
Comparative Analysis of Viromes Identified in Multiple Macrofungi


Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection, DNA and RNA Extraction
2.2. High-Throughput Sequencing, Analysis and RT-PCR Confirmation
2.3. Phylogenetic Analysis
2.4. Viral Diversity
3. Results
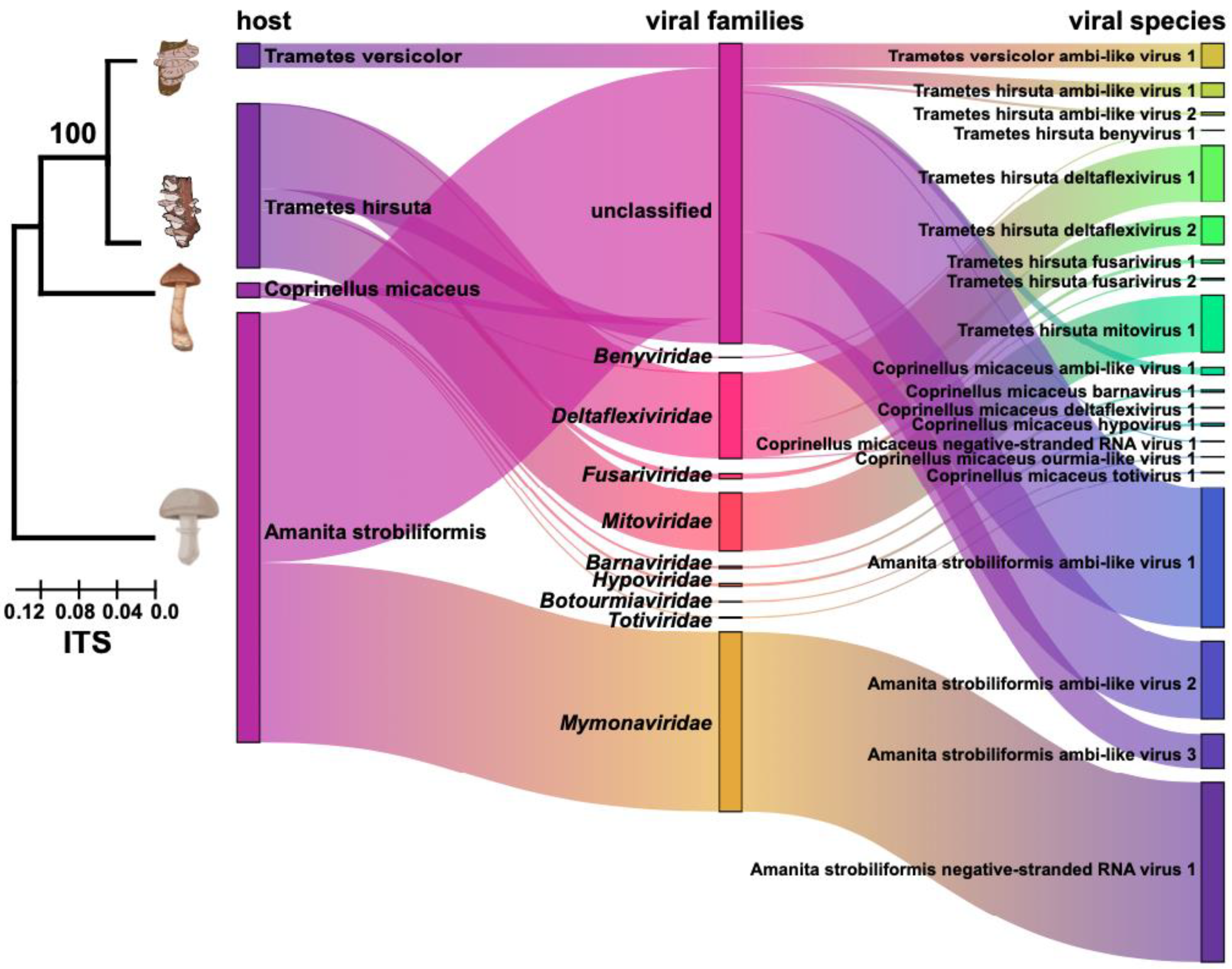
3.1. Metatranscriptomic Identification of Mycoviruses in Four Species of Macrofungi
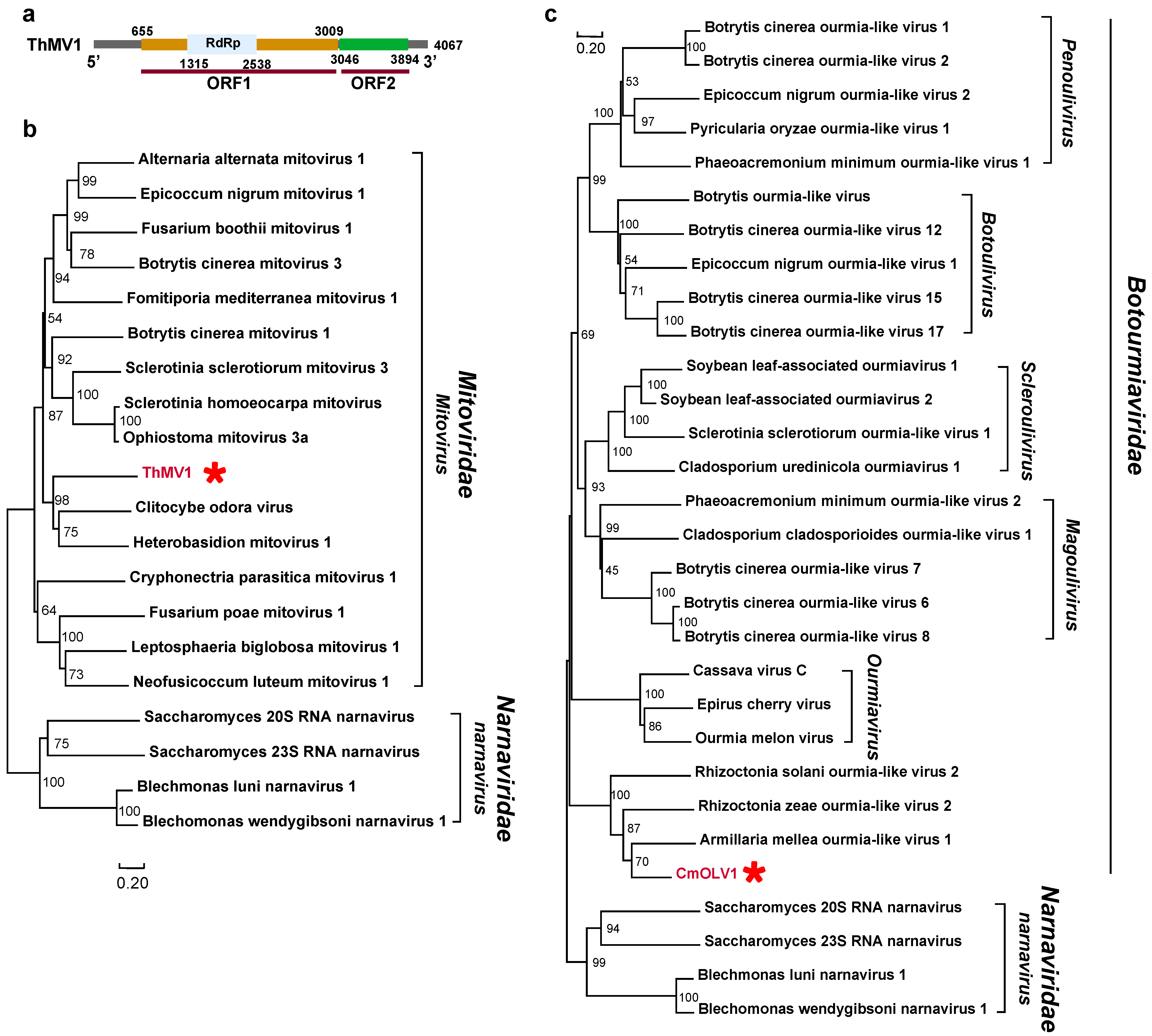
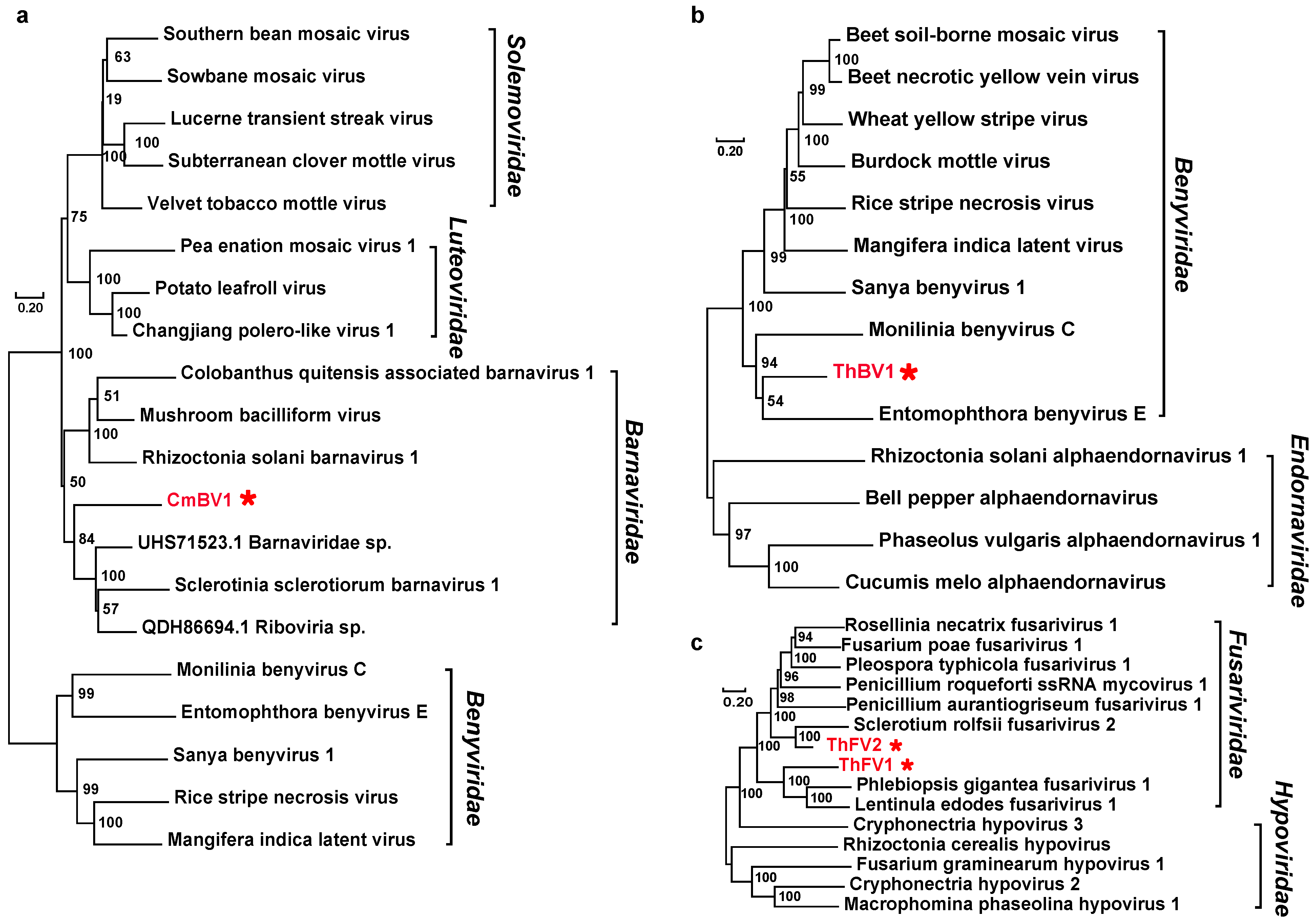
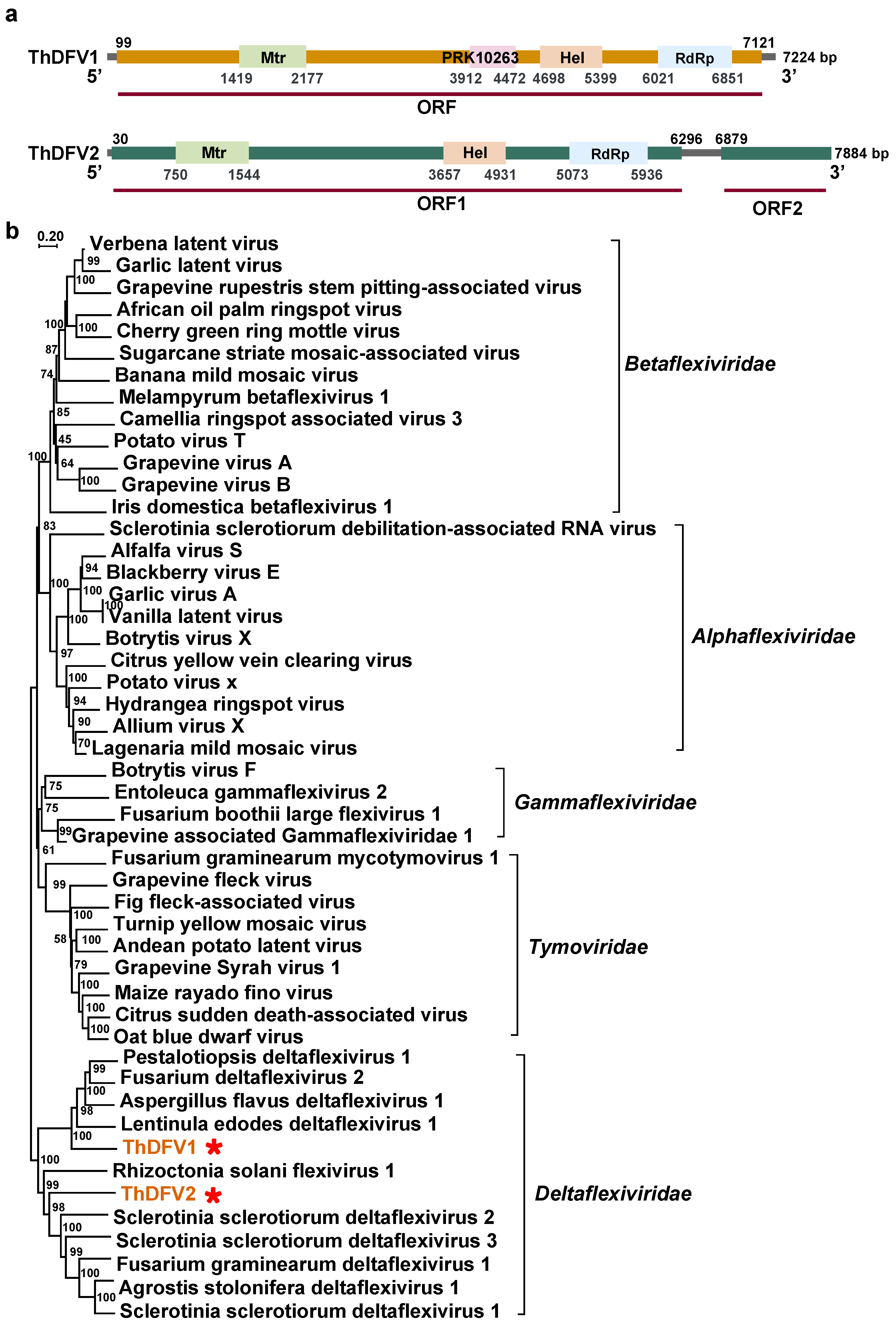
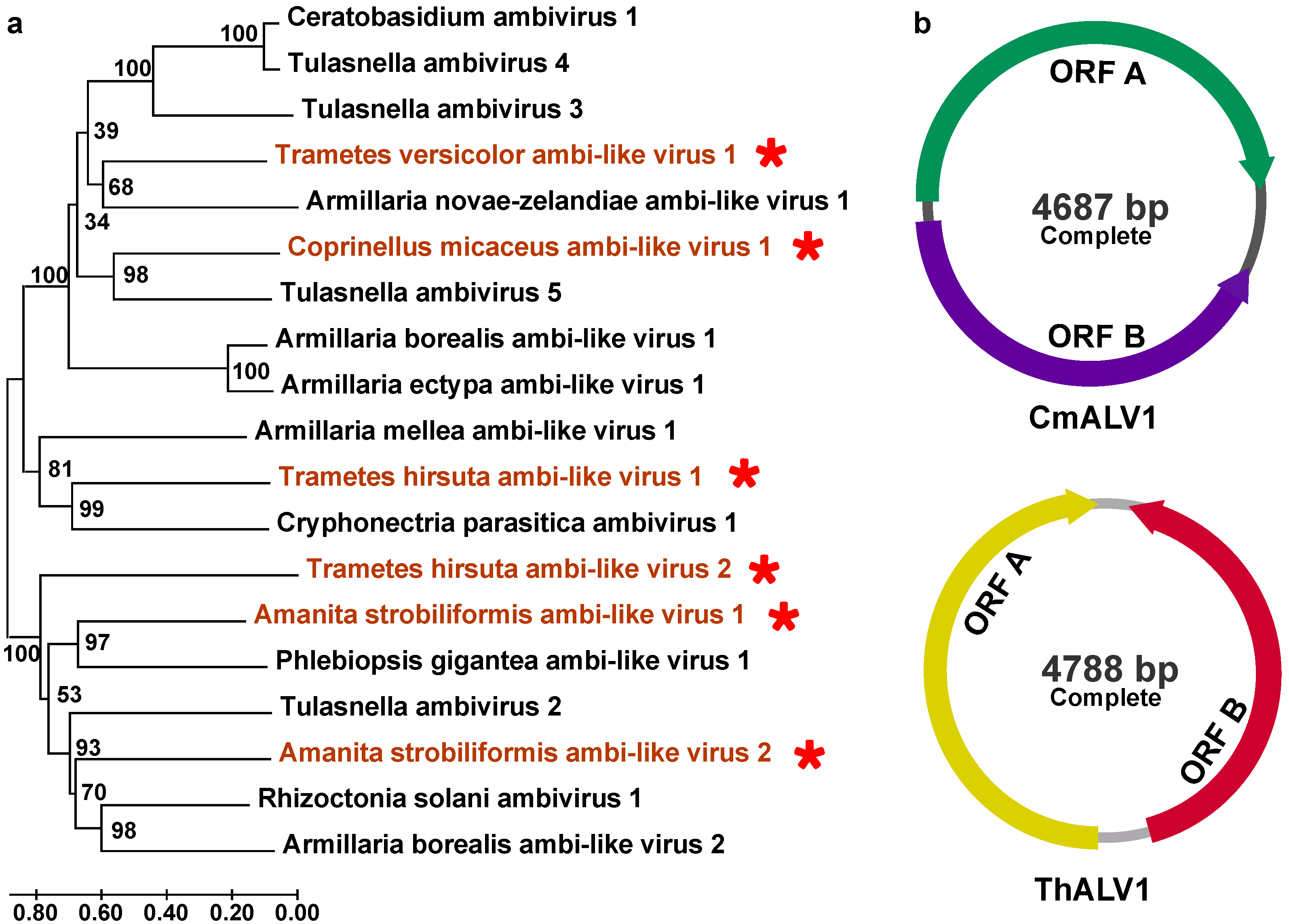
3.2. Positive, Single-Stranded RNA Mycoviruses
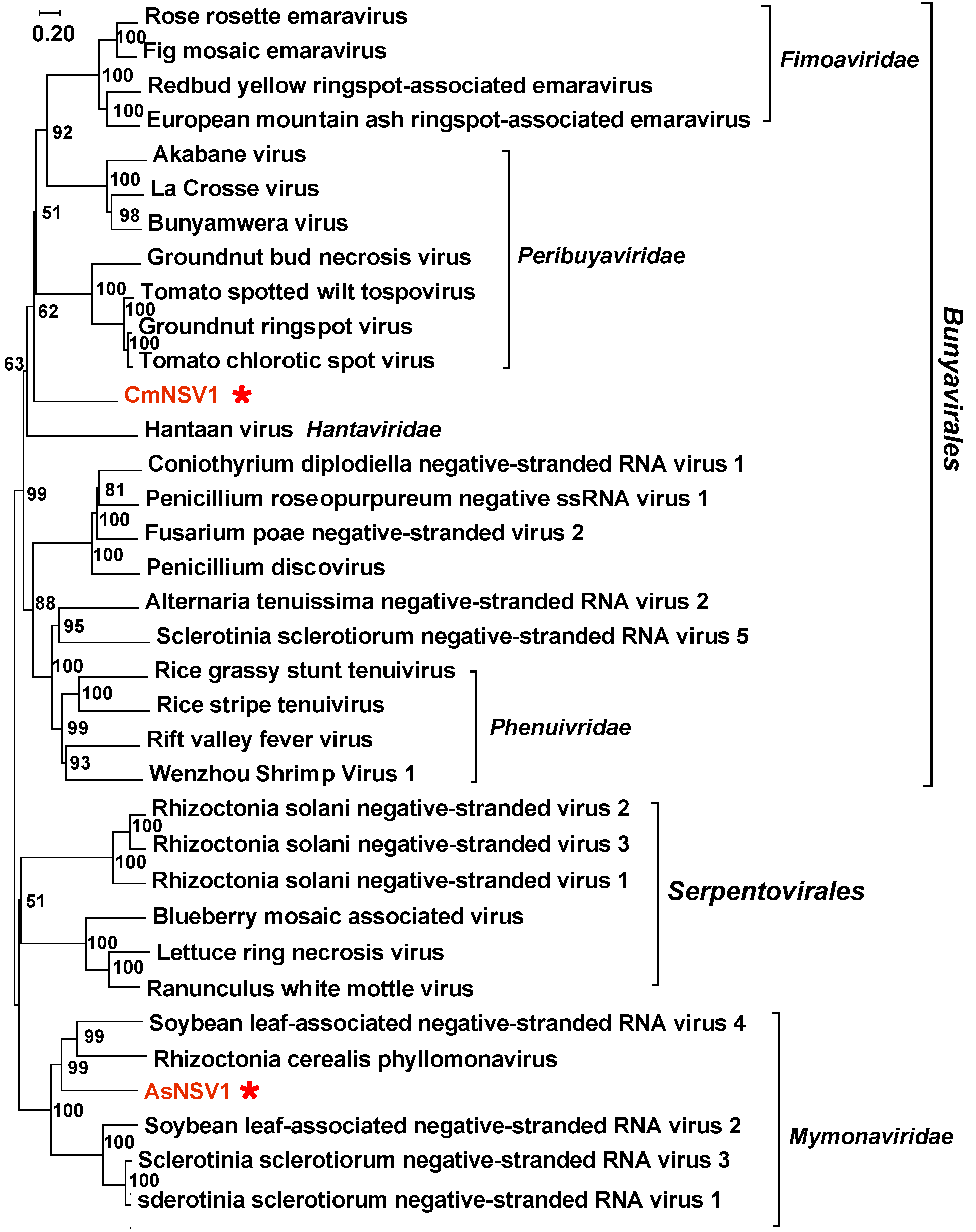
3.3. Negative Single-Stranded RNA Mycoviruses
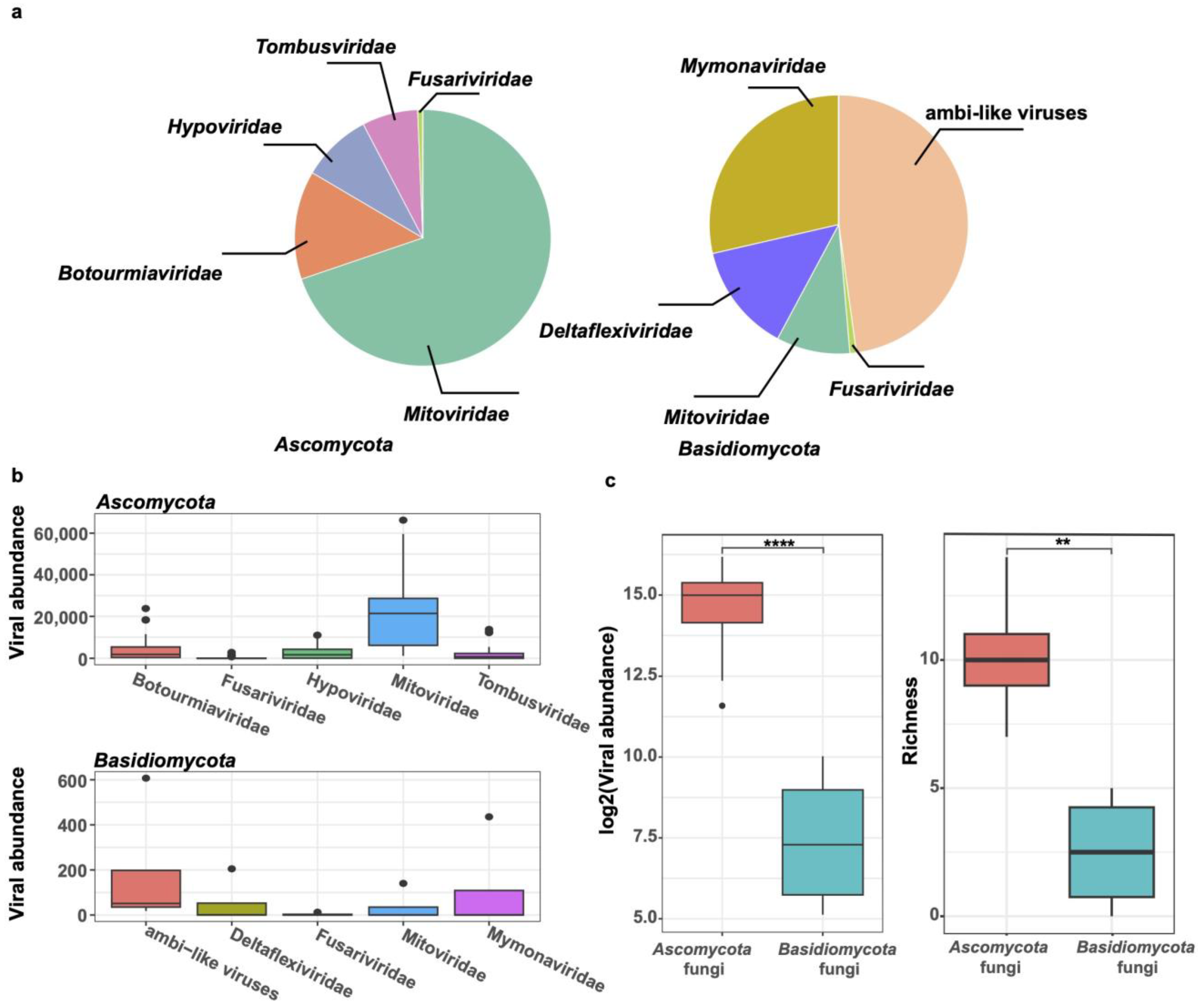
3.4. Comparison of Viromes from the Phyla Ascomycota and Basidiomycota
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tang, X.; Mi, F.; Zhang, Y.; He, X.; Cao, Y.; Wang, P.; Liu, C.; Yang, D.; Dong, J.; Zhang, K.; et al. Diversity, population genetics, and evolution of macrofungi associated with animals. Mycology 2015, 6, 94–109. [Google Scholar] [CrossRef][Green Version]
- Mueller, G.M.; Schmit, J.P.; Leacock, P.R.; Buyck, B.; Cifuentes, J.; Desjardin, D.E.; Halling, R.E.; Hjortstam, K.; Iturriaga, T.; Larsson, K.H. Global diversity and distribution of macrofungi. Biodivers. Conserv. 2007, 16, 37–48. [Google Scholar] [CrossRef]
- Chang, S.; Buswell, J. Medicinal Mushrooms: Past, Present and Future. Adv. Biochem. Eng. Biotechnol. 2023, 184, 1–27. [Google Scholar] [PubMed]
- Xiao, H.; Zhong, J.J. Production of Useful Terpenoids by Higher-Fungus Cell Factory and Synthetic Biology Approaches. Trends Biotechnol. 2016, 34, 242–255. [Google Scholar] [CrossRef] [PubMed]
- Nikšić, M.; Podgornik, B.B.; Berovic, M. Farming of Medicinal Mushrooms. Adv. Biochem. Eng. Biotechnol. 2023, 184, 29–76. [Google Scholar]
- Wu, F.; Zhou, L.W.; Yang, Z.L.; Zhu, L.B.; Li, T.; Tai, H.D.; Yu, C. Resource diversity of Chinese macrofungi: Edible, medicinal and poisonous species. Fungal Divers. 2019, 98, 1–76. [Google Scholar]
- Ghabrial, S.A.; Castón, J.R.; Jiang, D.; Nibert, M.L.; Suzuki, N. 50-plus years of fungal viruses. Virology 2015, 356, 479–480. [Google Scholar] [CrossRef]
- Hillman, B.I.; Annisa, A.; Suzuki, N. Viruses of Plant-Interacting Fungi. Adv. Virus Res. 2018, 100, 99–116. [Google Scholar] [PubMed]
- Hollings, M. Viruses associated with a die-back disease of cultivated mushroom. Nature 1962, 196, 962–965. [Google Scholar] [CrossRef]
- Yu, X.; Li, B.; Fu, Y.; Jiang, D.; Ghabrial, S.A.; Li, G.; Peng, Y.; Xie, J.; Cheng, J.; Huang, J.; et al. A geminivirus-related DNA mycovirus that confers hypovirulence to a plant pathogenic fungus. Proc. Natl. Acad. Sci. USA 2010, 107, 8387–8392. [Google Scholar] [CrossRef]
- Li, P.; Wang, S.; Zhang, L.; Qiu, D.; Zhou, X.; Guo, L. A tripartite ssDNA mycovirus from a plant pathogenic fungus is infectious as cloned DNA and purified virions. Sci. Adv. 2020, 6, eaay9634. [Google Scholar] [CrossRef] [PubMed]
- Hao, F.M.; Wu, M.D.; Li, G.Q. Characterization of a novel genomovirus in the phytopathogenic fungus Botrytis cinerea. Virology 2021, 553, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Padilla, A.; Rodríguez-Romero, J.; Gómez-Cid, I.; Pacifico, D.; Ayllón, M.A. Novel mycoviruses discovered in the mycovirome of a necrotrophic fungus. mBio 2021, 12, e03705-20. [Google Scholar] [CrossRef]
- Kondo, H.; Botella, L.; Suzuki, N. Mycovirus diversity and evolution revealed/inferred from recent studies. Annu. Rev. Phytopathol. 2022, 60, 307–336. [Google Scholar] [CrossRef]
- Ayllón, M.A.; Vainio, E.J. Mycoviruses as a part of the global virome: Diversity, evolutionary links and lifestyle. Adv. Virus Res. 2023, 115, 1–86. [Google Scholar]
- Rumbou, A.; Vainio, E.J.; Büttner, C. Towards the Forest Virome: High-Throughput Sequencing Drastically Expands Our Understanding on Virosphere in Temperate Forest Ecosystems. Microorganisms 2021, 9, 1730. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Fu, Y.; Jiang, D.; Mu, F.; Cheng, J.; Lin, Y.; Li, B.; Marzano, S.L.; Xie, J. Interannual dynamics, diversity and evolution of the virome in Sclerotinia sclerotiorum from a single crop field. Virus Evol. 2021, 7, veab032. [Google Scholar] [CrossRef] [PubMed]
- Forgia, M.; Navarro, B.; Daghino, S.; Cervera, A.; Gisel, A.; Perotto, S.; Aghayeva, D.N.; Akinyuwa, M.F.; Gobbi, E.; Zheludev, I.N.; et al. Hybrids of RNA viruses and viroid-like elements replicate in fungi. Nat. Commun. 2023, 14, 2591. [Google Scholar] [CrossRef]
- Nuss, D.L. Hypovirulence: Mycoviruses at the fungal–plant interface. Nat. Rev. Microbiol. 2005, 3, 632–642. [Google Scholar] [CrossRef]
- Vainio, E.J.; Rumbou, A.; Diez, J.J.; Büttner, C. Forest Tree Virome as a Source of Tree Diseases and Biological Control Agents. Curr. For. Rep. 2024. [Google Scholar] [CrossRef]
- Nuss, D.L. Biological control of chestnut blight: An example of virus-mediated attenuation of fungal pathogenesis. Microbiol. Rev. 1992, 56, 561–576. [Google Scholar] [CrossRef]
- Anagnostakis, S.L. Biological control of chestnut blight. Science 1982, 215, 466–471. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Li, B.; Fu, Y.; Xie, J.; Cheng, J.; Ghabrial, S.A.; Li, G.; Yi, X.; Jiang, D. Extracellular transmission of a DNA mycovirus and its use as a natural fungicide. Proc. Natl. Acad. Sci. USA 2013, 110, 1452–1457. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Suzuki, N. Continued mycovirus discovery expanding our understanding of virus lifestyles, symptom expression, and host defense. Curr. Opin. Microbiol. 2023, 75, 102337. [Google Scholar] [CrossRef] [PubMed]
- Mowna Sundari, T.; Alwin Prem Anand, A.; Jenifer, P.; Shenbagarathai, R. Bioprospection of Basidiomycetes and molecular phylogenetic analysis using internal transcribed spacer (ITS) and 5.8S rRNA gene sequence. Sci. Rep. 2018, 8, 10720. [Google Scholar] [CrossRef] [PubMed]
- Mu, F.; Li, B.; Cheng, S.; Jia, J.; Jiang, D.; Fu, Y.; Cheng, J.; Lin, Y.; Chen, T.; Xie, J. Nine viruses from eight lineages exhibiting new evolutionary modes that co-infect a hypovirulent phytopathogenic fungus. PLoS Pathog. 2021, 17, e1009823. [Google Scholar] [CrossRef] [PubMed]
- Möller, E.M.; Bahnweg, G.; Sandermann, H.; Geiger, H.H. A simple and efficient protocol for isolation of high molecular weight DNA from filamentous fungi, fruit bodies, and infected plant tissues. Nucleic Acids Res. 1992, 20, 6115–6116. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Zhou, K.; Wu, M.; Zhang, J.; Yang, L.; Chen, W.; Li, G. Viral cross-class transmission results in disease of a phytopathogenic fungus. ISME J. 2022, 16, 2763–2774. [Google Scholar] [CrossRef] [PubMed]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. meta-SPAdes: A new versatile metagenomic assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Sun, A.; Zhao, L.; Sun, Y.; Chen, Y.; Li, C.; Dong, W.; Yang, G. Horizontal and Vertical Transmission of a Mycovirus Closely Related to the Partitivirus RhsV717 That Confers Hypovirulence in Rhizoctonia solani. Viruses 2023, 15, 2088. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Wille, M.; Harvey, E.; Shi, M.; Gonzalez-Acuña, D.; Holmes, E.C.; Hurt, A.C. Sustained RNA virome diversity in Antarctic penguins and their ticks. ISME J. 2020, 14, 1768–1782. [Google Scholar] [CrossRef] [PubMed]
- Wille, M.; Shi, M.; Klaassen, M.; Hurt, A.C.; Holmes, E.C. Virome heterogeneity and connectivity in waterfowl and shorebird communities. ISME J. 2019, 13, 2603–2616. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Guillaume, B.F.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; O’Hara, R.B.; Solymos, P.; Stevens, M.H.H.; Szoecs, E.; et al. Vegan: Community Ecology Package. R Package 2022, Version 2.6-2. Available online: https://CRAN.R-project.org/package=vegan (accessed on 13 December 2023).
- O’Brien, H.E.; Parrent, J.L.; Jackson, J.A.; Moncalvo, J.M.; Vilgalys, R. Fungal community analysis by large-scale sequencing of environmental samples. Appl. Environ. Microbiol. 2005, 71, 5544–5550. [Google Scholar] [CrossRef] [PubMed]
- Sahin, E.; Akata, I. Viruses infecting macrofungi. Virus Dis. 2018, 29, 1–18. [Google Scholar] [CrossRef]
- Vainio, E.J. Mitoviruses in the conifer root rot pathogens Heterobasidion annosum and H. parviporum. Virus Res. 2019, 271, 197681. [Google Scholar] [CrossRef]
- Ohkit, S.; Lee, Y.; Nguyen, Q.; Ikeda, K.; Suzukib, N.; Nakayashiki, H. Three ourmia-like viruses and their associated RNAs in Pyricularia oryzae. Virology 2019, 534, 25–35. [Google Scholar] [CrossRef]
- Xu, Z.Y.; Wu, S.S.; Liu, L.J.; Cheng, J.S.; Fu, Y.P.; Jiang, D.H.; Xie, J.T. A mitovirus related to plant mitochondrial gene confers hypovirulence on the phytopathogenic fungus Sclerotinia sclerotiorum. Virus Res. 2015, 197, 127–136. [Google Scholar] [CrossRef]
- Hong, Y.; Cole, T.E.; Brasier, C.M.; Buck, K.W. Evolutionary relationships among putative RNA-dependent RNA polymerases encoded by a mitochondrial virus-like RNA in the Dutch elm disease fungus, Ophiostoma novoulmi, by other viruses and virus-like RNAs and by the Arabidopsis mitochondrial genome. Virology 1998, 246, 158–169. [Google Scholar] [CrossRef]
- Polashock, J.J.; Hillman, B.I. A small mitochondrial double-stranded (ds) RNA element associated with a hypovirulent strain of the chestnut blight fungus and ancestrally related to yeast cytoplasmic T and W dsRNAs. Proc. Natl. Acad. Sci. USA 1994, 91, 8680–8684. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Dover, S.L.; Cole, T.E.; Brasier, C.M.; Buck, K.W. Multiple mitochondrial viruses in an isolate of the Dutch elm disease fungus Ophiostoma novoulmi. Virology 1999, 258, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Martelli, G.P.; Adams, M.J.; Kreuze, J.F.; Dolja, V.V. Family Flexiviridae: A case study in virion and genome plasticity. Annu. Rev. Phytopathol. 2007, 45, 73–100. [Google Scholar] [CrossRef] [PubMed]
- Hamid, M.R.; Xie, J.; Wu, S.; Maria, S.K.; Zheng, D.; Assane Hamidou, A.; Wang, Q.; Cheng, J.; Fu, Y.; Jiang, D. A Novel deltaflexivirus that infects the plant fungal pathogen, Sclerotinia sclerotiorum, can be transmitted among host vegetative incompatible strains. Viruses 2018, 10, 295. [Google Scholar] [CrossRef] [PubMed]
- Li, K.F.; Zheng, D.; Cheng, J.S.; Chen, T.; Fu, Y.P.; Jiang, D.H.; Xie, J.T. Characterization of a novel Sclerotinia sclerotiorum RNA virus as the prototype of a new proposed family within the order Tymovirales. Virus Res. 2016, 219, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Sutela, S.; Forgia, M.; Vainio, E.J.; Chiapello, M.; Daghino, S.; Vallino, M.; Martino, E.; Girlanda, M.; Perotto, S.; Turina, M. The virome from a collection of endomycorrhizal fungi reveals new viral taxa with unprecedented genome organization. Virus Evol. 2020, 6, veaa076. [Google Scholar] [CrossRef] [PubMed]
- Forgia, M.; Isgandarli, E.; Aghayeva, D.N.; Huseynova, I.; Turina, M. Virome characterization of Cryphonectria parasitica isolates from Azerbaijan unveiled a new mymonavirus and a putative new RNA virus unrelated to described viral sequences. Virology 2021, 553, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Linnakoski, R.; Sutela, S.; Coetzee, M.P.A.; Duong, T.A.; Pavlov, I.N.; Litovka, Y.A.; Hantula, J.; Wingfield, B.D.; Vainio, E.J. Armillaria root rot fungi host single-stranded RNA viruses. Sci. Rep. 2021, 11, 7336. [Google Scholar] [CrossRef]
- Sutela, S.; Piri, T.; Vainio, E.J. Discovery and community dynamics of novel ssRNA mycoviruses in the conifer pathogen Heterobasidion parviporum. Front. Microbiol. 2021, 12, 770787. [Google Scholar] [CrossRef]
- Tonka, T.; Walterová, L.; Čurn, V. Development of RT-PCR for rapid detection of ssRNA ambi-like mycovirus in a root rot fungus (Armillaria spp.). Acta Virol. 2022, 66, 287–289. [Google Scholar] [CrossRef] [PubMed]
- Melzer, M.S.; Ikeda, S.S.; Boland, G.J. Interspecific transmission of double-stranded RNA and hypovirulence from Sclerotinia sclerotiorum to S. minor. Phytopathology 2002, 92, 780–784. [Google Scholar] [CrossRef] [PubMed]
- Coenen, A.; Kevei, F.; Hoekstra, R.F. Factors affecting the spread of double-stranded RNA viruses in Aspergillus nidulans. Genet. Res. 1997, 69, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.C.; Linder-Basso, D.; Hillman, B.I.; Kaneko, S.; Milgroom, M.G. Evidence for inter-species transmission of viruses in natural populations of filamentous fungi in the genus Cryphonectria. Mol. Ecol. 2003, 12, 1619–1628. [Google Scholar] [CrossRef] [PubMed]
- Shahi, S.; Eusebio-Cope, A.; Kondo, H.; Hillman, B.I.; Suzuki, N. Investigation of host range of and host defense against a mitochondrially replicating mitovirus. J. Virol. 2019, 93, e01503-18. [Google Scholar] [CrossRef]
- Wu, T.; Mao, H.; Hai, D.; Cheng, J.; Fu, Y.; Lin, Y.; Jiang, D.; Xie, J. Molecular characterization of a novel fungal alphaflexivirus reveals potential inter-species horizontal gene transfer. Virus Res. 2023, 334, 199151. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Viral Name | Length (nt) | Best Match | Identity (aa) | Family | Genome | Accession Number |
|---|---|---|---|---|---|---|
| Amanita strobiliformis ambi-like virus 1 | 4503 | UJT31805.1 Phlebiopsis gigantea ambi-like virus 1 | 40% | unclassified | +ssRNA | PP105116 |
| Amanita strobiliformis ambi-like virus 2 | 3116 | QMP84024.1 Rhizoctonia solani ambivirus 1 | 35% | unclassified | +ssRNA | PP105117 |
| Amanita strobiliformis ambi-like virus 3 | 1258 | QPB44667.1 Tulasnella ambivirus 2 | 26% | unclassified | +ssRNA | PP105118 |
| Amanita strobiliformis negative-stranded RNA virus 1 | 2421 | YP_010784559.1 Soybean leaf-associated negative-stranded RNA virus 4 | 34% | Mymonaviridae | −ssRNA | PP105119 |
| Trametes versicolor ambi-like virus 1 | 3426 | WNH24528.1 Heterobasidion ambi-like virus 15 | 37% | unclassified | +ssRNA | PP105120 |
| Coprinellus micaceus ambi-like virus 1 | 4687 | QPB44674.1 Tulasnella ambivirus 5 | 40% | unclassified | +ssRNA | PP105121 |
| Coprinellus micaceus hypovirus 1 | 3416 | WMI40060.1 Rhizoctonia cerealis hypovirus | 40% | Hypoviridae | +ssRNA | PP105122 |
| Coprinellus micaceus barnavirus 1 | 2292 | UHS71731.1 Sclerotinia sclerotiorum barnavirus 1 | 36% | Barnaviridae | +ssRNA | PP105123 |
| Coprinellus micaceus totivirus 1 | 1544 | WPH57541.1 Rhizoctonia solani toti-like virus 1 | 48% | Totiviridae | dsRNA | PP105124 |
| Coprinellus micaceus deltaflexivirus 1 | 1273 | QTH80200.1 Pestalotiopsis deltaflexivirus 1 | 47% | Deltaflexiviridae | +ssRNA | PP105125 |
| Coprinellus micaceus negative stranded RNA virus 1 | 802 | QTT60994.1 Phytophthora condilina negative stranded RNA virus 2 | 40% | unclassified | −ssRNA | PP105126 |
| Coprinellus micaceus ourmia-like virus 1 | 784 | YP_010805005.1 Armillaria mellea ourmia-like virus 1 | 45% | Botourmiaviridae | +ssRNA | PP105127 |
| Trametes hirsuta deltaflexivirus 1 | 7224 | QOX06047.1 Lentinula edodes deltaflexivirus 1 | 39% | Deltaflexiviridae | +ssRNA | PP105128 |
| Trametes hirsuta deltaflexivirus 2 | 7884 | UUW06602.1 Cat Tien Macrotermes Deltaflexi-like virus | 32% | Deltaflexiviridae | +ssRNA | PP105129 |
| Trametes hirsuta ambi-like virus 1 | 4788 | WPS91567.1 Fusarium graminearum ambivirus 1 | 32% | unclassified | +ssRNA | PP105130 |
| Trametes hirsuta ambi-like virus 2 | 2980 | WNA22195.1 Downy mildew lesion associated ambivirus 2 | 31% | unclassified | +ssRNA | PP105132 |
| Trametes hirsuta mitovirus 1 | 4067 | QOX06058.1 Lentinula edodes mitovirus 1 | 43% | Mitoviridae | +ssRNA | PP105131 |
| Trametes hirsuta fusarivirus 1 | 4953 | UJT31800.1 Phlebiopsis gigantea fusarivirus 1 | 44% | Fusariviridae | +ssRNA | PP105133 |
| Trametes hirsuta fusarivirus 2 | 2275 | YP_010799383.1 Sclerotium rolfsii fusarivirus 2 | 57% | Fusariviridae | +ssRNA | PP105134 |
| Trametes hirsuta benyvirus 1 | 671 | QWC36503.1 Bemisia tabaci beny-like virus 3 | 42% | Benyviridae | +ssRNA | PP105135 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, K.; Zhang, F.; Deng, Y. Comparative Analysis of Viromes Identified in Multiple Macrofungi. Viruses 2024, 16, 597. https://doi.org/10.3390/v16040597
Zhou K, Zhang F, Deng Y. Comparative Analysis of Viromes Identified in Multiple Macrofungi. Viruses. 2024; 16(4):597. https://doi.org/10.3390/v16040597
Chicago/Turabian StyleZhou, Kang, Fan Zhang, and Yue Deng. 2024. "Comparative Analysis of Viromes Identified in Multiple Macrofungi" Viruses 16, no. 4: 597. https://doi.org/10.3390/v16040597
APA StyleZhou, K., Zhang, F., & Deng, Y. (2024). Comparative Analysis of Viromes Identified in Multiple Macrofungi. Viruses, 16(4), 597. https://doi.org/10.3390/v16040597