
Mechanisms of Hepatitis B Virus cccDNA and Minichromosome Formation and HBV Gene Transcription


{kind=link}
{kind=link}
Abstract
:1. Introduction
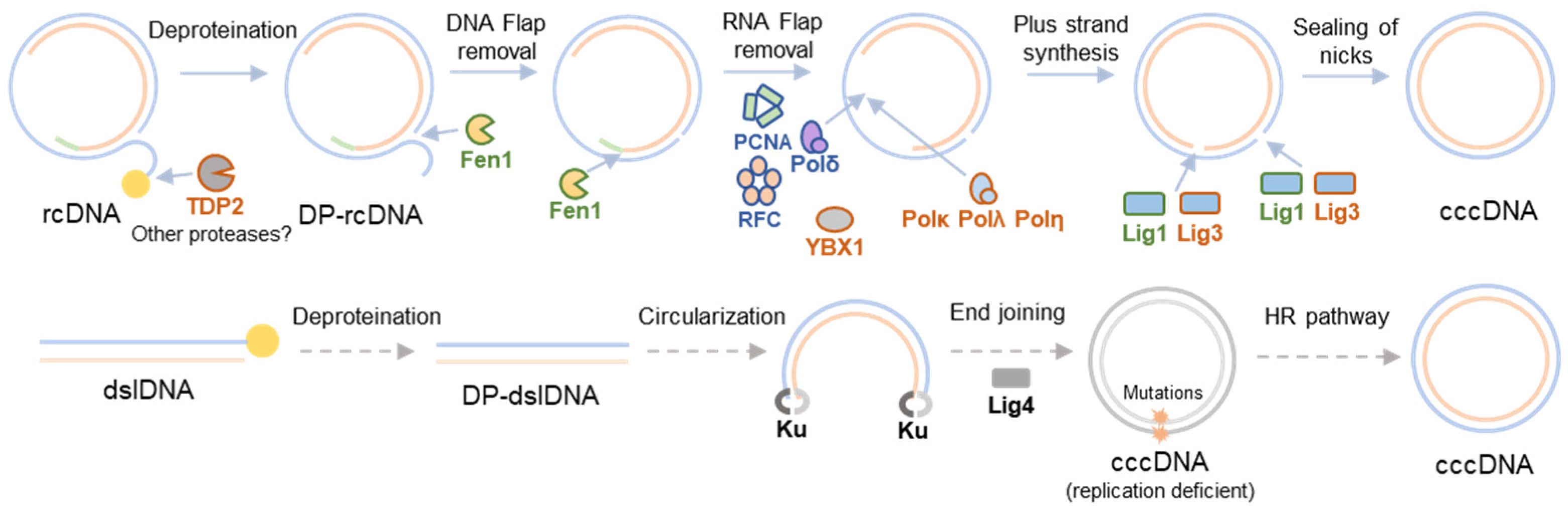
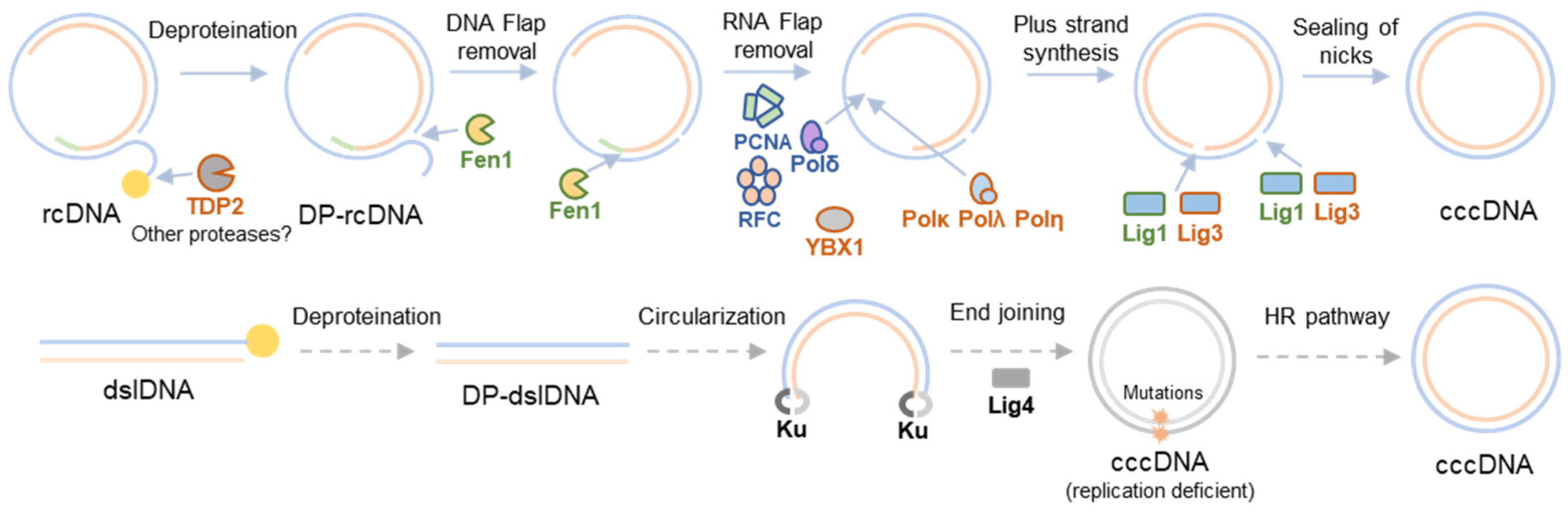
2. HBV cccDNA Formation
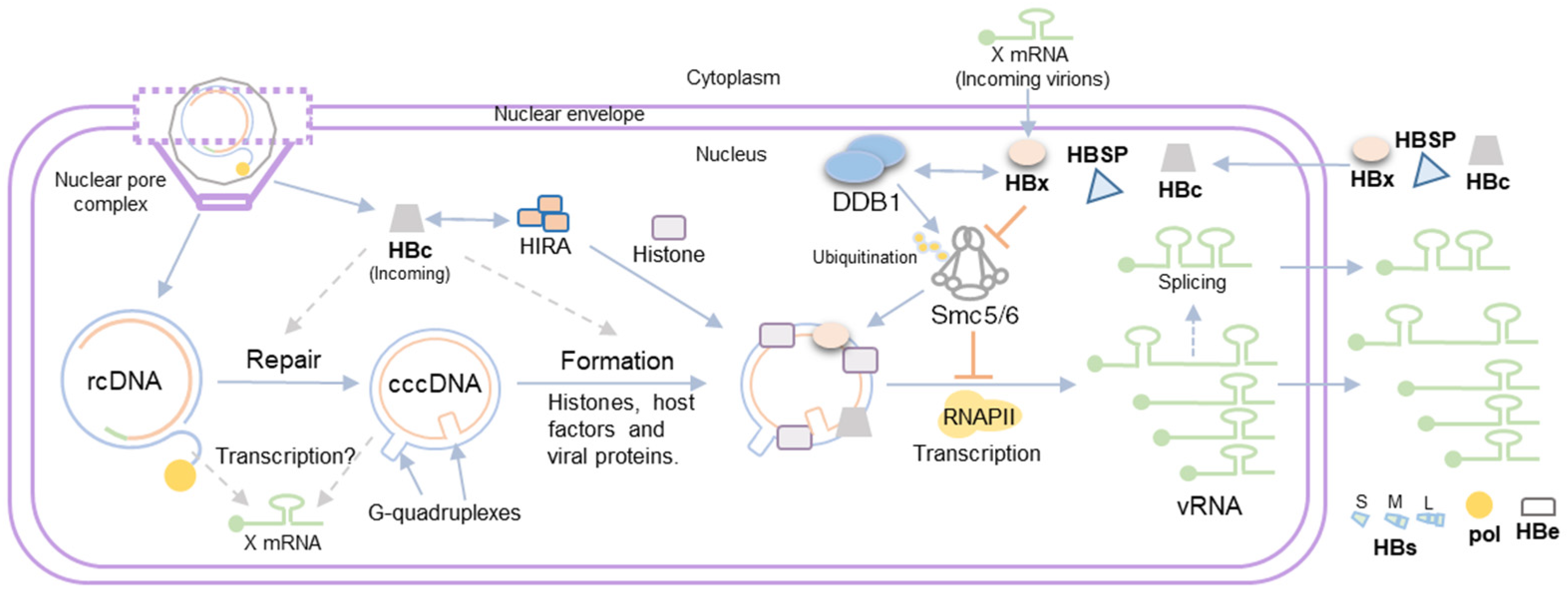
3. HBV Minichromosome Formation
4. Viral Gene Expression from the HBV Minichromosome
5. Role of HBV Proteins in cccDNA Transcription
6. HBV Post-Transcriptional Modifications
7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chisari, F.V.; Ferrari, C. Hepatitis B Virus Immunopathogenesis. Annu. Rev. Immunol. 1995, 13, 29–60. [Google Scholar] [CrossRef] [PubMed]
- Winer, B.Y.; Ploss, A. Determinants of Hepatitis B and Delta Virus Host Tropism. Curr. Opin. Virol. 2015, 13, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Ott, J.J.; Stevens, G.A.; Groeger, J.; Wiersma, S.T. Global Epidemiology of Hepatitis B Virus Infection: New Estimates of Age-Specific HBsAg Seroprevalence and Endemicity. Vaccine 2012, 30, 2212–2219. [Google Scholar] [CrossRef] [PubMed]
- Seeger, C.; Mason, W.S. Molecular Biology of Hepatitis B Virus Infection. Virology 2015, 479–480, 672–686. [Google Scholar]
- Ozer, A.; Khaoustov, V.I.; Mearns, M.; Lewis, D.E.; Genta, R.M.; Darlington, G.J.; Yoffe, B. Effect of Hepatocyte Proliferation and Cellular DNA Synthesis on Hepatitis B Virus Replication. Gastroenterology 1996, 110, 1519–1528. [Google Scholar] [CrossRef] [PubMed]
- Summers, J.; Mason, W.S. Replication of the Genome of a Hepatitis B-like Virus by Reverse Transcription of an RNA Intermediate. Cell 1982, 29, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Schulze, A.; Gripon, P.; Urban, S. Hepatitis B Virus Infection Initiates with a Large Surface Protein-Dependent Binding to Heparan Sulfate Proteoglycans. Hepatology 2007, 46, 1759–1768. [Google Scholar] [CrossRef]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium Taurocholate Cotransporting Polypeptide Is a Functional Receptor for Human Hepatitis B and D Virus. eLife 2012, 2012, e00049. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, M.; Saso, W.; Sugiyama, R.; Ishii, K.; Ohki, M.; Nagamori, S.; Suzuki, R.; Aizaki, H.; Ryo, A.; Yun, J.-H.; et al. Epidermal Growth Factor Receptor Is a Host-Entry Cofactor Triggering Hepatitis B Virus Internalization. Proc. Natl. Acad. Sci. USA 2019, 116, 8487–8492. [Google Scholar] [CrossRef]
- Huang, H.-C.; Chen, C.-C.; Chang, W.-C.; Tao, M.-H.; Huang, C. Entry of Hepatitis B Virus into Immortalized Human Primary Hepatocytes by Clathrin-Dependent Endocytosis. J. Virol. 2012, 86, 9443–9453. [Google Scholar] [CrossRef]
- Rabe, B.; Vlachou, A.; Panté, N.; Helenius, A.; Kann, M. Nuclear Import of Hepatitis B Virus Capsids and Release of the Viral Genome. Proc. Natl. Acad. Sci. USA 2003, 100, 9849–9854. [Google Scholar] [CrossRef]
- Ghaemi, Z.; Gruebele, M.; Tajkhorshid, E. Molecular Mechanism of Capsid Disassembly in Hepatitis B Virus. Proc. Natl. Acad. Sci. USA 2021, 118, e2102530118. [Google Scholar] [CrossRef] [PubMed]
- Newbold, J.E.; Xin, H.; Tencza, M.; Sherman, G.; Dean, J.; Bowden, S.; Locarnini, S. The Covalently Closed Duplex Form of the Hepadnavirus Genome Exists in Situ as a Heterogeneous Population of Viral Minichromosomes. J. Virol. 1995, 69, 3350–3357. [Google Scholar] [CrossRef] [PubMed]
- Bartenschlager, R.; Schaller, H. Hepadnaviral Assembly Is Initiated by Polymerase Binding to the Encapsidation Signal in the Viral RNA Genome. EMBO J. 1992, 11, 3413–3420. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Seeger, C. Expression and Characterization of Hepadnavirus Reverse Transcriptases. Methods Enzymol. 1996, 275, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Staprans, S.; Loeb, D.D.; Ganem, D. Mutations Affecting Hepadnavirus Plus-Strand DNA Synthesis Dissociate Primer Cleavage from Translocation and Reveal the Origin of Linear Viral DNA. J. Virol. 1991, 65, 1255–1262. [Google Scholar] [CrossRef] [PubMed]
- Tuttleman, J.S.; Pourcel, C.; Summers, J. Formation of the Pool of Covalently Closed Circular Viral DNA in Hepadnavirus-Infected Cells. Cell 1986, 47, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Sorensen, E.M.; Naito, A.; Schott, M.; Kim, S.; Ahlquist, P. Involvement of Host Cellular Multivesicular Body Functions in Hepatitis B Virus Budding. Proc. Natl. Acad. Sci. USA 2007, 104, 10205–10210. [Google Scholar] [CrossRef] [PubMed]
- Ning, X.; Nguyen, D.; Mentzer, L.; Adams, C.; Lee, H.; Ashley, R.; Hafenstein, S.; Hu, J. Secretion of Genome-Free Hepatitis B Virus--Single Strand Blocking Model for Virion Morphogenesis of Para-Retrovirus. PLoS Pathog. 2011, 7, e1002255. [Google Scholar] [CrossRef]
- Chen, M.T.; Billaud, J.N.; Sällberg, M.; Guidotti, L.G.; Chisari, F.V.; Jones, J.; Hughes, J.; Milich, D.R. A Function of the Hepatitis B Virus Precore Protein Is to Regulate the Immune Response to the Core Antigen. Proc. Natl. Acad. Sci. USA 2004, 101, 14913–14918. [Google Scholar] [CrossRef]
- Vanlandschoot, P.; Leroux-Roels, G. Viral Apoptotic Mimicry: An Immune Evasion Strategy Developed by the Hepatitis B Virus? Trends Immunol. 2003, 24, 144–147. [Google Scholar] [CrossRef]
- Tu, T.; Budzinska, M.A.; Shackel, N.A.; Urban, S. HBV DNA Integration: Molecular Mechanisms and Clinical Implications. Viruses 2017, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Ploss, A. Mechanism of Hepatitis B Virus cccDNA Formation. Viruses 2021, 13, 1463. [Google Scholar] [CrossRef] [PubMed]
- KöNiger, C.; Wingert, I.; Marsmann, M.; Rösler, C.; Beck, J.; Nassal, M. Involvement of the Host DNA-Repair Enzyme TDP2 in Formation of the Covalently Closed Circular DNA Persistence Reservoir of Hepatitis B Viruses. Proc. Natl. Acad. Sci. USA 2014, 111, E4244–E4253. [Google Scholar] [CrossRef]
- Kitamura, K.; Que, L.; Shimadu, M.; Koura, M.; Ishihara, Y.; Wakae, K.; Nakamura, T.; Watashi, K.; Wakita, T.; Muramatsu, M. Flap Endonuclease 1 Is Involved in CccDNA Formation in the Hepatitis B Virus. PLoS Pathog. 2018, 14, e1007124. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Ploss, A. Core Components of DNA Lagging Strand Synthesis Machinery Are Essential for Hepatitis B Virus CccDNA Formation. Nat. Microbiol. 2020, 5, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Ploss, A. Hepatitis B Virus CccDNA Is Formed through Distinct Repair Processes of Each Strand. Nat. Commun. 2021, 12, 1591. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Gao, Z.; Xu, G.; Peng, B.; Liu, C.; Yan, H.; Yao, Q.; Sun, G.; Liu, Y.; Tang, D.; et al. DNA Polymerase κ Is a Key Cellular Factor for the Formation of Covalently Closed Circular DNA of Hepatitis B Virus. PLoS Pathog. 2016, 12, e1005893. [Google Scholar] [CrossRef] [PubMed]
- Verrier, E.R.; Ligat, G.; Heydmann, L.; Doernbrack, K.; Miller, J.; Maglott-Roth, A.; Jühling, F.; El Saghire, H.; Heuschkel, M.J.; Fujiwara, N.; et al. Cell-Based CccDNA Reporter Assay Combined with Functional Genomics Identifies YBX1 as HBV CccDNA Host Factor and Antiviral Candidate Target. Gut 2022, 72, 1745–1757. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Summers, J. Illegitimate Replication of Linear Hepadnavirus DNA through Nonhomologous Recombination. J. Virol. 1995, 69, 4029–4036. [Google Scholar] [CrossRef]
- Yang, W.; Summers, J. Infection of Ducklings with Virus Particles Containing Linear Double-Stranded Duck Hepatitis B Virus DNA: Illegitimate Replication and Reversion. J. Virol. 1998, 72, 8710–8717. [Google Scholar] [CrossRef]
- Guo, H.; Xu, C.; Zhou, T.; Block, T.M.; Guo, J.T. Characterization of the Host Factors Required for Hepadnavirus Covalently Closed Circular (Ccc) DNA Formation. PLoS ONE 2012, 7, e43270. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R. The Mechanism of Double-Strand DNA Break Repair by the Nonhomologous DNA End-Joining Pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Summers, J. Integration of Hepadnavirus DNA in Infected Liver: Evidence for a Linear Precursor. J. Virol. 1999, 73, 9710–9717. [Google Scholar] [CrossRef] [PubMed]
- Bock, C.T.; Schwinn, S.; Locarnini, S.; Fyfe, J.; Manns, M.P.; Trautwein, C.; Zentgraf, H. Structural Organization of the Hepatitis B Virus Minichromosome. J. Mol. Biol. 2001, 307, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Andrisani, O. Epigenetic Mechanisms in Hepatitis B Virus-Associated Hepatocellular Carcinoma. Hepatoma Res. 2021, 7, 12. [Google Scholar] [CrossRef] [PubMed]
- Dandri, M. Epigenetic Modulation in Chronic Hepatitis B Virus Infection. Semin. Immunopathol. 2020, 42, 173–185. [Google Scholar] [CrossRef]
- Locatelli, M.; Quivy, J.-P.; Chapus, F.; Michelet, M.; Fresquet, J.; Maadadi, S.; Aberkane, A.N.; Diederichs, A.; Lucifora, J.; Rivoire, M.; et al. HIRA Supports Hepatitis B Virus Minichromosome Establishment and Transcriptional Activity in Infected Hepatocytes. Cell. Mol. Gastroenterol. Hepatol. 2022, 14, 527–551. [Google Scholar] [CrossRef]
- Tagami, H.; Ray-Gallet, D.; Almouzni, G.; Nakatani, Y. Histone H3.1 and H3.3 Complexes Mediate Nucleosome Assembly Pathways Dependent or Independent of DNA Synthesis. Cell 2004, 116, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Ray-Gallet, D.; Woolfe, A.; Vassias, I.; Pellentz, C.; Lacoste, N.; Puri, A.; Schultz, D.C.; Pchelintsev, N.A.; Adams, P.D.; Jansen, L.E.T.; et al. Dynamics of Histone H3 Deposition In Vivo Reveal a Nucleosome Gap-Filling Mechanism for H3.3 to Maintain Chromatin Integrity. Mol. Cell 2011, 44, 928–941. [Google Scholar] [CrossRef]
- Rai, T.S.; Glass, M.; Cole, J.J.; Rather, M.I.; Marsden, M.; Neilson, M.; Brock, C.; Humphreys, I.R.; Everett, R.D.; Adams, P.D. Histone Chaperone HIRA Deposits Histone H3.3 onto Foreign Viral DNA and Contributes to Anti-Viral Intrinsic Immunity. Nucleic Acids Res. 2017, 45, 11673–11683. [Google Scholar] [CrossRef]
- Adam, S.; Polo, S.E.; Almouzni, G. Transcription Recovery after DNA Damage Requires Chromatin Priming by the H3.3 Histone Chaperone HIRA. Cell 2013, 155, 94–106. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Astudillo, F.; Garrido, D.; Varas-Godoy, M.; Gutiérrez, J.L.; Villanueva, R.A.; Loyola, A. The Histone Variant H3.3 Regulates the Transcription of the Hepatitis B Virus. Ann. Hepatol. 2021, 21, 100261. [Google Scholar] [CrossRef] [PubMed]
- Lucifora, J.; Pastor, F.; Charles, É.; Pons, C.; Auclair, H.; Fusil, F.; Rivoire, M.; Cosset, F.-L.; Durantel, D.; Salvetti, A. Evidence for Long-Term Association of Virion-Delivered HBV Core Protein with CccDNA Independently of Viral Protein Production. JHEP reports Innov. Hepatol. 2021, 3, 100330. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Feng, J.; Liu, Y.; Zhao, M.; Yuan, Y.; Yuan, H.; Yun, H.; Sun, M.; Bu, Y.; Liu, L.; et al. HAT1 Signaling Confers to Assembly and Epigenetic Regulation of HBV CccDNA Minichromosome. Theranostics 2019, 9, 7345–7358. [Google Scholar] [CrossRef] [PubMed]
- Vachon, A.; Seo, G.E.; Patel, N.H.; Coffin, C.S.; Marinier, E.; Eyras, E.; Osiowy, C. Hepatitis B Virus Serum RNA Transcript Isoform Composition and Proportion in Chronic Hepatitis B Patients by Nanopore Long-Read Sequencing. Front. Microbiol. 2023, 14, 1233178. [Google Scholar] [CrossRef]
- Lim, C.S.; Sozzi, V.; Littlejohn, M.; Yuen, L.K.W.; Warner, N.; Betz-Stablein, B.; Luciani, F.; Revill, P.A.; Brown, C.M. Quantitative Analysis of the Splice Variants Expressed by the Major Hepatitis B Virus Genotypes. Microb. Genom. 2021, 7, 000492. [Google Scholar] [CrossRef]
- Kremsdorf, D.; Lekbaby, B.; Bablon, P.; Sotty, J.; Augustin, J.; Schnuriger, A.; Pol, J.; Soussan, P. Alternative Splicing of Viral Transcripts: The Dark Side of HBV. Gut 2021, 70, 2373–2382. [Google Scholar] [CrossRef] [PubMed]
- Ito, N.; Nakashima, K.; Sun, S.; Ito, M.; Suzuki, T. Cell Type Diversity in Hepatitis B Virus RNA Splicing and Its Regulation. Front. Microbiol. 2019, 10, 207. [Google Scholar] [CrossRef]
- Soussan, P.; Tuveri, R.; Nalpas, B.; Garreau, F.; Zavala, F.; Masson, A.; Pol, S.; Brechot, C.; Kremsdorf, D. The Expression of Hepatitis B Spliced Protein (HBSP) Encoded by a Spliced Hepatitis B Virus RNA Is Associated with Viral Replication and Liver Fibrosis. J. Hepatol. 2003, 38, 343–348. [Google Scholar] [CrossRef]
- Sozzi, V.; McCoullough, L.; Mason, H.; Littlejohn, M.; Revill, P.A. The in Vitro Replication Phenotype of Hepatitis B Virus (HBV) Splice Variant Sp1. Virology 2022, 574, 65–70. [Google Scholar] [CrossRef]
- Chung, C.-Y.; Sun, C.-P.; Tao, M.-H.; Wu, H.-L.; Wang, S.-H.; Yeh, S.-H.; Zheng, Q.-B.; Yuan, Q.; Xia, N.-S.; Ogawa, K.; et al. Major HBV Splice Variant Encoding a Novel Protein Important for Infection. J. Hepatol. 2024, in press. [Google Scholar] [CrossRef]
- Wang, H.; Liu, K.; Fang, B.A.M.; Wu, H.; Li, F.; Xiang, X.; Tang, W.; Zhao, G.; Lin, L.; Bao, S.; et al. Identification of Acetyltransferase Genes (HAT1 and KAT8) Regulating HBV Replication by RNAi Screening. Cell Biosci. 2015, 5, 66. [Google Scholar] [CrossRef] [PubMed]
- Alarcon, V.; Hernández, S.; Rubio, L.; Alvarez, F.; Flores, Y.; Varas-Godoy, M.; De Ferrari, G.V.; Kann, M.; Villanueva, R.A.; Loyola, A. The Enzymes LSD1 and Set1A Cooperate with the Viral Protein HBx to Establish an Active Hepatitis B Viral Chromatin State. Sci. Rep. 2016, 6, 25901. [Google Scholar] [CrossRef] [PubMed]
- Benhenda, S.; Ducroux, A.; Rivière, L.; Sobhian, B.; Ward, M.D.; Dion, S.; Hantz, O.; Protzer, U.; Michel, M.-L.; Benkirane, M.; et al. Methyltransferase PRMT1 Is a Binding Partner of HBx and a Negative Regulator of Hepatitis B Virus Transcription. J. Virol. 2013, 87, 4360–4371. [Google Scholar] [CrossRef]
- Decorsière, A.; Mueller, H.; Van Breugel, P.C.; Abdul, F.; Gerossier, L.; Beran, R.K.; Livingston, C.M.; Niu, C.; Fletcher, S.P.; Hantz, O.; et al. Hepatitis B Virus X Protein Identifies the Smc5/6 Complex as a Host Restriction Factor. Nature 2016, 531, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Vivekanandan, P.; Thomas, D.; Torbenson, M. Hepatitis B Viral DNA Is Methylated in Liver Tissues. J. Viral Hepat. 2008, 15, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Xie, W. The Role of 3D Genome Organization in Development and Cell Differentiation. Nat. Rev. Mol. Cell Biol. 2019, 20, 535–550. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Zhao, H.; Wu, Y.; Peng, B.; Gao, Z.; Sun, Y.; Duan, J.; Qi, Y.; Li, Y.; Zhou, Z.; et al. Transcriptionally Inactive Hepatitis B Virus Episome DNA Preferentially Resides in the Vicinity of Chromosome 19 in 3D Host Genome upon Infection. Cell Rep. 2021, 35, 109288. [Google Scholar] [CrossRef] [PubMed]
- Niu, C.; Livingston, C.M.; Li, L.; Beran, R.K.; Daffis, S.; Ramakrishnan, D.; Burdette, D.; Peiser, L.; Salas, E.; Ramos, H.; et al. The Smc5/6 Complex Restricts HBV When Localized to ND10 without Inducing an Innate Immune Response and Is Counteracted by the HBV X Protein Shortly after Infection. PLoS ONE 2017, 12, e0169648. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.; Peng, B.; Li, C.; Li, X.; Chen, M.; Zhou, Z.; Tang, D.; He, J.; Wu, Y.; Sun, Y.; et al. SLF2 Interacts with the SMC5/6 Complex to Direct Hepatitis B Virus Episomal DNA to Promyelocytic Leukemia Bodies for Transcriptional Repression. J. Virol. 2023, 97, e0032823. [Google Scholar] [CrossRef]
- Abdul, F.; Diman, A.; Baechler, B.; Ramakrishnan, D.; Kornyeyev, D.; Beran, R.K.; Fletcher, S.P.; Strubin, M. Smc5/6 Silences Episomal Transcription by a Three-Step Function. Nat. Struct. Mol. Biol. 2022, 29, 922–931. [Google Scholar] [CrossRef]
- Huppert, J.L. Four-Stranded Nucleic Acids: Structure, Function and Targeting of G-Quadruplexes. Chem. Soc. Rev. 2008, 37, 1375. [Google Scholar] [CrossRef] [PubMed]
- Giraud, G.; Rodà, M.; Huchon, P.; Michelet, M.; Maadadi, S.; Jutzi, D.; Montserret, R.; Ruepp, M.-D.; Parent, R.; Combet, C.; et al. G-Quadruplexes Control Hepatitis B Virus Replication by Promoting CccDNA Transcription and Phase Separation in Hepatocytes. Nucleic Acids Res. 2023, 52, 2290–2305. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Escribano, P.; Hormeño, S.; Madariaga-Marcos, J.; Solé-Soler, R.; O’Reilly, F.J.; Morris, K.; Aicart-Ramos, C.; Aramayo, R.; Montoya, A.; Kramer, H.; et al. Purified Smc5/6 Complex Exhibits DNA Substrate Recognition and Compaction. Mol. Cell 2020, 80, 1039–1054.e6. [Google Scholar] [CrossRef] [PubMed]
- Belloni, L.; Pollicino, T.; De Nicola, F.; Guerrieri, F.; Raffa, G.; Fanciulli, M.; Raimondo, G.; Levrero, M. Nuclear HBx Binds the HBV Minichromosome and Modifies the Epigenetic Regulation of CccDNA Function. Proc. Natl. Acad. Sci. USA 2009, 106, 19975–19979. [Google Scholar] [CrossRef] [PubMed]
- Tu, T.; Zehnder, B.; Qu, B.; Urban, S. De Novo Synthesis of Hepatitis B Virus Nucleocapsids Is Dispensable for the Maintenance and Transcriptional Regulation of CccDNA. JHEP Rep. 2021, 3, 100195. [Google Scholar] [CrossRef]
- Zhong, Y.; Wu, C.; Xu, Z.; Teng, Y.; Zhao, L.; Zhao, K.; Wang, J.; Wang, W.; Zhan, Q.; Zhu, C.; et al. Hepatitis B Virus Core Protein Is Not Required for Covalently Closed Circular DNA Transcriptional Regulation. J. Virol. 2022, 96, e0136222. [Google Scholar] [CrossRef]
- Wu, M.; Wang, C.; Shi, B.; Fang, Z.; Qin, B.; Zhou, X.; Zhang, X.; Yuan, Z. A Novel Recombinant CccDNA-Based Mouse Model with Long Term Maintenance of RcccDNA and Antigenemia. Antivir. Res. 2020, 180, 104826. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Zhou, B.; Cai, D.; Zong, Y.; Wu, Y.; Liu, S.; Mercier, A.; Guo, H.; Hou, J.; Colonno, R.; et al. Rapid Turnover of Hepatitis B Virus Covalently Closed Circular DNA Indicated by Monitoring Emergence and Reversion of Signature-Mutation in Treated Chronic Hepatitis B Patients. Hepatology 2021, 73, 41–52. [Google Scholar] [CrossRef]
- Kornyeyev, D.; Ramakrishnan, D.; Voitenleitner, C.; Livingston, C.M.; Xing, W.; Hung, M.; Kwon, H.J.; Fletcher, S.P.; Beran, R.K. Spatiotemporal Analysis of Hepatitis B Virus X Protein in Primary Human Hepatocytes. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- van Breugel, P.C.; Robert, E.I.; Mueller, H.; Decorsière, A.; Zoulim, F.; Hantz, O.; Strubin, M. Hepatitis B Virus X Protein Stimulates Gene Expression Selectively from Extrachromosomal DNA Templates. Hepatology 2012, 56, 2116–2124. [Google Scholar] [CrossRef]
- Leupin, O.; Bontron, S.; Schaeffer, C.; Strubin, M. Hepatitis B Virus X Protein Stimulates Viral Genome Replication via a DDB1-Dependent Pathway Distinct from That Leading to Cell Death. J. Virol. 2005, 79, 4238–4245. [Google Scholar] [CrossRef] [PubMed]
- Angers, S.; Li, T.; Yi, X.; MacCoss, M.J.; Moon, R.T.; Zheng, N. Molecular Architecture and Assembly of the DDB1-CUL4A Ubiquitin Ligase Machinery. Nature 2006, 443, 590–593. [Google Scholar] [CrossRef] [PubMed]
- Stadelmayer, B.; Diederichs, A.; Chapus, F.; Rivoire, M.; Neveu, G.; Alam, A.; Fraisse, L.; Carter, K.; Testoni, B.; Zoulim, F. Full-Length 5′RACE Identifies All Major HBV Transcripts in HBV-Infected Hepatocytes and Patient Serum. J. Hepatol. 2020, 73, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Serrano, D.; Cordero, G.; Kawamura, R.; Sverzhinsky, A.; Sarker, M.; Roy, S.; Malo, C.; Pascal, J.M.; Marko, J.F.; D’Amours, D. The Smc5/6 Core Complex Is a Structure-Specific DNA Binding and Compacting Machine. Mol. Cell 2020, 80, 1025–1038.e5. [Google Scholar] [CrossRef]
- Berggren, K.A.; Schwartz, R.E.; Kleiner, R.E.; Ploss, A. The Impact of Epitranscriptomic Modifications on Liver Disease. Trends Endocrinol. Metab. 2024, 35, 331–346. [Google Scholar] [CrossRef]
- Roundtree, I.A.; Evans, M.E.; Pan, T.; He, C. Dynamic RNA Modifications in Gene Expression Regulation. Cell 2017, 169, 1187–1200. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.; Liu, J.; He, C. RNA N6-Methyladenosine Methylation in Post-Transcriptional Gene Expression Regulation. Genes Dev. 2015, 29, 1343–1355. [Google Scholar] [CrossRef] [PubMed]
- Imam, H.; Khan, M.; Gokhale, N.S.; McIntyre, A.B.R.; Kim, G.-W.; Jang, J.Y.; Kim, S.-J.; Mason, C.E.; Horner, S.M.; Siddiqui, A. N6-Methyladenosine Modification of Hepatitis B Virus RNA Differentially Regulates the Viral Life Cycle. Proc. Natl. Acad. Sci. USA 2018, 115, 8829–8834. [Google Scholar] [CrossRef]
- Kim, G.-W.; Moon, J.-S.; Siddiqui, A. N6-Methyladenosine Modification of the 5′ Epsilon Structure of the HBV Pregenome RNA Regulates Its Encapsidation by the Viral Core Protein. Proc. Natl. Acad. Sci. USA 2022, 119, e2120485119. [Google Scholar] [CrossRef]
- Kim, G.-W.; Siddiqui, A. Hepatitis B Virus X Protein Recruits Methyltransferases to Affect Cotranscriptional N6-Methyladenosine Modification of Viral/Host RNAs. Proc. Natl. Acad. Sci. USA 2021, 118, e2019455118. [Google Scholar] [CrossRef]
- Liang, G.; Kitamura, K.; Wang, Z.; Liu, G.; Chowdhury, S.; Fu, W.; Koura, M.; Wakae, K.; Honjo, T.; Muramatsu, M. RNA Editing of Hepatitis B Virus Transcripts by Activation-Induced Cytidine Deaminase. Proc. Natl. Acad. Sci. USA 2013, 110, 2246–2251. [Google Scholar] [CrossRef] [PubMed]
- Martin-Vilchez, S.; Lara-Pezzi, E.; Trapero-Marugán, M.; Moreno-Otero, R.; Sanz-Cameno, P. The Molecular and Pathophysiological Implications of Hepatitis B X Antigen in Chronic Hepatitis B Virus Infection. Rev. Med. Virol. 2011, 21, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.; Zlotnick, A. Asymmetric Modification of Hepatitis B Virus (HBV) Genomes by an Endogenous Cytidine Deaminase inside HBV Cores Informs a Model of Reverse Transcription. J. Virol. 2018, 92. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gómez-Moreno, A.; Ploss, A. Mechanisms of Hepatitis B Virus cccDNA and Minichromosome Formation and HBV Gene Transcription. Viruses 2024, 16, 609. https://doi.org/10.3390/v16040609
Gómez-Moreno A, Ploss A. Mechanisms of Hepatitis B Virus cccDNA and Minichromosome Formation and HBV Gene Transcription. Viruses. 2024; 16(4):609. https://doi.org/10.3390/v16040609
Chicago/Turabian StyleGómez-Moreno, Andoni, and Alexander Ploss. 2024. "Mechanisms of Hepatitis B Virus cccDNA and Minichromosome Formation and HBV Gene Transcription" Viruses 16, no. 4: 609. https://doi.org/10.3390/v16040609
APA StyleGómez-Moreno, A., & Ploss, A. (2024). Mechanisms of Hepatitis B Virus cccDNA and Minichromosome Formation and HBV Gene Transcription. Viruses, 16(4), 609. https://doi.org/10.3390/v16040609