
The Effect of Global Spread, Epidemiology, and Control Strategies on the Evolution of the GI-19 Lineage of Infectious Bronchitis Virus
 , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Dataset
2.2. Population Dynamics and Evolution Reconstruction
2.3. Selective Pressure Analyses
2.4. Homology Modeling
3. Results
3.1. Dataset
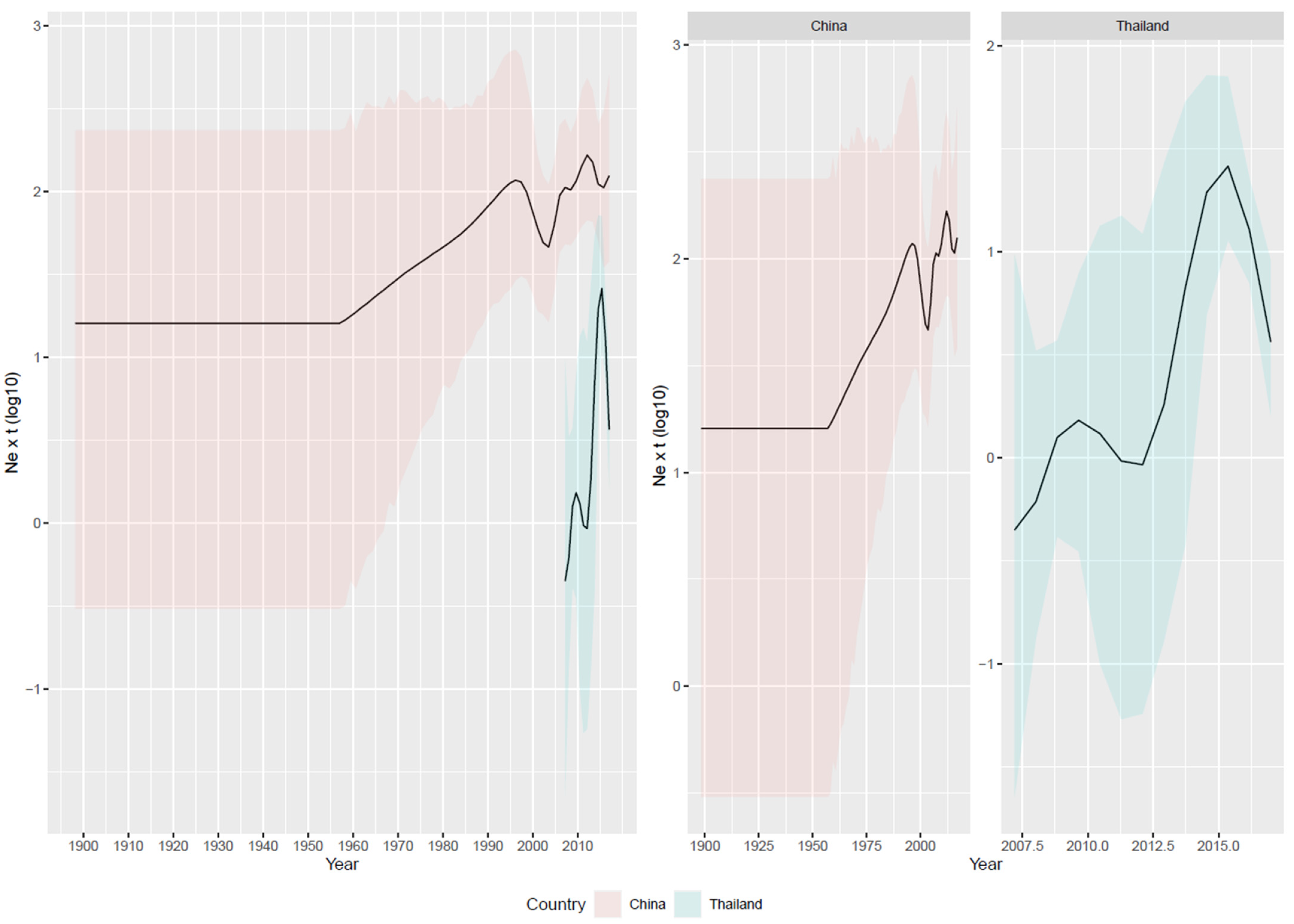
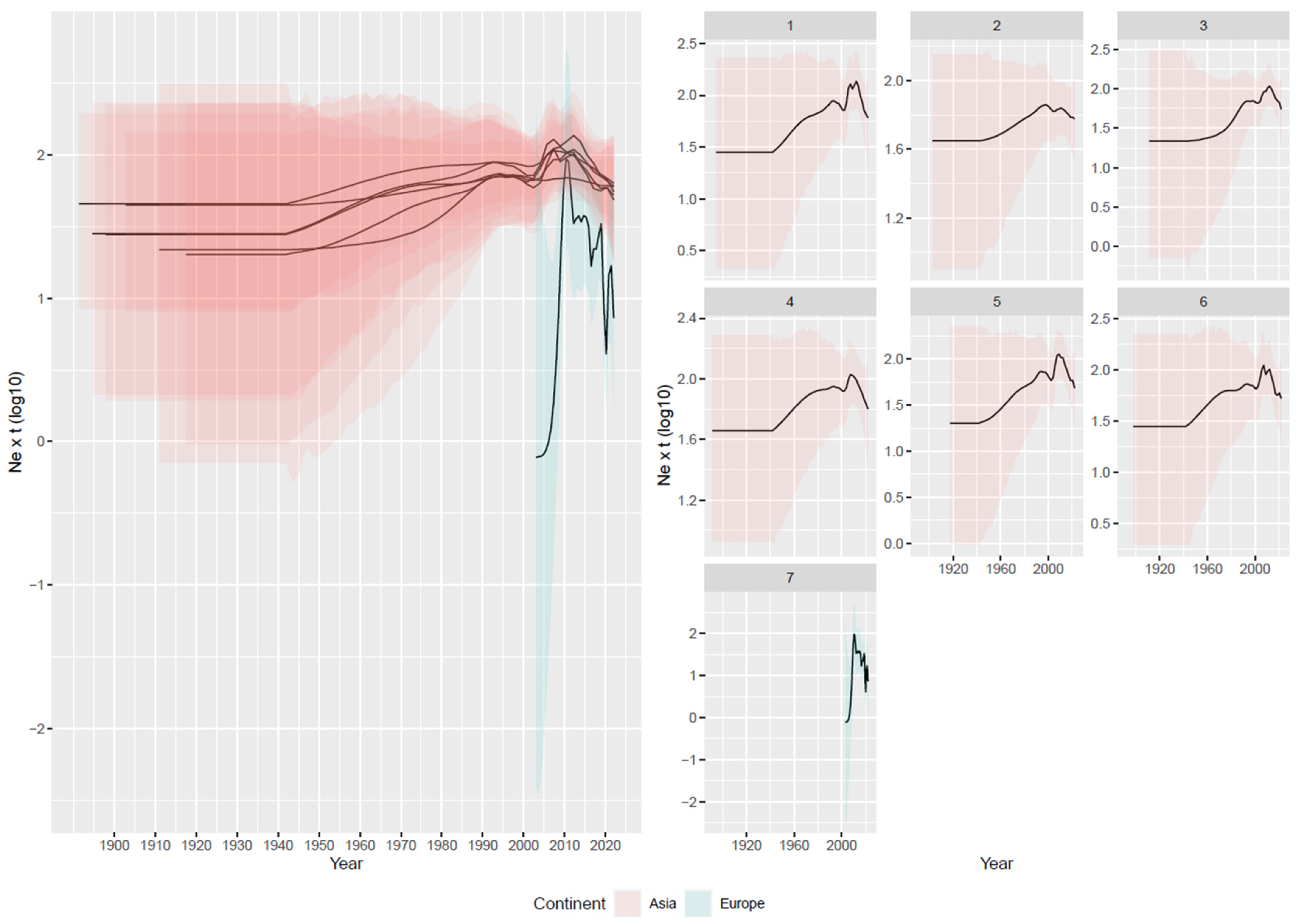
3.2. Reconstruction of Viral Population Dynamics and Evolution
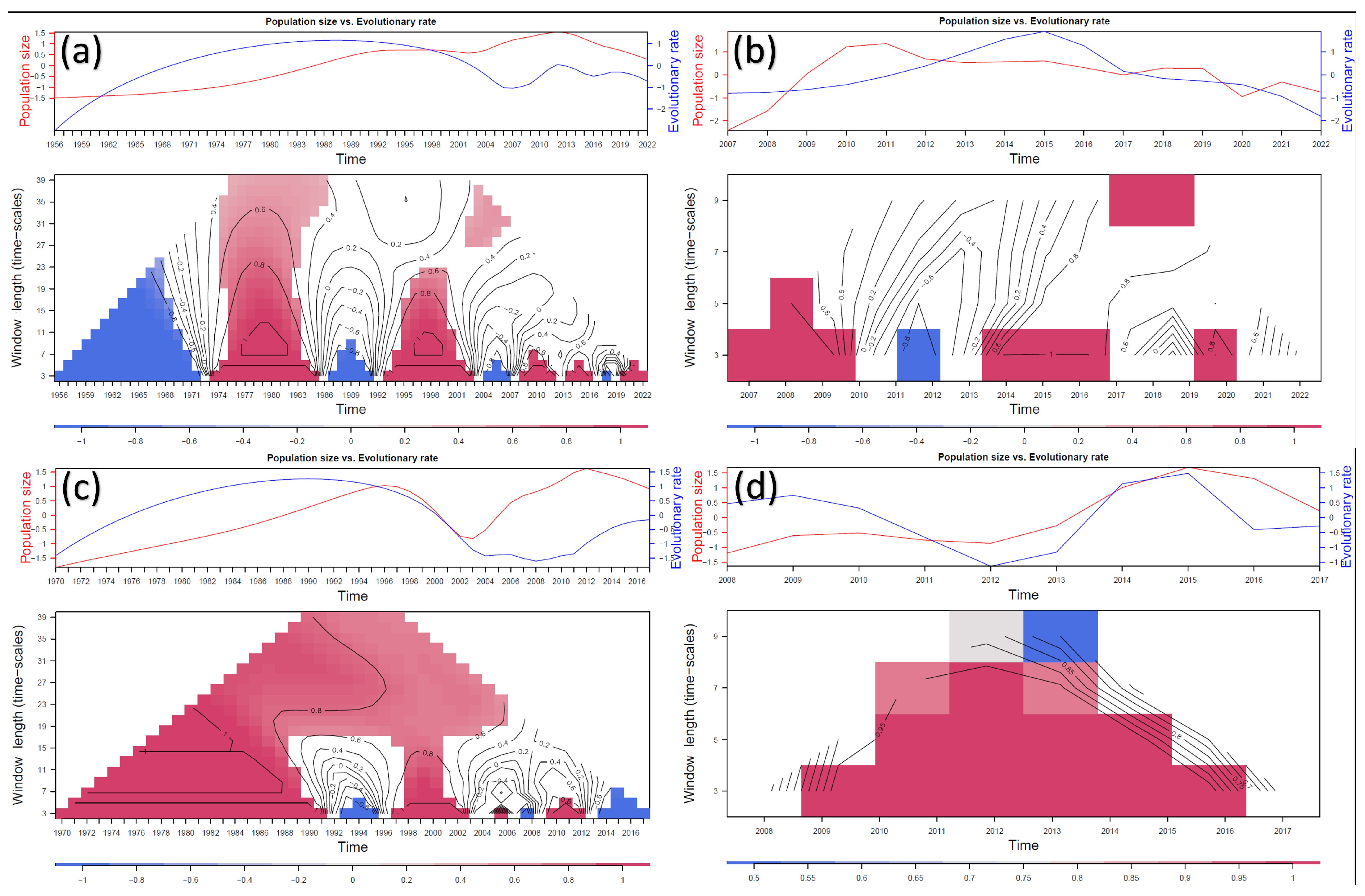
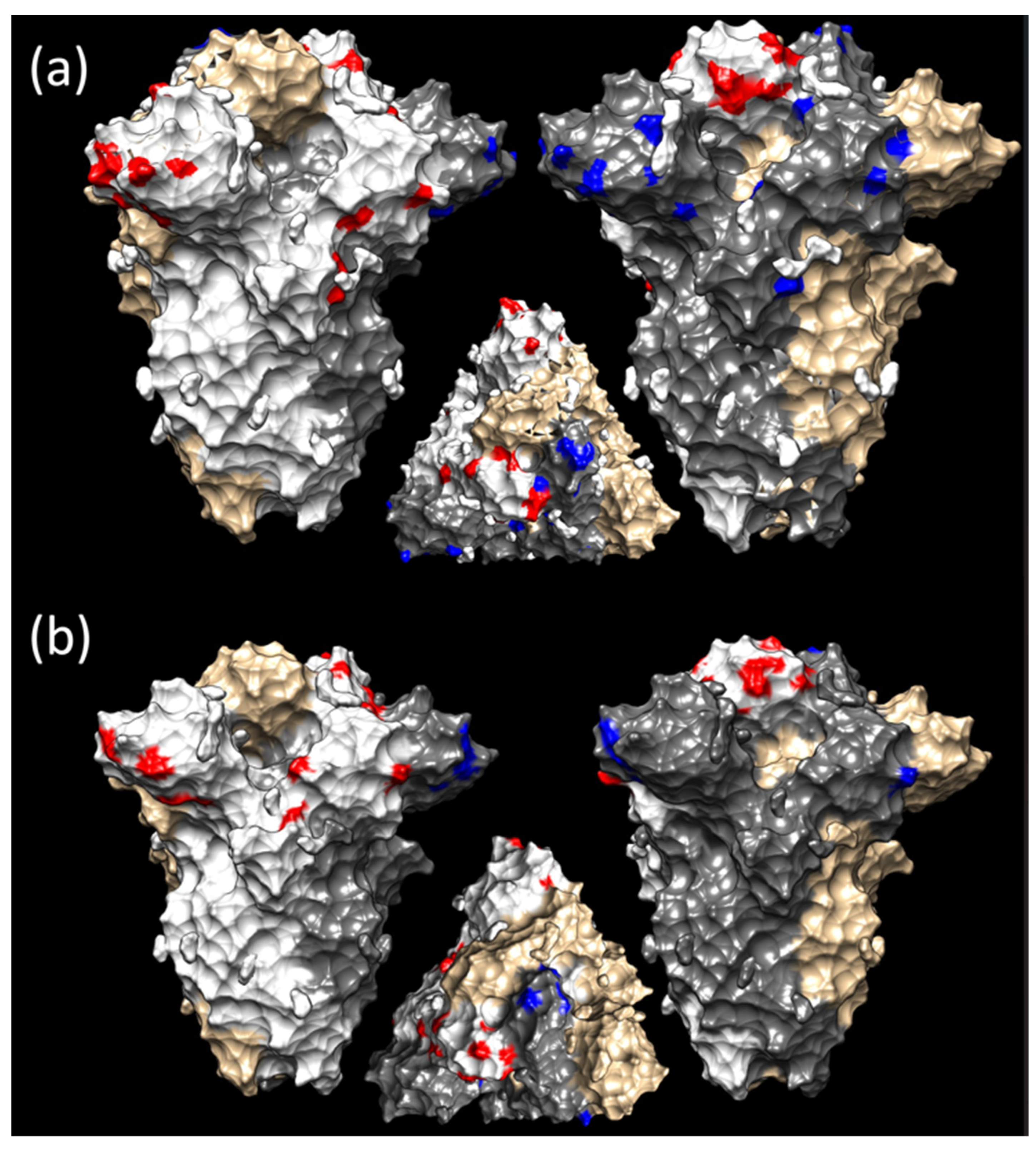
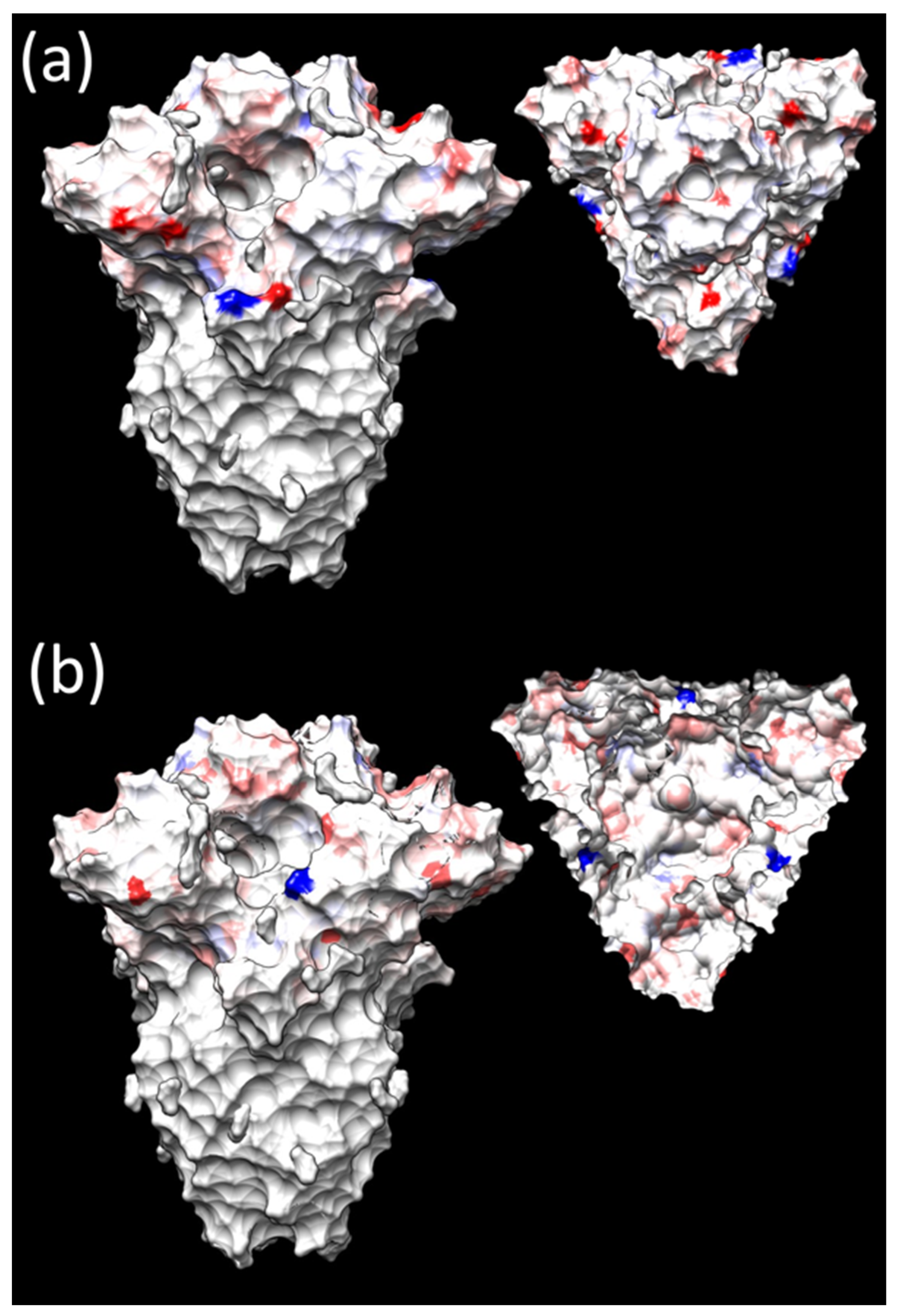
3.3. Selective Pressures Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Duffy, S. Why Are RNA Virus Mutation Rates so Damn High? PLoS Biol. 2018, 16, e3000003. [Google Scholar] [CrossRef] [PubMed]
- Simon-Loriere, E.; Holmes, E.C. Why Do RNA Viruses Recombine? Nat. Rev. Microbiol. 2011, 9, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Geoghegan, J.L.; Senior, A.M.; Holmes, E.C. Pathogen Population Bottlenecks and Adaptive Landscapes: Overcoming the Barriers to Disease Emergence. Proc. R. Soc. B Biol. Sci. 2016, 283, 20160727. [Google Scholar] [CrossRef]
- Watabe, T.; Kishino, H. Structural Considerations in the Fitness Landscape of a Virus. Mol. Biol. Evol. 2010, 27, 1782–1791. [Google Scholar] [CrossRef] [PubMed]
- Legnardi, M.; Tucciarone, C.M.; Franzo, G.; Cecchinato, M. Infectious Bronchitis Virus Evolution, Diagnosis and Control. Vet. Sci. 2020, 7, 79. [Google Scholar] [CrossRef] [PubMed]
- Jackwood, M.W.; Hall, D.; Handel, A. Molecular Evolution and Emergence of Avian Gammacoronaviruses. Infect. Genet. Evol. 2012, 12, 1305–1311. [Google Scholar] [CrossRef]
- Shang, J.; Zheng, Y.; Yang, Y.; Liu, C.; Geng, Q.; Luo, C. Cryo-EM Structure of Infectious Bronchitis Coronavirus Spike Protein Reveals Structural and Functional Evolution of Coronavirus Spike Proteins. PLoS Pathog. 2018, 14, e1007009. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Liao, Y.; Fan, J.; Zhang, Y.; Mao, X.; Sun, Y.; Song, C.; Qiu, X.; Meng, C.; Ding, C. Prediction and Identification of Novel IBV S1 Protein Derived CTL Epitopes in Chicken. Vaccine 2016, 34, 380–386. [Google Scholar] [CrossRef]
- Valastro, V.; Holmes, E.C.; Britton, P.; Fusaro, A.; Jackwood, M.W.; Cattoli, G.; Monne, I. S1 Gene-Based Phylogeny of Infectious Bronchitis Virus: An Attempt to Harmonize Virus Classification. Infect. Genet. Evol. 2016, 39, 349–364. [Google Scholar] [CrossRef]
- Wickramasinghe, I.N.A.; van Beurden, S.J.; Weerts, E.A.W.S.; Verheije, M.H. The Avian Coronavirus Spike Protein. Virus Res. 2014, 194, 37–48. [Google Scholar] [CrossRef]
- Hewson, K.A.; Noormohammadi, A.H.; Devlin, J.M.; Browning, G.F.; Schultz, B.K.; Ignjatovic, J. Evaluation of a Novel Strain of Infectious Bronchitis Virus Emerged as a Result of Spike Gene Recombination between Two Highly Diverged Parent Strains. Avian Pathol. 2014, 43, 249–257. [Google Scholar] [CrossRef]
- Thor, S.W.; Hilt, D.A.; Kissinger, J.C.; Paterson, A.H.; Jackwood, M.W. Recombination in Avian Gamma-Coronavirus Infectious Bronchitis Virus. Viruses 2011, 3, 1777–1799. [Google Scholar] [CrossRef]
- Franzo, G.; Massi, P.; Tucciarone, C.M.; Barbieri, I.; Tosi, G.; Fiorentini, L.; Ciccozzi, M.; Lavazza, A.; Cecchinato, M.; Moreno, A. Think Globally, Act Locally: Phylodynamic Reconstruction of Infectious Bronchitis Virus (IBV) QX Genotype (GI-19 Lineage) Reveals Different Population Dynamics and Spreading Patterns When Evaluated on Different Epidemiological Scales. PLoS ONE 2017, 12, e0184401. [Google Scholar] [CrossRef] [PubMed]
- Franzo, G.; Tucciarone, C.M.; Blanco, A.; Nofrarías, M.; Biarnés, M.; Cortey, M.; Majó, N.; Catelli, E.; Cecchinato, M. Effect of Different Vaccination Strategies on IBV QX Population Dynamics and Clinical Outbreaks. Vaccine 2016, 34, 5670–5676. [Google Scholar] [CrossRef]
- Legnardi, M.; Franzo, G.; Koutoulis, K.C.; Wiśniewski, M.; Catelli, E.; Tucciarone, C.M.; Cecchinato, M. Vaccine or Field Strains: The Jigsaw Pattern of Infectious Bronchitis Virus Molecular Epidemiology in Poland. Poult. Sci. 2019, 98, 6388–6392. [Google Scholar] [CrossRef] [PubMed]
- Franzo, G.; Legnardi, M.; Tucciarone, C.M.; Drigo, M.; Martini, M.; Cecchinato, M. Evolution of Infectious Bronchitis Virus in the Field after Homologous Vaccination Introduction. Vet. Res. 2019, 50, 92. [Google Scholar] [CrossRef] [PubMed]
- Abascal, F.; Zardoya, R.; Telford, M.J. TranslatorX: Multiple Alignment of Nucleotide Sequences Guided by Amino Acid Translations. Nucleic Acids Res. 2010, 38, W7–W13. [Google Scholar] [CrossRef] [PubMed]
- Standley, K. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability (Outlines Version 7). Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Kosakovsky Pond, S.L.; Posada, D.; Gravenor, M.B.; Woelk, C.H.; Frost, S.D.W. GARD: A Genetic Algorithm for Recombination Detection. Bioinformatics 2006, 22, 3096–3098. [Google Scholar] [CrossRef]
- Weaver, S.; Shank, S.D.; Spielman, S.J.; Li, M.; Muse, S.V.; Kosakovsky Pond, S.L. Datamonkey 2.0: A Modern Web Application for Characterizing Selective and Other Evolutionary Processes. Mol. Biol. Evol. 2018, 35, 773–777. [Google Scholar] [CrossRef] [PubMed]
- Hill, V.; Baele, G. Bayesian Estimation of Past Population Dynamics in BEAST 1.10 Using the Skygrid Coalescent Model. Mol. Biol. Evol. 2019, 36, 2620–2628. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. JModelTest 2: More Models, New Heuristics and Parallel Computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Baele, G.; Lemey, P.; Bedford, T.; Rambaut, A.; Suchard, M.A.; Alekseyenko, A.V. Improving the Accuracy of Demographic and Molecular Clock Model Comparison While Accommodating Phylogenetic Uncertainty. Mol. Biol. Evol. 2012, 29, 2157–2167. [Google Scholar] [CrossRef]
- Hall, M.D.; Woolhouse, M.E.J.; Rambaut, A. The Effects of Sampling Strategy on the Quality of Reconstruction of Viral Population Dynamics Using Bayesian Skyline Family Coalescent Methods: A Simulation Study. Virus Evol. 2016, 2, vew003. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Ginestet, C. Ggplot2: Elegant Graphics for Data Analysis. J. R. Stat. Soc. Ser. A Stat. Soc. 2011, 174, 245–246. [Google Scholar] [CrossRef]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.Y. Ggtree: An R Package for Visualization and Annotation of Phylogenetic Trees with Their Covariates and Other Associated Data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Polanco-martínez, J.M.; López-martínez, J.L. NonParRolCor: An R Package for Estimating Rolling Correlation for Two Regular Time Series. SoftwareX 2023, 22, 101353. [Google Scholar] [CrossRef]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Kosakovsky Pond, S.L.; Scheffler, K. FUBAR: A Fast, Unconstrained Bayesian AppRoximation for Inferring Selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Kosakovsky Pond, S.L. Detecting Individual Sites Subject to Episodic Diversifying Selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef] [PubMed]
- Kosakovsky Pond, S.L.; Wisotsky, S.R.; Escalante, A.; Magalis, B.R.; Weaver, S. Contrast-FEL—A Test for Differences in Selective Pressures at Individual Sites among Clades and Sets of Branches. Mol. Biol. Evol. 2021, 38, 1184–1198. [Google Scholar] [CrossRef] [PubMed]
- Kosakovsky Pond, S.L.; Frost, S.D.W.; Muse, S.V. HyPhy: Hypothesis Testing Using Phylogenies. Bioinformatics 2005, 21, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology Modelling of Protein Structures and Complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Cook, J.K.A.; Jackwood, M.; Jones, R.C. The Long View: 40 Years of Infectious Bronchitis Research. Avian Pathol. 2012, 41, 239–250. [Google Scholar] [CrossRef]
- Jackwood, M.W. Review of Infectious Bronchitis Virus around the World. Avian Dis. 2012, 56, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Houta, M.H.; Hassan, K.E.; Legnardi, M.; Tucciarone, C.M.; Abdel-Moneim, A.S.; Cecchinato, M.; El-Sawah, A.A.; Ali, A.; Franzo, G. Phylodynamic and Recombination Analyses of Avian Infectious Bronchitis Gi-23 Reveal a Widespread Recombinant Cluster and New among-Countries Linkages. Animals 2021, 11, 3182. [Google Scholar] [CrossRef]
- Franzo, G.; Cecchinato, M.; Tosi, G.; Fiorentini, L.; Faccin, F.; Tucciarone, C.M.; Trogu, T.; Barbieri, I.; Massi, P.; Moreno, A. GI-16 Lineage (624/I or Q1), There and Back Again: The History of One of the Major Threats for Poultry Farming of Our Era. PLoS ONE 2018, 13, e0203513. [Google Scholar] [CrossRef]
- Chen, H. H5N1 Avian Influenza in China. Sci. China C Life Sci. 2009, 52, 419–427. [Google Scholar] [CrossRef]
- Sarkar, M.; Bandyopadhyay, M. Avian Influenza “Bird Flu” Factsheet. Bull RGKMC 2003, 8, 16–17. [Google Scholar] [CrossRef]
- Awad, F.; Hutton, S.; Forrester, A.; Baylis, M.; Ganapathy, K. Heterologous Live Infectious Bronchitis Virus Vaccination in Day-Old Commercial Broiler Chicks: Clinical Signs, Ciliary Health, Immune Responses and Protection against Variant Infectious Bronchitis Viruses. Avian Pathol. 2016, 45, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Kutle, L.; Ljuma Skupnjak, L.; Vrdoljak, A.; Jankovic, D.; Boelm, G.J.; Kelemen, F.; Zorman Rojs, O.; Millecam, J. Efficacy of Infectious Bronchitis GI-13 (793B) Vaccine Candidate Tested According to the Current European Union Requirements and for Cross-Protection against Heterologous QX-Like Challenge. Viral Immunol. 2020, 33, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Terregino, C.; Toffan, A.; Serena Beato, M.; De Nardi, R.; Vascellari, M.; Meini, A.; Ortali, G.; Mancin, M.; Capua, I. Pathogenicity of a QX Strain of Infectious Bronchitis Virus in Specific Pathogen Free and Commercial Broiler Chickens, and Evaluation of Protection Induced by a Vaccination Programme Based on the Ma5 and 4/91 Serotypes. Avian Pathol. 2008, 37, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Munyahongse, S.; Pohuang, T.; Nonthabenjawan, N.; Sasipreeyajan, J.; Thontiravong, A. Genetic Characterization of Infectious Bronchitis Viruses in Thailand, 2014–2016: Identification of a Novel Recombinant Variant. Poult. Sci. 2020, 99, 1888–1895. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.M.; Jackwood, M.W.; Hilt, D.A. Identification of Amino Acids Involved in a Serotype and Neutralization Specific Epitope within the S1 Subunit of Avian Infectious Bronchitis Virus. Arch. Virol. 1997, 142, 2249–2256. [Google Scholar] [CrossRef] [PubMed]
- Kant, A.; Koch, G.; Van Roozelaar, D.J.; Kusters, J.G.; Poelwijk, F.A.J.; Van der Zeijst, B.A.M. Location of Antigenic Sites Defined by Neutralizing Monoclonal Antibodies on the S1 Avian Infectious Bronchitis Virus Glycopolypeptide. J. Gen. Virol. 1992, 73 Pt 3, 591–596. [Google Scholar] [CrossRef]
- Cavanagh, D.; Davis, P.J.; Mockett, A.P.A. Amino Acids within Hypervariable Region 1 of Avian Coronavirus IBV (Massachusetts Serotype) Spike Glycoprotein Are Associated with Neutralization Epitopes. Virus Res. 1988, 11, 141–150. [Google Scholar] [CrossRef]
- Niesters, H.G.M.; Lenstra, J.A.; Spaan, W.J.M.; Zijderveld, A.J.; Bleumink-Pluym, N.M.C.; Hong, F.; van Scharrenburg, G.J.M.; Horzinek, M.C.; van der Zeijst, B.A.M. The Peplomer Protein Sequence of the M41 Strain of Coronavirus IBV and Its Comparison with Beaudette Strains. Virus Res. 1986, 5, 253–263. [Google Scholar] [CrossRef]
- Franzo, G.; Tucciarone, C.M.; Cecchinato, M.; Drigo, M. Porcine Circovirus Type 2 (PCV2) Evolution before and after the Vaccination Introduction: A Large Scale Epidemiological Study. Sci. Rep. 2016, 6, 39458. [Google Scholar] [CrossRef]
- Read, A.F.; Mackinnon, M.J. Pathogen Evolution in a Vaccinated World. Evol. Health Dis. 2010, 2, 139–152. [Google Scholar] [CrossRef]
- Weidt, G.; Deppert, W.; Hlen, O.U.; Heukeshoven, J.; Lehmann-Grube, F. Emergence of Virus Escape Mutants after Immunization with Epitope Vaccine. J. Virol. 1995, 69, 7147–7151. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franzo, G.; Faustini, G.; Tucciarone, C.M.; Poletto, F.; Tonellato, F.; Cecchinato, M.; Legnardi, M. The Effect of Global Spread, Epidemiology, and Control Strategies on the Evolution of the GI-19 Lineage of Infectious Bronchitis Virus. Viruses 2024, 16, 481. https://doi.org/10.3390/v16030481
Franzo G, Faustini G, Tucciarone CM, Poletto F, Tonellato F, Cecchinato M, Legnardi M. The Effect of Global Spread, Epidemiology, and Control Strategies on the Evolution of the GI-19 Lineage of Infectious Bronchitis Virus. Viruses. 2024; 16(3):481. https://doi.org/10.3390/v16030481
Chicago/Turabian StyleFranzo, Giovanni, Giulia Faustini, Claudia Maria Tucciarone, Francesca Poletto, Francesca Tonellato, Mattia Cecchinato, and Matteo Legnardi. 2024. "The Effect of Global Spread, Epidemiology, and Control Strategies on the Evolution of the GI-19 Lineage of Infectious Bronchitis Virus" Viruses 16, no. 3: 481. https://doi.org/10.3390/v16030481
APA StyleFranzo, G., Faustini, G., Tucciarone, C. M., Poletto, F., Tonellato, F., Cecchinato, M., & Legnardi, M. (2024). The Effect of Global Spread, Epidemiology, and Control Strategies on the Evolution of the GI-19 Lineage of Infectious Bronchitis Virus. Viruses, 16(3), 481. https://doi.org/10.3390/v16030481