
Dengue Fever Surveillance in Mato Grosso do Sul: Insights from Genomic Analysis and Implications for Public Health Strategies


 ,
,  , , , , , ,
, , , , , ,  , and add
Show full author list
, and add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Collection of Samples and Molecular Testing
2.3. Genome Sequencing and cDNA synthesis
2.4. Generation of Consensus Sequences
2.5. Phylogenetic Reconstruction
2.6. Epidemic Curves Based on Cases of Dengue Fever Recorded in Mato Grosso do Sul
3. Results
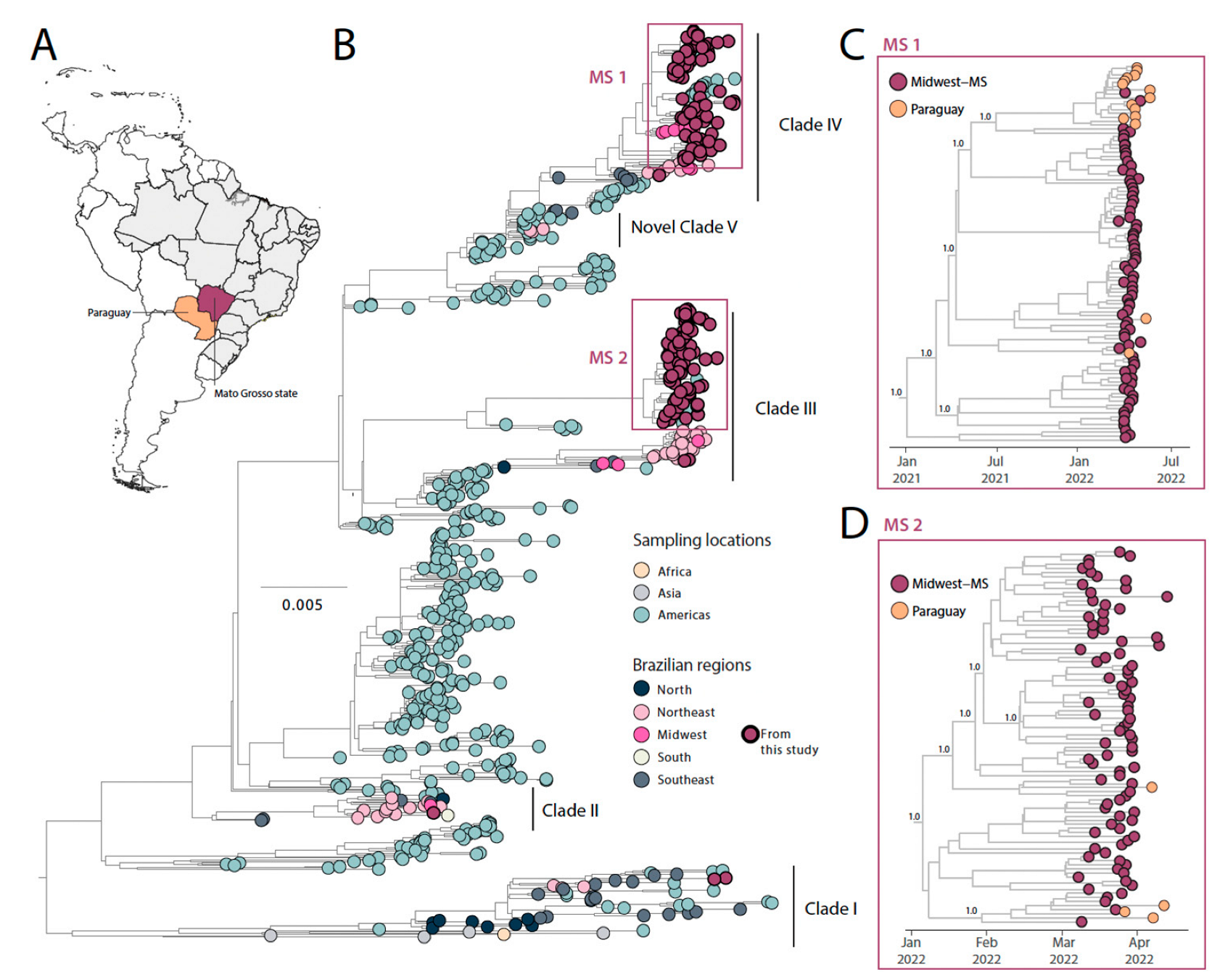
3.1. DENV1 Phylodynamics in the State of Mato Grosso do Sul, Midwest Brazil
3.2. DENV2 Phylodynamics in the State of Mato Grosso do Sul, Midwest Brazil
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Adelino, T.É.R.; Giovanetti, M.; Fonseca, V.; Xavier, J.; de Abreu, Á.S.; do Nascimento, V.A.; Demarchi, L.H.F.; Oliveira, M.A.A.; da Silva, V.L.; e Silva de Mello, A.L.; et al. Field and classroom initiatives for portable sequence-based monitoring of dengue virus in Brazil. Nat Commu. 2021, 12, 2296. [Google Scholar] [CrossRef] [PubMed]
- Brito, A.F.; Machado, L.C.; Oidtman, R.J.; Siconelli, M.J.L.; Tran, Q.M.; Fauver, J.R.; de Oliveira Carvalho, R.D.; Dezordi, F.Z.; Pereira, M.R.; de Castro-Jorge, L.A.; et al. Lying in wait: The resurgence of dengue virus after the Zika epidemic in Brazil. Nat. Commun. 2021, 12, 2616. [Google Scholar] [CrossRef] [PubMed]
- Brazil, Ministério da Saúde. Health Surveillance Secretariat and Department of Communicable Disease Surveillance; 2016. Dengue: Diagnosis and Clinical Management: Adult and Child. Available online: https://bvsms.saude.gov.br/bvs/publicacoes/dengue_diagnosticomanejoclinicoadulto.pdf (accessed on 7 April 2023).
- Brasil, C.A.; Cavalcante Araújo, E.D.; Andrade, H.J.; de Araújo, L.K.L. Classical dengue: Comparative analysis of the epidemiological profile of morbidity and mortality in Bahia and Brazil. Braz. J. Dev. 2023, 9, 2460–2472. [Google Scholar] [CrossRef]
- Vishwakarma, K.; Goyal, M. Assessment of clinical profile of children with dengue fever. J. Adv. Med. Dent. Sci. Res. 2021, 9, 32–35. [Google Scholar] [CrossRef]
- Pinheiro, M.J.S.; Dos Santos, J.S.G.; Dantas, L.A. Use of paracetamol in the treatment of dengue and liver damage: Review. Braz. J. Sci. 2023, 2, 32–40. [Google Scholar] [CrossRef]
- Hanan, F.; Ahmad, J.; Hadi, S.; Ali, I.; Fahim, M.; Qayum, A. Analysis of dengue virus genotypes and further investigations for mixed infections by RT-PCR in patients with classical dengue in Pakistan. Int. J. Pathol. 2022, 20, 72–78. [Google Scholar]
- Tang, K.F.; Ooi, E.E. Dengue diagnosis: An update. Expert Rev. Anti Infect. Ther. 2012, 10, 895–907. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Dengue and Severe Dengue. Available online: http://www.who.int/mediacentre/factsheets/fs117/en/ (accessed on 7 April 2023).
- World Health Organization. Geographic Expansion of Dengue and Chikungunya Cases beyond Historical Transmission Areas in the Region of the Americas. Available online: https://www.who.int/emergencies/disease-outbreak-news/item/2023-DON448 (accessed on 20 July 2023).
- Giovanetti, M.; Faria, N.R.; Lourenço, J.; de Jesus, J.G.; Xavier, J.; Claro, I.M.; Kraemer, M.U.G.; Fonseca, V.; Dellicour, S.; Thezé, J.; et al. Genomic and Epidemiological Surveillance of Zika Virus in the Amazon Region. Cell Rep. 2020, 30, 2275–2283. [Google Scholar] [CrossRef] [PubMed]
- Giovanetti, M.; Cantarelli, V.V.; Tosta, S.; Nardy, V.B.; Reboredo de Oliveira da Silva, L.; Gómez, M.K.A.; Lima, J.G.; Ribeiro, A.A.; Guimarães, N.R.; Watanabe, L.T.; et al. Genomic epidemiology of the SARS-CoV-2 epidemic in Brazil. Nat. Microbiol. 2022, 7, 1490–1500. [Google Scholar] [CrossRef] [PubMed]
- Filho, L.C.F.; Ribeiro, R.B.; Coelho, M.S.; Cardoso, J.F.; Lemos, P.S.; dos Reis, A.S.; Favacho, J.F.R.; More, N.R.F.S.; Nunes, M.R.T. Dengue virus serotype 4 genome sequencing in a bat (Platyrrhinus helleri) brain sample from the Brazilian Amazon. Infect. Genet. Evol. 2023, 109, 105407. [Google Scholar] [CrossRef] [PubMed]
- Brazil. Ministério da Saúde. Secretary of Health Surveillance. General Coordination of Surveillance of Arboviruses of the Department of Immunization and Communicable Diseases. Epidemiological Bulletin: Monitoring of Cases of Arboviruses until Epidemiological Week 50 of 2022. 2022; 53(47). Available online: https://www.gov.br/saude/pt-br/centrais-de-conteudo/publicacoes/boletins/epidemiologicos/edicoes/2022/boletim-epidemiologico-vol-53-no47/view (accessed on 7 April 2023).
- Vazquez, C.; Alcantara, L.C.J.; Fonseca, V.; Lima, M.; Xavier, J.; Adelino, T.; Fritsch, H.; Castro, E.; de Oliveira, C.; Schuab, G.; et al. Retrospective Spatio-Temporal Dynamics of Dengue Virus 1, 2 and 4 in Paraguay. Vírus 2023, 15, 1275. [Google Scholar] [CrossRef] [PubMed]
- Vilsker, M.; Moosa, Y.; Nooij, S.; Fonseca, V.; Ghysens, Y.; Dumon, K.; Pauwels, R.; Alcantara, L.C.; Eynden, E.V.; Vandamme, A.-M.; et al. Genome Detective: An automated system for virus identification from high- throughput sequencing data. Bioinformatics 2019, 35, 871–873. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of 28 recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Lam, T.T.; Carvalho, L.M.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [PubMed]
- Suchard, M.A.; Lemey, P.; Baele, G.; Aires, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [PubMed]
- Baele, G.; Li, W.L.S.; Drummond, A.J.; Suchard, M.A.; Lemey, P. Accurate model selection of relaxed molecular clocks in Bayesian phylogenetics. Mol. Biol. Evol. 2012, 30, 239–243. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castilho de Arruda, L.D.; Giovanetti, M.; Fonseca, V.; Zardin, M.C.S.U.; Lichs, G.G.d.C.; Asato, S.; Esposito, A.O.P.; Tokeshi Müller, M.; Xavier, J.; Fritsch, H.; et al. Dengue Fever Surveillance in Mato Grosso do Sul: Insights from Genomic Analysis and Implications for Public Health Strategies. Viruses 2023, 15, 1790. https://doi.org/10.3390/v15091790
Castilho de Arruda LD, Giovanetti M, Fonseca V, Zardin MCSU, Lichs GGdC, Asato S, Esposito AOP, Tokeshi Müller M, Xavier J, Fritsch H, et al. Dengue Fever Surveillance in Mato Grosso do Sul: Insights from Genomic Analysis and Implications for Public Health Strategies. Viruses. 2023; 15(9):1790. https://doi.org/10.3390/v15091790
Chicago/Turabian StyleCastilho de Arruda, Larissa Domingues, Marta Giovanetti, Vagner Fonseca, Marina Castilhos Souza Umaki Zardin, Gislene Garcia de Castro Lichs, Silvia Asato, Ana Olivia Pascoto Esposito, Miriam Tokeshi Müller, Joilson Xavier, Hegger Fritsch, and et al. 2023. "Dengue Fever Surveillance in Mato Grosso do Sul: Insights from Genomic Analysis and Implications for Public Health Strategies" Viruses 15, no. 9: 1790. https://doi.org/10.3390/v15091790
APA StyleCastilho de Arruda, L. D., Giovanetti, M., Fonseca, V., Zardin, M. C. S. U., Lichs, G. G. d. C., Asato, S., Esposito, A. O. P., Tokeshi Müller, M., Xavier, J., Fritsch, H., Lima, M., de Oliveira, C., Santos, E. V., Maziero, L. d. M. A., Frias, D. F. R., Ahad das Neves, D., Ferreira da Silva, L., Rodrigues Barretos, E. C., Tsuha Oshiro, P. E., ... Cavalheiro Maymone Gonçalves, C. (2023). Dengue Fever Surveillance in Mato Grosso do Sul: Insights from Genomic Analysis and Implications for Public Health Strategies. Viruses, 15(9), 1790. https://doi.org/10.3390/v15091790