
Molecular Epidemiological Analysis of the Origin and Transmission Dynamics of the HIV-1 CRF01_AE Sub-Epidemic in Bulgaria

 , ,
, ,  , , ,
, , , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design and Patient Samples
2.2. Dataset and Sequence Analyses
2.3. Statistical Analysis
3. Results
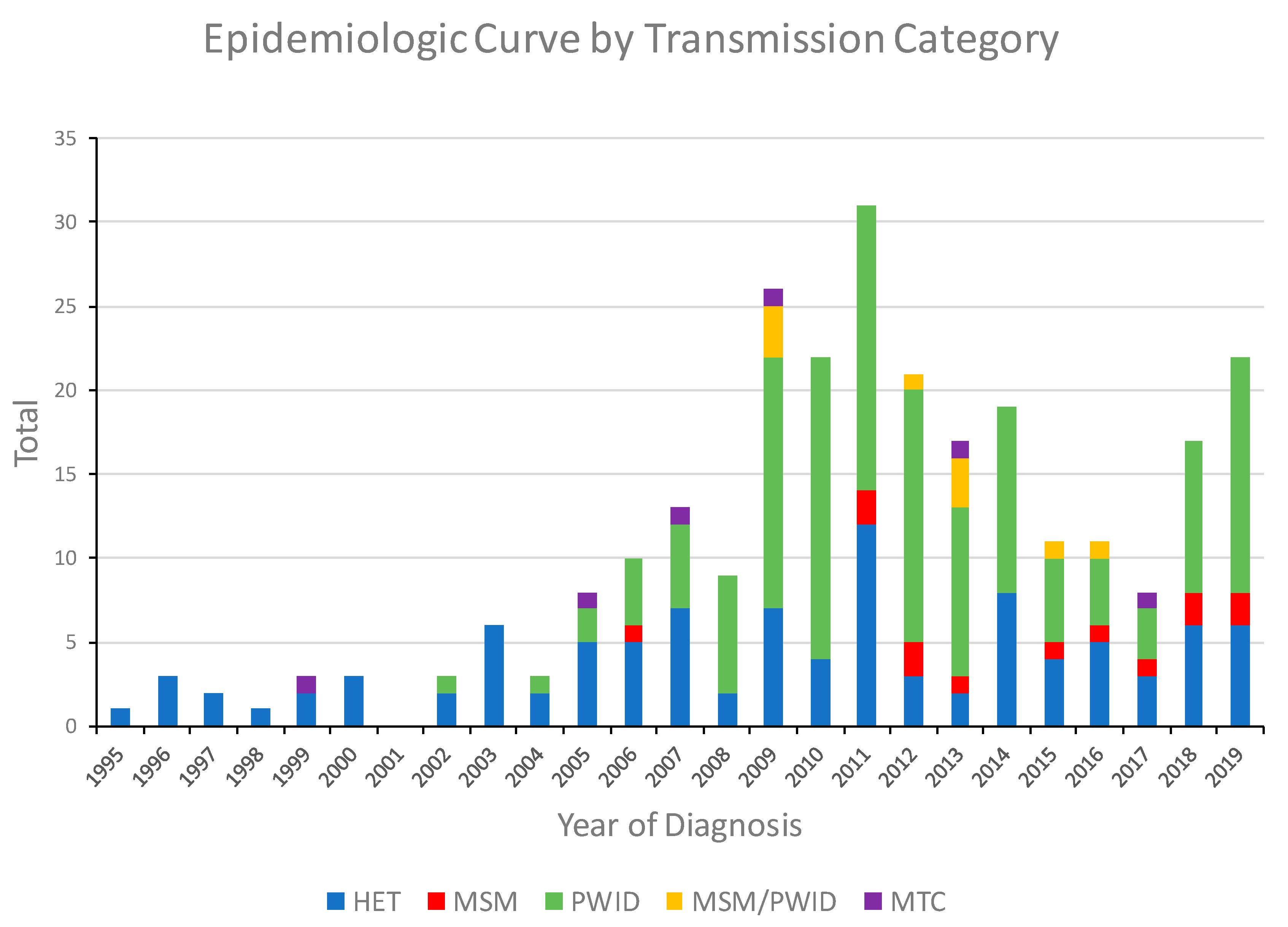
3.1. Characteristics of the HIV-1 CRF01_AE Infections in Bulgaria
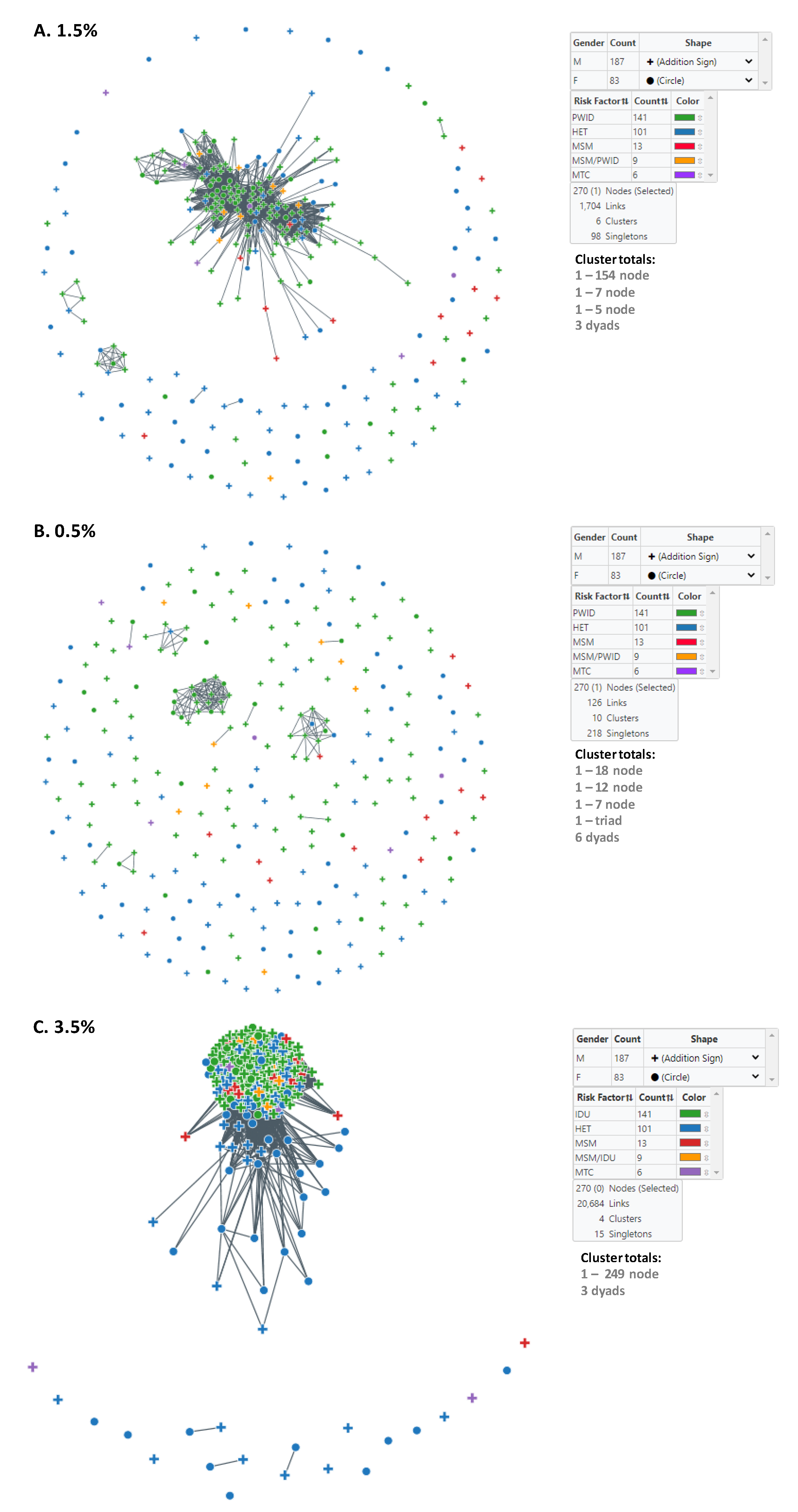
3.2. Identification and Characterization of HIV-1 Subtype CRF01_AE Transmission Clusters in Bulgaria
3.3. Assortative Mixing of Pairs, Sex, Similar Ages, Transmission Category, and Geographic Location in the HIV-1 CRF01_AE Transmission Networks
3.4. Origin of HIV-1 CRF01_AE Infections in Bulgaria
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Leitner, T. The Puzzle of HIV Neutral and Selective Evolution. Mol. Biol. Evol. 2018, 35, 1355–1358. [Google Scholar] [CrossRef] [PubMed]
- Coffin, J.M. HIV population dynamics in vivo: Implications for genetic variation, pathogenesis, and therapy. Science 1995, 267, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.X.; Taylor, J. Template switching by reverse transcriptase during DNA synthesis. J. Virol. 1990, 64, 4321–4328. [Google Scholar] [CrossRef]
- Sharp, P.M.; Hahn, B.H. Origins of HIV and the AIDS Pandemic. Cold Spring Harb. Perspect. Med. 2011, 1, a006841. [Google Scholar] [CrossRef] [PubMed]
- Hemelaar, J.; Elangovan, R.; Yun, J.; Dickson-Tetteh, L.; Fleminger, I.; Kirtley, S.; Williams, B.; Gouws-Williams, E.; Ghys, P.D.; Abimiku, A.G.; et al. Global and regional molecular epidemiology of HIV-1, 1990–2015: A systematic review, global survey, and trend analysis. Lancet Infect. Dis. 2019, 19, 143–155. [Google Scholar] [CrossRef]
- Carr, J.K.; O Salminen, M.; Koch, C.; Gotte, D.; Artenstein, A.W.; A Hegerich, P.; Louis, D.S.; Burke, D.S.; E McCutchan, F. Full-length sequence and mosaic structure of a human immunodeficiency virus type 1 isolate from Thailand. J. Virol. 1996, 70, 5935–5943. [Google Scholar] [CrossRef]
- McCutchan, F.E. Global epidemiology of HIV. J. Med Virol. 2006, 78, S7–S12. [Google Scholar] [CrossRef]
- Junqueira, D.M.; Wilkinson, E.; Vallari, A.S.; Deng, X.; Achari, A.; Yu, G.; McArthur, C.P.; Kaptue, L.; Mbanya, D.N.; Chiu, C.Y.; et al. New Genomes from the Congo Basin Expand History of CRF01_AE Origin and Dissemination. AIDS Res. Hum. Retrovir. 2020, 36, 574–582. [Google Scholar] [CrossRef]
- Hofstra, L.M.; Sauvageot, N.; Albert, J.; Alexiev, I.; Garcia, F.P.; Struck, D.; Van De Vijver, D.A.M.C.; Åsjö, B.; Beshkov, D.; Coughlan, S.; et al. Transmission of HIV Drug Resistance and the Predicted Effect on Current First-line Regimens in Europe. Clin. Infect. Dis. 2016, 62, 655–663. [Google Scholar] [CrossRef]
- Stanojevic, M.; Alexiev, I.; Beshkov, D.; Gokengin, D.; Mezei, M.; Minarovits, J.; Otelea, D.; Paraschiv, S.; Poljak, M.; Zidovec, S.L.; et al. HIV1 Molecular Epidemiology in the Balkans-A Melting Pot for High Genetic Diversity. Aids. Rev. 2012, 14, 28–36. [Google Scholar] [PubMed]
- Alexiev, I.; Beshkov, D.; Shankar, A.; Hanson, D.L.; Paraskevis, D.; Georgieva, V.; Karamacheva, L.; Taskov, H.; Varleva, T.; Elenkov, I.; et al. Detailed Molecular Epidemiologic Characterization of HIV-1 Infection in Bulgaria Reveals Broad Diversity and Evolving Phylodynamics. PLoS ONE 2013, 8, e59666. [Google Scholar] [CrossRef]
- Alexiev, I.; Shankar, A.; Wensing, A.M.J.; Beshkov, D.; Elenkov, I.; Stoycheva, M.; Nikolova, D.; Nikolova, M.; Switzer, W.M. Low HIV-1 transmitted drug resistance in Bulgaria against a background of high clade diversity. J. Antimicrob. Chemother. 2015, 70, 1874–1880. [Google Scholar] [CrossRef] [PubMed]
- Alexiev, I.; Shankar, A.; Dimitrova, R.; Gancheva, A.; Kostadinova, A.; Teoharov, P.; Golkocheva, E.; Nikolova, M.; Muhtarova, M.; Elenkov, I.; et al. Origin and spread of HIV-1 in persons who inject drugs in Bulgaria. Infect. Genet. Evol. 2016, 46, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Struck, D.; Lawyer, G.; Ternes, A.-M.; Schmit, J.-C.; Bercoff, D.P. COMET: Adaptive context-based modeling for ultrafast HIV-1 subtype identification. Nucleic Acids Res. 2014, 42, e144. [Google Scholar] [CrossRef] [PubMed]
- Pineda-Pena, A.; Faria, N.R.; Imbrechts, S.; Libin, P.; Abecasis, A.B.; Deforche, K.; Gómez-López, A.; Camacho, R.J.; De Oliveira, T.; Vandamme, A.-M. Automated subtyping of HIV-1 genetic sequences for clinical and surveillance purposes: Performance evaluation of the new REGA version 3 and seven other tools. Infect. Genet. Evol. 2013, 19, 337–348. [Google Scholar] [CrossRef]
- Schultz, A.-K.; Zhang, M.; Bulla, I.; Leitner, T.; Korber, B.; Morgenstern, B.; Stanke, M. jpHMM: Improving the reliability of recombination prediction in HIV-1. Nucleic Acids Res. 2009, 37, W647–W651. [Google Scholar] [CrossRef]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-Length Human Immunodeficiency Virus Type 1 Genomes from Subtype C-Infected Seroconverters in India, with Evidence of Intersubtype Recombination. J. Virol. 1999, 73, 152–160. [Google Scholar] [CrossRef]
- Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef]
- Kuraku, S.; Zmasek, C.M.; Nishimura, O.; Katoh, K. aLeaves facilitates on-demand exploration of metazoan gene family trees on MAFFT sequence alignment server with enhanced interactivity. Nucleic Acids Res. 2013, 41, W22–W28. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2-Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; A Neher, R. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [PubMed]
- Sagulenko, P.; Puller, V.; Neher, R.A. TreeTime: Maximum-likelihood phylodynamic analysis. Virus Evol. 2018, 4, vex042. [Google Scholar] [CrossRef] [PubMed]
- Campbell, E.; Boylesm, A.; Shankar, A.; Kim, J.; Knyazev, S.; Switzer, W.M. MicrobeTrace: Retooling Molecular Epidemiology for Rapid Public Health Response. bioRxiv 2020. [Google Scholar] [CrossRef]
- Attribute Assortativity. Available online: https://github.com/Sergey-Knyazev/attribute_assortativity (accessed on 28 October 2020).
- NetworkX. Available online: https://networkx.github.io (accessed on 28 October 2020).
- Hai-Long, H.; Jian, Z.; Ping-Ping, Y.; Liang, C.; Xun, L.; Shan, Z.Z.; Yan-Sheng, Y. Genetic Characterization of CRF01_AE Full-Length Human Immunodeficiency Virus Type 1 Sequences from Fujian, China. AIDS Res. Hum. Retrovir. 2007, 23, 569–574. [Google Scholar] [CrossRef]
- Billings, E.A.; Heipertz, R.A.; Varleva, T.; Sanders-Buell, E.; O’Sullivan, A.M.; Bose, M.; Howell, S.; Kijak, G.H.; Taskov, H.; Elenkov, I.; et al. HIV-1 genetic diversity and demographic characteristics in Bulgaria. PLoS ONE 2019, 14, e0217063. [Google Scholar] [CrossRef]
- Alexiev, I.; Campbell, E.; Knyazev, S.; Pan, Y.; Grigorova, L.; Dimitrova, R.; Partsuneva, A.; Gancheva, A.; Kostadinova, A.; Seguin-Devaux, C.; et al. Molecular Epidemiology of the HIV-1 Subtype B Sub-Epidemic in Bulgaria. Viruses 2020, 12, 441. [Google Scholar] [CrossRef]
- Campbell, E.M.; Jia, H.; Shankar, A.; Hanson, D.; Luo, W.; Masciotra, S.; Owen, S.M.; Oster, A.M.; Galang, R.R.; Spiller, M.W.; et al. Detailed Transmission Network Analysis of a Large Opiate-Driven Outbreak of HIV Infection in the United States. J. Infect. Dis. 2017, 216, 1053–1062. [Google Scholar] [CrossRef]
- Hassan, A.S.; Pybus, O.G.; Sanders, E.J.; Albert, J.; Esbjörnsson, J. Defining HIV-1 transmission clusters based on sequence data. AIDS 2017, 31, 1211–1222. [Google Scholar] [CrossRef]
- Oster, A.M.; France, A.M.; Panneer, N.; Ocfemia, M.C.B.; Campbell, E.; Dasgupta, S.; Switzer, W.M.; Wertheim, J.O.; Hernandez, A.L. Identifying Clusters of Recent and Rapid HIV Transmission Through Analysis of Molecular Surveillance Data. JAIDS J. Acquir. Immune Defic. Syndr. 2018, 79, 543–550. [Google Scholar] [CrossRef]
- Rich, S.; Richards, V.L.; Mavian, C.; Switzer, W.M.; Magalis, B.R.; Poschman, K.; Geary, S.; Broadway, S.E.; Bennett, S.B.; Blanton, J.; et al. Employing Molecular Phylodynamic Methods to Identify and Forecast HIV Transmission Clusters in Public Health Settings: A Qualitative Study. Viruses 2020, 12, 921. [Google Scholar] [CrossRef]
- Paraskevis, D.; Nikolopoulos, G.; Tsiara, C.; Paraskeva, D.; Antoniadou, A.; Lazanas, M.; Gargalianos, P.; Psychogiou, M.; Malliori, M.; Kremastinou, J.; et al. HIV-1 outbreak among injecting drug users in Greece, 2011: A preliminary report. Eurosurveillance 2011, 16, 19962. [Google Scholar] [CrossRef]
- Arendt, V.; Guillorit, L.; Origer, A.; Sauvageot, N.; Vaillant, M.; Fischer, A.; Goedertz, H.; François, J.-H.; Alexiev, I.; Staub, T.; et al. Injection of cocaine is associated with a recent HIV outbreak in people who inject drugs in Luxembourg. PLoS ONE 2019, 14, e0215570. [Google Scholar] [CrossRef] [PubMed]
- Hightower, G.K.; May, S.J.; Pérez-Santiago, J.; Pacold, M.E.; Wagner, G.A.; Little, S.J.; Richman, D.D.; Mehta, S.R.; Smith, D.M.; Pond, S.L.K. HIV-1 Clade B pol Evolution following Primary Infection. PLoS ONE 2013, 8, e68188. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Detecting and Responding to HIV Transmission Clusters. A Guide for Health Departments. June 2018. Available online: https://www.cdc.gov/hiv/pdf/funding/announcements/ps18-1802/CDC-HIV-PS18-1802-AttachmentE-Detecting-Investigating-and-Responding-to-HIV-Transmission-Clusters.pdf (accessed on 28 October 2020).
- Campbell, E.; Patala, A.; Shankar, A.; Li, J.-F.; Johnson, J.A.; Westheimer, E.; Gay, C.L.; Cohen, S.E.; Switzer, W.M.; Peters, P.J. Phylodynamic Analysis Complements Partner Services by Identifying Acute and Unreported HIV Transmission. Viruses 2020, 12, 145. [Google Scholar] [CrossRef] [PubMed]
- Novitsky, V.; Steingrimsson, J.A.; Howison, M.; Gillani, F.S.; Li, Y.; Manne, A.; Fulton, J.; Spence, M.; Parillo, Z.; Marak, T.; et al. Empirical comparison of analytical approaches for identifying molecular HIV-1 clusters. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; He, X.; Hsi, J.H.; Li, F.; Li, X.; Wang, Q.; Ruan, Y.; Xing, H.; Lam, T.T.-Y.; Pybus, O.G.; et al. The rapidly expanding CRF01_AE epidemic in China is driven by multiple lineages of HIV-1 viruses introduced in the 1990s. AIDS 2013, 27, 1793–1802. [Google Scholar] [CrossRef]
- Li, X.; Xue, Y.; Lin, Y.; Gai, J.; Zhang, L.; Cheng, H.; Ning, Z.; Zhou, L.; Zhu, K.; Vanham, G.; et al. Evolutionary Dynamics and Complicated Genetic Transmission Network Patterns of HIV-1 CRF01_AE among MSM in Shanghai, China. Sci. Rep. 2016, 6, 34729. [Google Scholar] [CrossRef]
- Li, X.; Liu, H.; Liu, L.; Feng, Y.; Kalish, M.L.; Ho, S.Y.W.; Shao, Y. Tracing the epidemic history of HIV-1 CRF01_AE clusters using near-complete genome sequences. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Angelis, K.; Albert, J.; Mamais, I.; Magiorkinis, G.; Hatzakis, A.; Hamouda, O.; Struck, D.; Vercauteren, J.; Wensing, A.M.J.; Alexiev, I.; et al. Global Dispersal Pattern of HIV Type 1 Subtype CRF01_AE: A Genetic Trace of Human Mobility Related to Heterosexual Sexual Activities Centralized in Southeast Asia. J. Infect. Dis. 2014, 211, 1735–1744. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, P.J.; Zhang, Y.; Fogel, J.M.; Guo, X.; Clarke, W.; Breaud, A.; Richardson, P.; Piwowar-Manning, E.; Hart, S.; Hamilton, E.L.; et al. HIV drug resistance in persons who inject drugs enrolled in an HIV prevention trial in Indonesia, Ukraine, and Vietnam: HPTN 074. PLoS ONE 2019, 14, e0223829. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.; Bozzi, G.; Franzetti, M.; Binda, F.; Simonetti, F.R.; De Luca, A.; Micheli, V.; Meraviglia, P.; Bagnarelli, P.; Di Biagio, A.; et al. HIV-1 A1 Subtype Epidemic in Italy Originated from Africa and Eastern Europe and Shows a High Frequency of Transmission Chains Involving Intravenous Drug Users. PLoS ONE 2016, 11, e0146097. [Google Scholar] [CrossRef] [PubMed]
- Paraschiv, S.; Bănică, L.; Nicolae, I.; Niculescu, I.; Abagiu, A.; Jipa, R.; Pineda-Pena, A.; Pingarilho, M.; Neaga, E.; Theys, K.; et al. Epidemic dispersion of HIV and HCV in a population of co-infected Romanian injecting drug users. PLoS ONE 2017, 12, e0185866. [Google Scholar] [CrossRef] [PubMed]
- Golden, M.R.; Lechtenberg, R.; Glick, S.N.; Dombrowski, J.; Duchin, J.; Reuer, J.R.; Dhanireddy, S.; Neme, S.; Buskin, S.E. Outbreak of Human Immunodeficiency Virus Infection Among Heterosexual Persons Who Are Living Homeless and Inject Drugs—Seattle, Washington, 2018. MMWR. Morb. Mortal. Wkly. Rep. 2019, 68, 344–349. [Google Scholar] [CrossRef]
- An, M.; Han, X.; Zhao, B.; English, S.; Frost, S.D.W.; Zhang, H.; Shang, H. Cross-Continental Dispersal of Major HIV-1 CRF01_AE Clusters in China. Front. Microbiol. 2020, 11, 61. [Google Scholar] [CrossRef]
- World Health Organization. HIV/AIDS Surveillance in Europe: 2019. Available online: https://www.ecdc.europa.eu/sites/default/files/documents/HIV-annual-surveillance-report-2019.pdf (accessed on 28 October 2020).




{kind=link}
{kind=link}
{kind=link}
| Characteristic | Subtype CRF01_AE n (%) | Other Subtypes n (%) | p Value |
|---|---|---|---|
| Total | 270 | 1413 | |
| Sex | <0.0001 | ||
| Men | 187 (69.3) | 1195 (84.6) | |
| Women | 83 (30.7) | 218 (15.4) | |
| Age (years) | 0.001 | ||
| ≤19 | 28 (10.4) | 68 (4.8) | |
| 20–29 | 108 (40.0) | 538 (38.1) | |
| 30–39 | 90 (33.3) | 493 (34.9) | |
| 40–49 | 35 (13.0) | 204 (14.4) | |
| ≥50 | 9 (3.3) | 110 (7.8) | |
| Country of Birth | 0.0018 | ||
| Bulgaria | 269 (99.6) | 1361 (96.3) | |
| Other country | 1 (0.4) | 52 (3.7) | |
| Likely Country of Infection | <0.0001 | ||
| Bulgaria | 251 (93.0) | 1176 (83.2) | |
| Other country 2 | 19 (7.0) | 237 (16.8) | |
| Region in Bulgaria | <0.0001 | ||
| Sofia | 164 (60.7) | 625 (44.2) | |
| Other regions | 106 (39.3) | 788 (55.8) | |
| Transmission category 3 | <0.0001 | ||
| HET | 101 (37.4) | 592 (41.9) | |
| MSM | 13 (4.8) | 630 (44.6) | |
| PWID | 141 (52.2) | 158 (11.2) | |
| Other | 15 (5.6) | 33 (2.3) | |
| Characteristic | Odds Ratio | 95% CI | p Value | |
|---|---|---|---|---|
| Sex (Women vs. Men) | 2.14 | 1.48 | 3.08 | <0.0001 |
| Age at diagnosis (in years) | 1.00 | 0.99 | 1.02 | 0.73 |
| Likely country of infection (Bulgaria vs. Other) | 1.90 | 1.11 | 3.27 | 0.02 |
| Region in Bulgaria (Sofia vs. Other) | 2.98 | 2.19 | 4.05 | <0.0001 |
| Transmission category | <0.0001 | |||
| MSM vs. HET | 0.13 | 0.07 | 0.24 | <0.0001 |
| PWID vs. HET | 6.40 | 4.41 | 9.28 | <0.0001 |
| Other vs. HET | 3.44 | 1.64 | 7.23 | 0.001 |
| Cluster Sizes at 1.5% | Male | Female | HET | MSM | MSM/PWID | PWID | Vertical | Mean/Median Age at Diagnosis | Diagnosis Date Range | Likely Country of Infection (Bulgaria) | Likely Country of Infection (Other) 2 | |
| 154 | 116 | 38 | 30 | 5 | 8 | 108 | 3 | 28.4/29.0 | 2002–2019 | 143 | 11 | |
| 7 | 5 | 2 | 2 | 0 | 0 | 5 | 0 | 38.9/39.0 | 2018–2019 | 6 | 1 | |
| 5 | 5 | 0 | 1 | 0 | 0 | 4 | 0 | 33.0/32.0 | 2010–2019 | 5 | 0 | |
| Three dyads (6 total) | 4 | 2 | 4 | 0 | 0 | 2 | 0 | 38.8/32.5 | 1999–2019 | 5 | 1 | |
| Singletons (98 total) | 57 | 41 | 64 | 8 | 1 | 22 | 3 | 31.2/31.0 | 1995–2019 | 92 | 6 | |
| Totals | 270 | 187 | 83 | 101 | 13 | 9 | 141 | 6 | 30.0/29.0 | 1995–2019 | 251 | 19 |
| Cluster Sizes at 0.5% | Male | Female | HET | MSM | MSM/PWID | PWID | Vertical | Mean/Median Age at Diagnosis | Diagnosis Date Range | Likely Country of Infection (Bulgaria) | Likely Country of Infection (Other) 2 | |
| 18 | 10 | 8 | 0 | 0 | 0 | 18 | 0 | 22.5/20.5 | 2009–2018 | 17 | 1 | |
| 12 | 9 | 3 | 2 | 1 | 0 | 9 | 0 | 30.3/31.0 | 2009–2015 | 10 | 2 | |
| 7 | 6 | 1 | 1 | 0 | 0 | 6 | 0 | 27.9/29.0 | 2011–2015 | 7 | 0 | |
| 3 | 2 | 1 | 0 | 0 | 0 | 3 | 0 | 38.0/38.0 | 2018–2019 | 2 | 1 | |
| Six dyads (12 total) | 9 | 3 | 0 | 0 | 2 | 9 | 1 | 29.2/29.5 | 2009–2019 | 12 | 0 | |
| Singletons (218 total) | 151 | 67 | 98 | 12 | 7 | 96 | 5 | 30.7/30.0 | 1995–2019 | 203 | 15 | |
| Totals | 270 | 187 | 83 | 101 | 13 | 9 | 141 | 6 | 30.0/29.0 | 1995–2019 | 251 | 19 |
| Cluster Sizes at 3.5% | Male | Female | HET | MSM | MSM/PWID | PWID | Vertical | Mean/Median Age at Diagnosis | Diagnosis Date Range | Likely Country of Infection (Bulgaria) | Likely Country of Infection (Other) 2 | |
| 249 | 176 | 73 | 83 | 12 | 9 | 141 | 4 | 30.0/29.0 | 1995–2019 | 234 | 15 | |
| Three dyads (6 total) | 3 | 3 | 6 | 0 | 0 | 0 | 0 | 30.3/30.0 | 2006–2019 | 4 | 2 | |
| Singletons (15 total) | 8 | 7 | 12 | 1 | 0 | 0 | 2 | 29.5/29.0 | 1998–2018 | 13 | 2 | |
| Totals | 270 | 187 | 83 | 101 | 13 | 9 | 141 | 6 | 30.0/29.0 | 1995–2019 | 251 | 19 |
| Cluster Size, Genetic Distance (d) Threshold, Total Persons | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Size | d 2 | Total 3 | Size | d | Total | Size | d | Total | Size | d | Total | Size | d | Total | Size | d | Total | Size | d | Total | Size | d | Total | |
| Cluster Characteristic | dyad | 0.5 | 12 | dyad | 1.5 | 6 | 3–9 | 0.5 | 10 | 3–9 | 1.5 | 12 | ≥10 | 0.5 | 30 | ≥10 | 1.5 | 154 | All | 0.5 | 52 | All | 1.5 | 172 |
| Region | 0.05 | 1 | 0.41 | −0.07 | 0.72 | 0.29 | 0.66 | 0.28 | ||||||||||||||||
| Sex | −0.33 | −0.5 | −0.24 | −0.07 | −0.03 | 0.01 | −0.05 | 0 | ||||||||||||||||
| Transmission category | −0.24 | 1 | −0.3 | −0.2 | −0.04 | 0.03 | −0.06 | 0.03 | ||||||||||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alexiev, I.; Campbell, E.M.; Knyazev, S.; Pan, Y.; Grigorova, L.; Dimitrova, R.; Partsuneva, A.; Gancheva, A.; Kostadinova, A.; Seguin-Devaux, C.; et al. Molecular Epidemiological Analysis of the Origin and Transmission Dynamics of the HIV-1 CRF01_AE Sub-Epidemic in Bulgaria. Viruses 2021, 13, 116. https://doi.org/10.3390/v13010116
Alexiev I, Campbell EM, Knyazev S, Pan Y, Grigorova L, Dimitrova R, Partsuneva A, Gancheva A, Kostadinova A, Seguin-Devaux C, et al. Molecular Epidemiological Analysis of the Origin and Transmission Dynamics of the HIV-1 CRF01_AE Sub-Epidemic in Bulgaria. Viruses. 2021; 13(1):116. https://doi.org/10.3390/v13010116
Chicago/Turabian StyleAlexiev, Ivailo, Ellsworth M. Campbell, Sergey Knyazev, Yi Pan, Lyubomira Grigorova, Reneta Dimitrova, Aleksandra Partsuneva, Anna Gancheva, Asya Kostadinova, Carole Seguin-Devaux, and et al. 2021. "Molecular Epidemiological Analysis of the Origin and Transmission Dynamics of the HIV-1 CRF01_AE Sub-Epidemic in Bulgaria" Viruses 13, no. 1: 116. https://doi.org/10.3390/v13010116
APA StyleAlexiev, I., Campbell, E. M., Knyazev, S., Pan, Y., Grigorova, L., Dimitrova, R., Partsuneva, A., Gancheva, A., Kostadinova, A., Seguin-Devaux, C., Elenkov, I., Yancheva, N., & Switzer, W. M. (2021). Molecular Epidemiological Analysis of the Origin and Transmission Dynamics of the HIV-1 CRF01_AE Sub-Epidemic in Bulgaria. Viruses, 13(1), 116. https://doi.org/10.3390/v13010116