTechOpen Limited: London, UK, 2011. [Google Scholar] [CrossRef] [Green Version]
- Pickles, R.J.; DeVincenzo, J.P. Respiratory syncytial virus (RSV) and its propensity for causing bronchiolitis. J. Pathol. 2015, 235, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Henderson, J.; Hilliard, T.N.; Sherriff, A.; Stalker, D.; Shammari, N.A.; Thomas, H.M.; ALSPAC Study Team. Hospitalization for RSV bronchiolitis before 12 months of age and subsequent asthma, atopy and wheeze: A longitudinal birth cohort study. Pediatr. Allergy Immunol. 2005, 16, 386–392. [Google Scholar] [CrossRef] [PubMed]
- Sigurs, N.; Aljassim, F.; Kjellman, B.; Robinson, P.D.; Sigurbergsson, F.; Bjarnason, R.; Gustafsson, P.M. Asthma and allergy patterns over 18 years after severe RSV bronchiolitis in the first year of life. Thorax 2010, 65, 1045. [Google Scholar] [CrossRef] [Green Version]
- Sigurs, N.; Bjarnason, R.; Sigurbergsson, F.; Kjellman, B. Respiratory Syncytial Virus Bronchiolitis in Infancy Is an Important Risk Factor for Asthma and Allergy at Age 7. Am. J. Respir. Crit. Care Med. 2000, 161, 1501–1507. [Google Scholar] [CrossRef] [PubMed]
- Sigurs, N.; Gustafsson, P.M.; Bjarnason, R.; Lundberg, F.; Schmidt, S.; Sigurbergsson, F.; Kjellman, B. Severe Respiratory Syncytial Virus Bronchiolitis in Infancy and Asthma and Allergy at Age 13. Am. J. Respir. Crit. Care Med. 2005, 171, 137–141. [Google Scholar] [CrossRef]
- Homaira, N.; Briggs, N.; Oei, J.L.; Hilder, L.; Bajuk, B.; Jaffe, A.; Omer, S.B. Association of Age at First Severe Respiratory Syncytial Virus Disease with Subsequent Risk of Severe Asthma: A Population-Based Cohort Study. J. Infect. Dis. 2019, 220, 550–556. [Google Scholar] [CrossRef]
- Rosenberg, H.F.; Domachowske, J.B. Inflammatory responses to respiratory syncytial virus (RSV) infection and the development of immunomodulatory pharmacotherapeutics. Curr. Med. Chem. 2012, 19, 1424–1431. [Google Scholar] [CrossRef]
- Choi, J.; Callaway, Z.; Kim, H.B.; Fujisawa, T.; Kim, C.K. The role of TNF-alpha in eosinophilic inflammation associated with RSV bronchiolitis. Pediatr. Allergy Immunol. 2010, 21, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Kitcharoensakkul, M.; Bacharier, L.B.; Yin-Declue, H.; Boomer, J.S.; Sajol, G.; Leung, M.K.; Wilson, B.; Schechtman, K.B.; Atkinson, J.P.; Green, J.M.; et al. Impaired tumor necrosis factor-α secretion by CD4 T cells during respiratory syncytial virus bronchiolitis associated with recurrent wheeze. Immun. Inflamm. Dis. 2020, 8, 30–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalliolias, G.D.; Ivashkiv, L.B. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat. Rev. Rheumatol. 2016, 12, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Arango Duque, G.; Descoteaux, A. Macrophage cytokines: Involvement in immunity and infectious diseases. Front. Immunol. 2014, 5, 491. [Google Scholar] [CrossRef] [Green Version]
- Horiuchi, T.; Mitoma, H.; Harashima, S.-i.; Tsukamoto, H.; Shimoda, T. Transmembrane TNF-alpha: Structure, function and interaction with anti-TNF agents. Rheumatology 2010, 49, 1215–1228. [Google Scholar] [CrossRef] [Green Version]
- Wajant, H.; Siegmund, D. TNFR1 and TNFR2 in the Control of the Life and Death Balance of Macrophages. Front. Cell Dev. Biol. 2019, 7, 91. [Google Scholar] [CrossRef]
- Brenner, D.; Blaser, H.; Mak, T.W. Regulation of tumour necrosis factor signalling: Live or let die. Nat. Rev. Immunol. 2015, 15, 362–374. [Google Scholar] [CrossRef]
- Faustman, D.; Davis, M. TNF receptor 2 pathway: Drug target for autoimmune diseases. Nat. Rev. Drug Discov. 2010, 9, 482–493. [Google Scholar] [CrossRef]
- Yang, S.; Wang, J.; Brand, D.D.; Zheng, S.G. Role of TNF–TNF Receptor 2 Signal in Regulatory T Cells and Its Therapeutic Implications. Front. Immunol. 2018, 9, 784. [Google Scholar] [CrossRef] [Green Version]
- Siegmund, D.; Kums, J.; Ehrenschwender, M.; Wajant, H. Activation of TNFR2 sensitizes macrophages for TNFR1-mediated necroptosis. Cell Death Dis. 2016, 7, e2375. [Google Scholar] [CrossRef]
- Park, H.H.; Lo, Y.C.; Lin, S.C.; Wang, L.; Yang, J.K.; Wu, H. The death domain superfamily in intracellular signaling of apoptosis and inflammation. Annu. Rev. Immunol. 2007, 25, 561–586. [Google Scholar] [CrossRef] [Green Version]
- Neuzil, K.M.; Tang, Y.W.; Graham, B.S. Protective Role of TNF-alpha in respiratory syncytial virus infection in vitro and in vivo. Am. J. Med. Sci. 1996, 311, 201–204. [Google Scholar] [CrossRef]
- Rutigliano, J.A.; Graham, B.S. Prolonged production of TNF-alpha exacerbates illness during respiratory syncytial virus infection. J. Immunol. 2004, 173, 3408–3417. [Google Scholar] [CrossRef] [Green Version]
- Hussell, T.; Pennycook, A.; Openshaw, P.J. Inhibition of tumor necrosis factor reduces the severity of virus-specific lung immunopathology. Eur. J. Immunol. 2001, 31, 2566–2573. [Google Scholar] [CrossRef]
- Nguyen, T.H.; Maltby, S.; Simpson, J.L.; Eyers, F.; Baines, K.J.; Gibson, P.G.; Foster, P.S.; Yang, M. TNF-alpha and Macrophages Are Critical for Respiratory Syncytial Virus-Induced Exacerbations in a Mouse Model of Allergic Airways Disease. J. Immunol. 2016, 196, 3547–3558. [Google Scholar] [CrossRef] [Green Version]
- McCann, F.E.; Perocheau, D.P.; Ruspi, G.; Blazek, K.; Davies, M.L.; Feldmann, M.; Dean, J.L.; Stoop, A.A.; Williams, R.O. Selective tumor necrosis factor receptor I blockade is antiinflammatory and reveals immunoregulatory role of tumor necrosis factor receptor II in collagen-induced arthritis. Arthritis Rheumatol. 2014, 66, 2728–2738. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.; Kontermann, R.E.; Maier, O. Targeting sTNF/TNFR1 Signaling as a New Therapeutic Strategy. Antibodies 2015, 4, 48–70. [Google Scholar] [CrossRef]
- Morris, D.; Ansar, M.; Speshock, J.; Ivanciuc, T.; Qu, Y.; Casola, A.; Garofalo, R. Antiviral and Immunomodulatory Activity of Silver Nanoparticles in Experimental RSV Infection. Viruses 2019, 11, 732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonaldi, T.; Talamo, F.; Scaffidi, P.; Ferrera, D.; Porto, A.; Bachi, A.; Rubartelli, A.; Agresti, A.; Bianchi, M.E. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003, 22, 5551–5560. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Nagarajan, A.; Uchil, P.D. Analysis of Cell Viability by the Lactate Dehydrogenase Assay. Cold Spring Harb. Protoc. 2018, 2018, pdb.prot095497. [Google Scholar] [CrossRef] [PubMed]
- Puthothu, B.; Bierbaum, S.; Kopp, M.V.; Forster, J.; Heinze, J.; Weckmann, M.; Krueger, M.; Heinzmann, A. Association of TNF-alpha with severe respiratory syncytial virus infection and bronchial asthma. Pediatr. Allergy Immunol. 2009, 20, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Erickson, E.N.; Bhakta, R.T.; Mendez, M.D. Pediatric Bronchiolitis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Mansbach, J.M.; Camargo, C.A., Jr. Respiratory viruses in bronchiolitis and their link to recurrent wheezing and asthma. Clin. Lab. Med. 2009, 29, 741–755. [Google Scholar] [CrossRef]
- Defer, N.; Azroyan, A.; Pecker, F.; Pavoine, C. TNFR1 and TNFR2 signaling interplay in cardiac myocytes. J. Biol. Chem. 2007, 282, 35564–35573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Y.; Fischer, R.; Naude, P.J.; Maier, O.; Nyakas, C.; Duffey, M.; Van der Zee, E.A.; Dekens, D.; Douwenga, W.; Herrmann, A.; et al. Essential protective role of tumor necrosis factor receptor 2 in neurodegeneration. Proc. Natl. Acad. Sci. USA 2016, 113, 12304–12309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, S.K.; Maier, O.; Fischer, R.; Fairless, R.; Hochmeister, S.; Stojic, A.; Pick, L.; Haar, D.; Musiol, S.; Storch, M.K.; et al. Antibody-mediated inhibition of TNFR1 attenuates disease in a mouse model of multiple sclerosis. PLoS ONE 2014, 9, e90117. [Google Scholar] [CrossRef]
- Müller, P.; Edelmann-Stephan, B.; Richter, F.; Pfizenmaier, K.; Fischer, T. TNFR1 Blockade Rather then TNFR2 Blockade Reduces Chronic Inflammation in JAK2+/V617F Mice. Blood 2019, 134, 2966. [Google Scholar] [CrossRef]
- Hunter, I.; Nixon, G.F. Spatial compartmentalization of tumor necrosis factor (TNF) receptor 1-dependent signaling pathways in human airway smooth muscle cells. Lipid rafts are essential for TNF-alpha-mediated activation of RhoA but dispensable for the activation of the NF-kappaB and MAPK pathways. J. Biol. Chem. 2006, 281, 34705–34715. [Google Scholar] [CrossRef] [Green Version]
- Muppidi, J.R.; Tschopp, J.; Siegel, R.M. Life and death decisions: Secondary complexes and lipid rafts in TNF receptor family signal transduction. Immunity 2004, 21, 461–465. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Tliba, O.; Van Besien, C.R.; Panettieri, R.A., Jr.; Amrani, Y. TNF-[alpha] modulates murine tracheal rings responsiveness to G-protein-coupled receptor agonists and KCl. J. Appl. Physiol. 2003, 95, 864–872. [Google Scholar] [CrossRef]
- Sezgin, E.; Levental, I.; Mayor, S.; Eggeling, C. The mystery of membrane organization: Composition, regulation and roles of lipid rafts. Nat. Rev. Mol. Cell Biol. 2017, 18, 361–374. [Google Scholar] [CrossRef] [Green Version]
- Yap, H.M.; Israf, D.A.; Harith, H.H.; Tham, C.L.; Sulaiman, M.R. Crosstalk between Signaling Pathways Involved in the Regulation of Airway Smooth Muscle Cell Hyperplasia. Front. Pharmacol. 2019, 10, 1148. [Google Scholar] [CrossRef]
- Legler, D.F.; Micheau, O.; Doucey, M.-A.; Tschopp, J.; Bron, C. Recruitment of TNF Receptor 1 to Lipid Rafts Is Essential for TNFα-Mediated NF-κB Activation. Immunity 2003, 18, 655–664. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Bhetwal, B.P.; Gunst, S.J. Rho kinase collaborates with p21-activated kinase to regulate actin polymerization and contraction in airway smooth muscle. J. Physiol. 2018, 596, 3617–3635. [Google Scholar] [CrossRef]
- Yoshimura, H.; Jones, K.A.; Perkins, W.J.; Kai, T.; Warner, D.O. Calcium sensitization produced by G protein activation in airway smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 281, L631–L638. [Google Scholar] [CrossRef] [Green Version]
- Anaparti, V.; Pascoe, C.D.; Jha, A.; Mahood, T.H.; Ilarraza, R.; Unruh, H.; Moqbel, R.; Halayko, A.J. Tumor necrosis factor regulates NMDA receptor-mediated airway smooth muscle contractile function and airway responsiveness. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L467–L480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amrani, Y.; Panettieri, R.A., Jr. Cytokines induce airway smooth muscle cell hyperresponsiveness to contractile agonists. Thorax 1998, 53, 713–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amrani, Y.; Chen, H.; Panettieri, R.A., Jr. Activation of tumor necrosis factor receptor 1 in airway smooth muscle: A potential pathway that modulates bronchial hyper-responsiveness in asthma? Respir. Res. 2000, 1, 49–53. [Google Scholar] [CrossRef] [Green Version]
- Tsai, M.H.; Jiang, M.J. Reactive oxygen species are involved in regulating alpha1-adrenoceptor-activated vascular smooth muscle contraction. J. Biomed. Sci. 2010, 17, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, I.T.; Lin, C.C.; Lee, C.Y.; Hsieh, P.W.; Yang, C.M. Protective effects of (-)-epigallocatechin-3-gallate against TNF-alpha-induced lung inflammation via ROS-dependent ICAM-1 inhibition. J. Nutr. Biochem. 2013, 24, 124–136. [Google Scholar] [CrossRef]
- Castro, S.M.; Guerrero-Plata, A.; Suarez-Real, G.; Adegboyega, P.A.; Colasurdo, G.N.; Khan, A.M.; Garofalo, R.P.; Casola, A. Antioxidant treatment ameliorates respiratory syncytial virus-induced disease and lung inflammation. Am. J. Respir. Crit. Care Med. 2006, 174, 1361–1369. [Google Scholar] [CrossRef] [Green Version]
- Ansar, M.; Ivanciuc, T.; Garofalo, R.P.; Casola, A. Increased Lung Catalase Activity Confers Protection against Experimental RSV Infection. Sci. Rep. 2020, 10, 3653. [Google Scholar] [CrossRef]
- Vieira, S.M.; Lemos, H.P.; Grespan, R.; Napimoga, M.H.; Dal-Secco, D.; Freitas, A.; Cunha, T.M.; Verri, W.A., Jr.; Souza-Junior, D.A.; Jamur, M.C.; et al. A crucial role for TNF-alpha in mediating neutrophil influx induced by endogenously generated or exogenous chemokines, KC/CXCL1 and LIX/CXCL5. Br. J. Pharmacol. 2009, 158, 779–789. [Google Scholar] [CrossRef] [Green Version]
- Deguine, J.; Wei, J.; Barbalat, R.; Gronert, K.; Barton, G.M. Local TNFR1 Signaling Licenses Murine Neutrophils for Increased TLR-Dependent Cytokine and Eicosanoid Production. J. Immunol. 2017, 198, 2865–2875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, Y.; Li, F.; Qin, Z. TNF Receptor 2 Makes Tumor Necrosis Factor a Friend of Tumors. Front. Immunol. 2018, 9, 1170. [Google Scholar] [CrossRef] [Green Version]
- Kolli, D.; Gupta, M.R.; Sbrana, E.; Velayutham, T.S.; Chao, H.; Casola, A.; Garofalo, R.P. Alveolar macrophages contribute to the pathogenesis of human metapneumovirus infection while protecting against respiratory syncytial virus infection. Am. J. Respir. Cell Mol. Biol. 2014, 51, 502–515. [Google Scholar] [CrossRef] [Green Version]
- Gimenez, M.A.; Sim, J.; Archambault, A.S.; Klein, R.S.; Russell, J.H. A tumor necrosis factor receptor 1-dependent conversation between central nervous system-specific T cells and the central nervous system is required for inflammatory infiltration of the spinal cord. Am. J. Pathol. 2006, 168, 1200–1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garofalo, R.P.; Patti, J.; Hintz, K.A.; Hill, V.; Ogra, P.L.; Welliver, R.C. Macrophage inflammatory protein-1alpha (not T helper type 2 cytokines) is associated with severe forms of respiratory syncytial virus bronchiolitis. J. Infect. Dis. 2001, 184, 393–399. [Google Scholar] [CrossRef]
- McNamara, P.S.; Flanagan, B.F.; Hart, C.A.; Smyth, R.L. Production of chemokines in the lungs of infants with severe respiratory syncytial virus bronchiolitis. J. Infect. Dis. 2005, 191, 1225–1232. [Google Scholar] [CrossRef] [PubMed]
- Sugai, K.; Kimura, H.; Miyaji, Y.; Tsukagoshi, H.; Yoshizumi, M.; Sasaki-Sakamoto, T.; Matsunaga, S.; Yamada, Y.; Kashiwakura, J.; Noda, M.; et al. MIP-1alpha level in nasopharyngeal aspirates at the first wheezing episode predicts recurrent wheezing. J. Allergy Clin. Immunol. 2016, 137, 774–781. [Google Scholar] [CrossRef] [Green Version]
- Mukaro, V.R.; Quach, A.; Gahan, M.E.; Boog, B.; Huang, Z.H.; Gao, X.; Haddad, C.; Mahalingam, S.; Hii, C.S.; Ferrante, A. Small tumor necrosis factor receptor biologics inhibit the tumor necrosis factor-p38 signalling axis and inflammation. Nat. Commun. 2018, 9, 1365. [Google Scholar] [CrossRef]
- Steeland, S.; Libert, C.; Vandenbroucke, R.E. A New Venue of TNF Targeting. Int. J. Mol. Sci. 2018, 19, 1442. [Google Scholar] [CrossRef] [Green Version]
- Proudfoot, A.; Bayliffe, A.; O’Kane, C.M.; Wright, T.; Serone, A.; Bareille, P.J.; Brown, V.; Hamid, U.I.; Chen, Y.; Wilson, R.; et al. Novel anti-tumour necrosis factor receptor-1 (TNFR1) domain antibody prevents pulmonary inflammation in experimental acute lung injury. Thorax 2018, 73, 723–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morris, D.R.; Ansar, M.; Ivanciuc, T.; Qu, Y.; Casola, A.; Garofalo, R.P. Selective Blockade of TNFR1 Improves Clinical Disease and Bronchoconstriction in Experimental RSV Infection. Viruses 2020, 12, 1176. https://doi.org/10.3390/v12101176
Morris DR, Ansar M, Ivanciuc T, Qu Y, Casola A, Garofalo RP. Selective Blockade of TNFR1 Improves Clinical Disease and Bronchoconstriction in Experimental RSV Infection. Viruses. 2020; 12(10):1176. https://doi.org/10.3390/v12101176
Chicago/Turabian StyleMorris, Dorothea R., Maria Ansar, Teodora Ivanciuc, Yue Qu, Antonella Casola, and Roberto P. Garofalo. 2020. "Selective Blockade of TNFR1 Improves Clinical Disease and Bronchoconstriction in Experimental RSV Infection" Viruses 12, no. 10: 1176. https://doi.org/10.3390/v12101176
APA StyleMorris, D. R., Ansar, M., Ivanciuc, T., Qu, Y., Casola, A., & Garofalo, R. P. (2020). Selective Blockade of TNFR1 Improves Clinical Disease and Bronchoconstriction in Experimental RSV Infection. Viruses, 12(10), 1176. https://doi.org/10.3390/v12101176