Prediction of Drug-Drug Interactions with Bupropion and Its Metabolites as CYP2D6 Inhibitors Using a Physiologically-Based Pharmacokinetic Model

Abstract
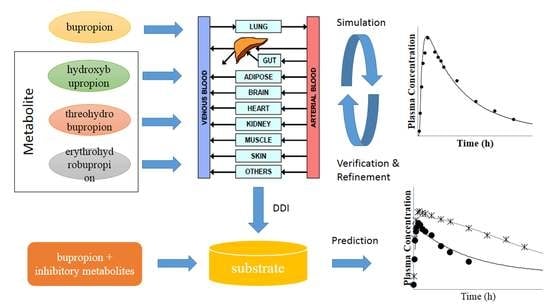
:
1. Introduction
2. Materials and Methods
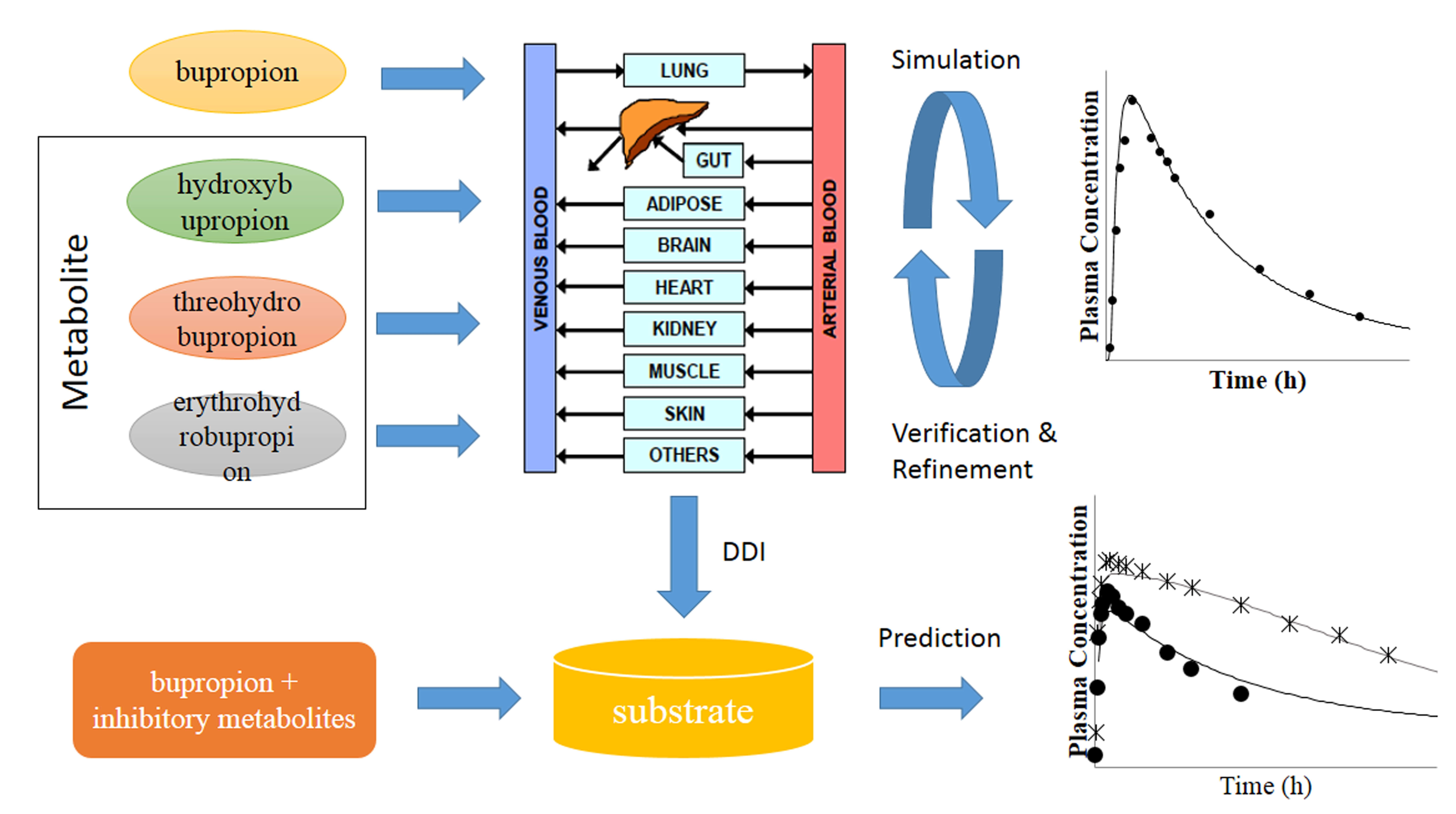
2.1. Physiologically-Based Pharmacokinetic (PBPK) Model Development
2.2. PBPK Model for Bupropion
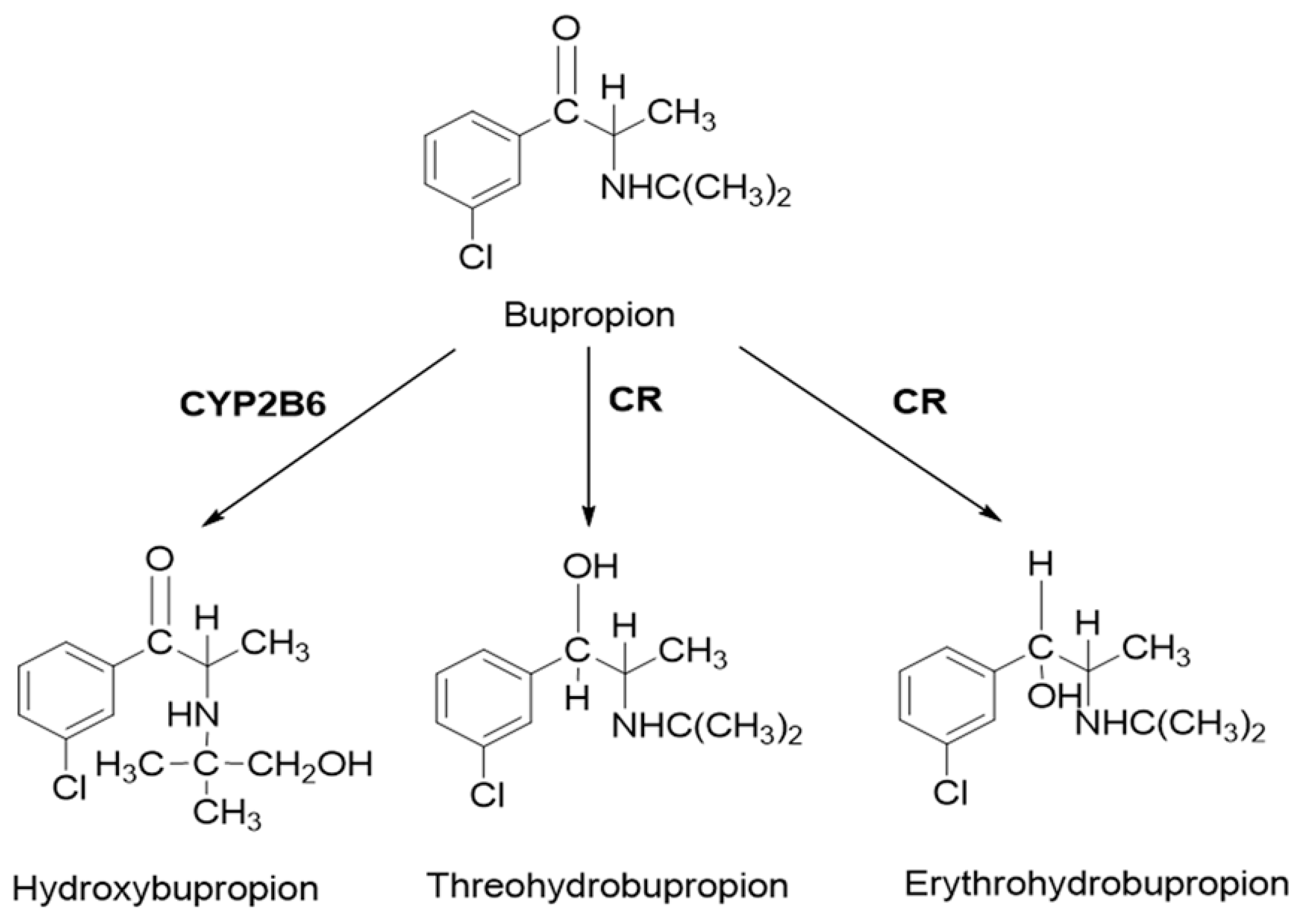
2.3. PBPK Model for Hydroxybupropion, Threohydrobupropion and Erythrohydrobupropion
2.4. PBPK Model for Venlafaxine
2.5. Simcyp Simulation
2.6. Simulation of Drug-Drug Interaction (DDI)
- (1)
- The subjects (10 trials × 15 subject, aged 20–50, female/male ratio 0) received 150 mg bupropion or matching placebo orally twice daily for 10 days, and on day 11 the subjects received a single oral dose of 50 mg desipramine. Plasma concentrations of bupropion and desipramine were simulated during the drug treatment period.
- (2)
- The subjects (10 trials × 18 subject, aged 20–50, female/male ratio 0.5) received bupropion (at a daily dose of 150 mg/day) with venlafaxine (at a daily dose of 75 mg/day) for 8 weeks. Plasma concentrations of bupropion and venlafaxine were simulated during the drug treatment period.
- (3)
- The subjects (10 trials × 13 subject, aged 21–64, female/male ratio 0.5) received 150 mg bupropion or matching placebo orally twice daily for 17 days, and on day 18 the subjects received a single oral dose of 30 mg dextromethorphan. Plasma concentrations of bupropion and dextromethorphan were simulated during the drug treatment period.
- (4)
- The subjects (10 trials × 10 subject, aged 20–56, female/male ratio 0.5) received bupropion (at a twice daily dose of 150 mg) with metoprolol (at a twice daily dose of 75 mg) for 12 days. Plasma concentrations of bupropion and metoprolol were simulated during the drug treatment period.
- (5)
- The subjects (10 trials × 10 subject, aged 20–50, female/male ratio 0.5) received 150 mg bupropion or matching placebo orally twice daily for 2 weeks, and on day 15 the subjects received a single oral dose of 20 mg bufuralol or 2 mg tolterodine. Plasma concentrations of bupropion, bufuralol and tolterodine were simulated during the drug treatment period.
2.7. PBPK Model for Stereo-Selective Bupropion and Its Metabolites
3. Results
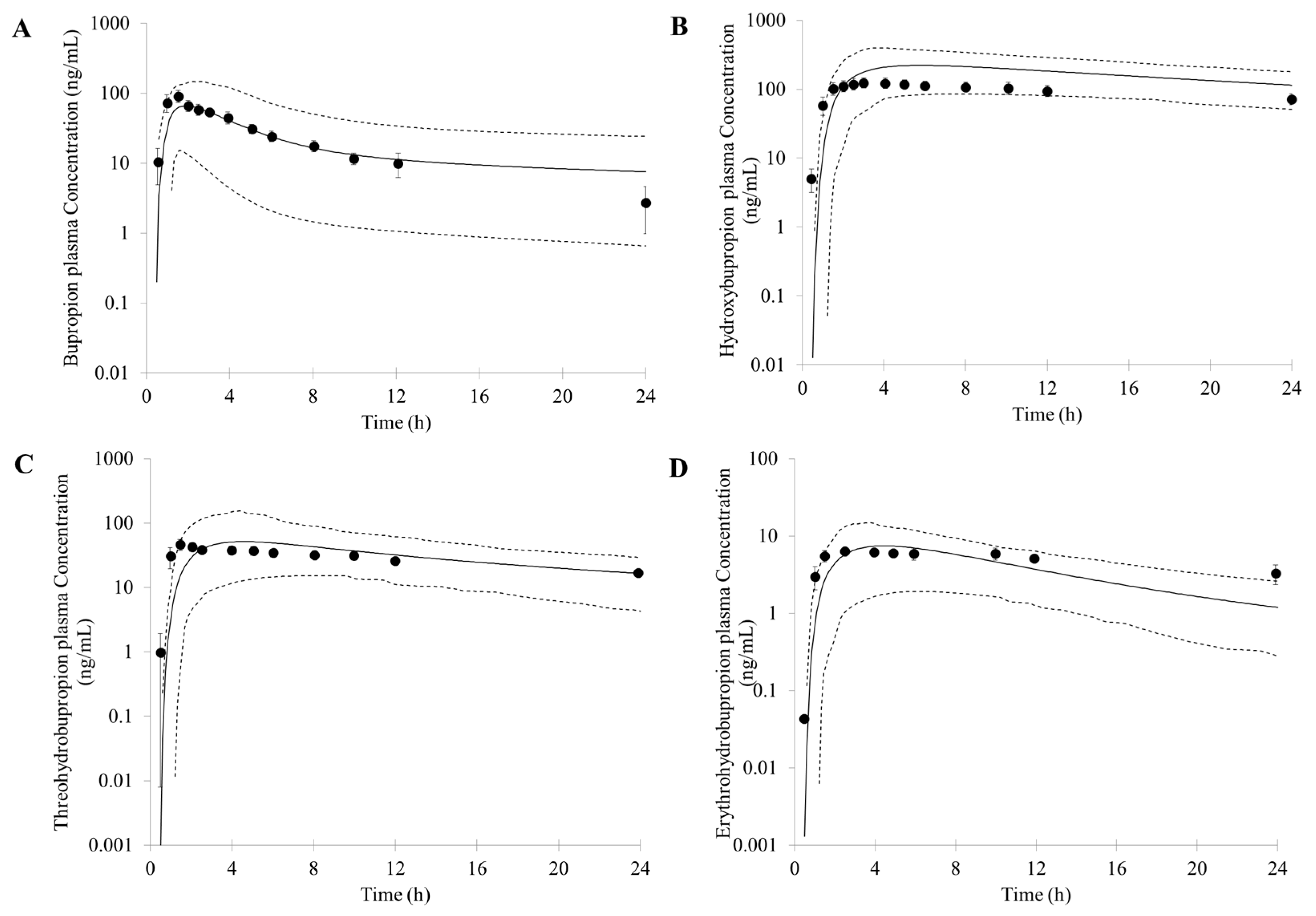
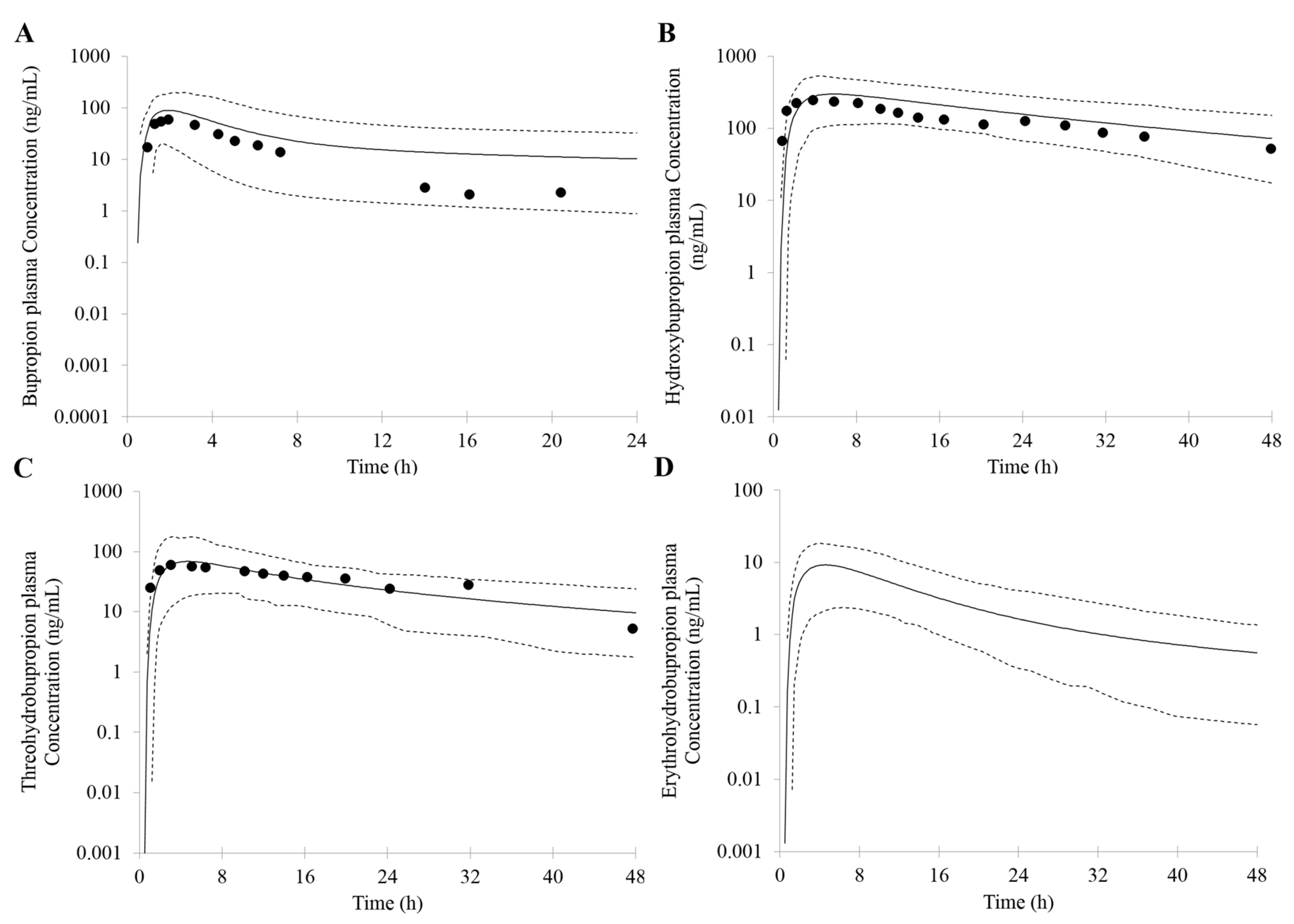
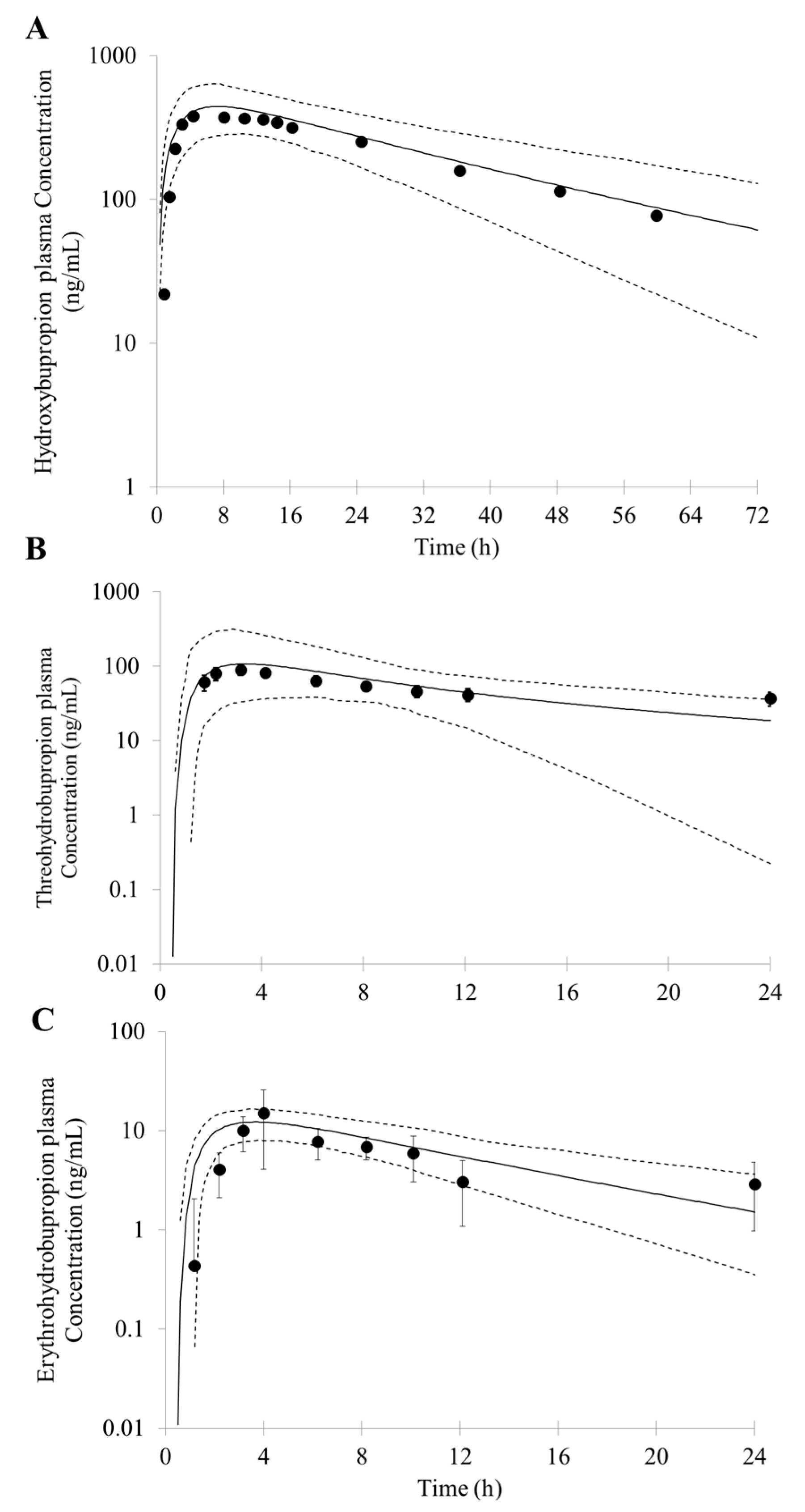
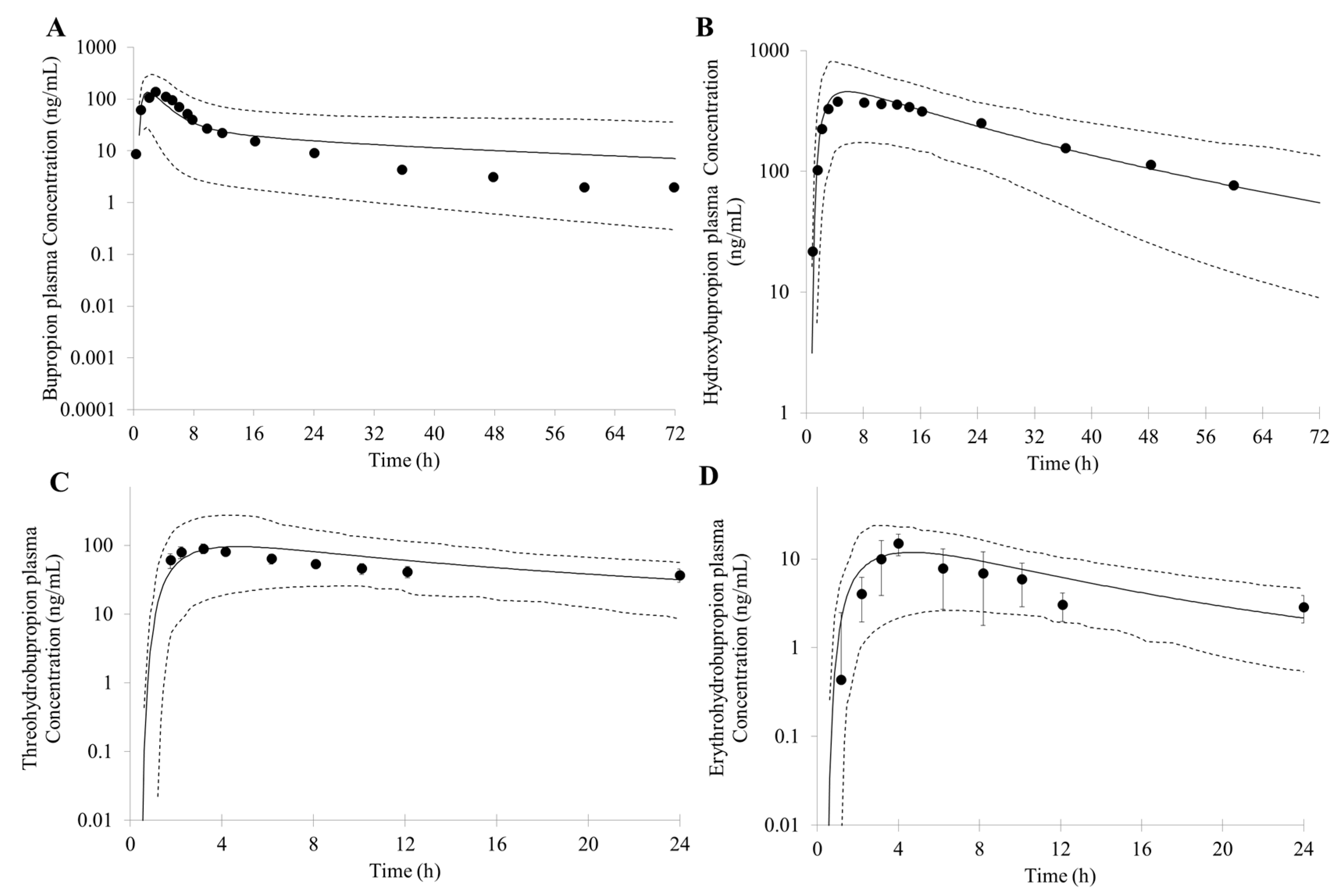
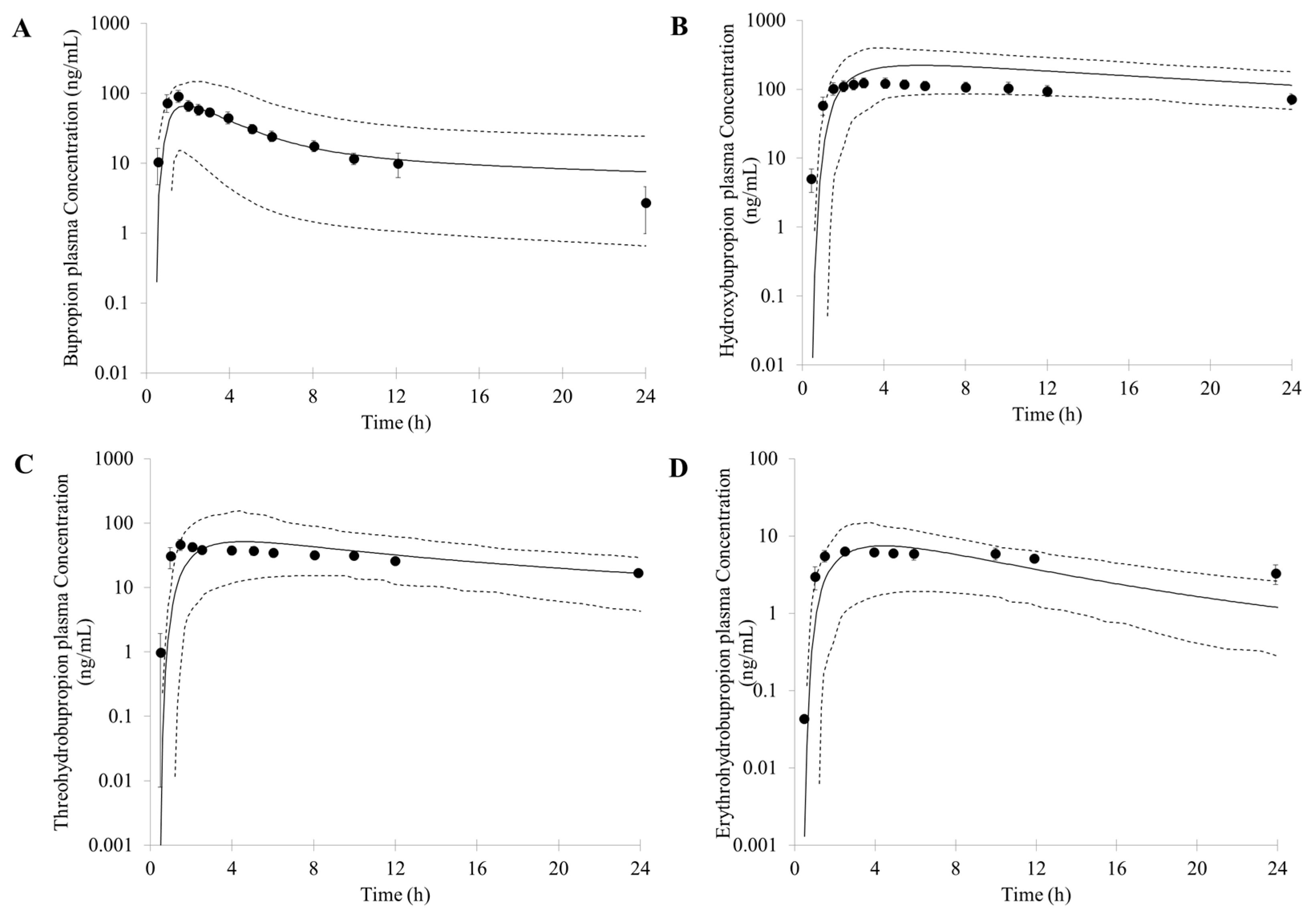
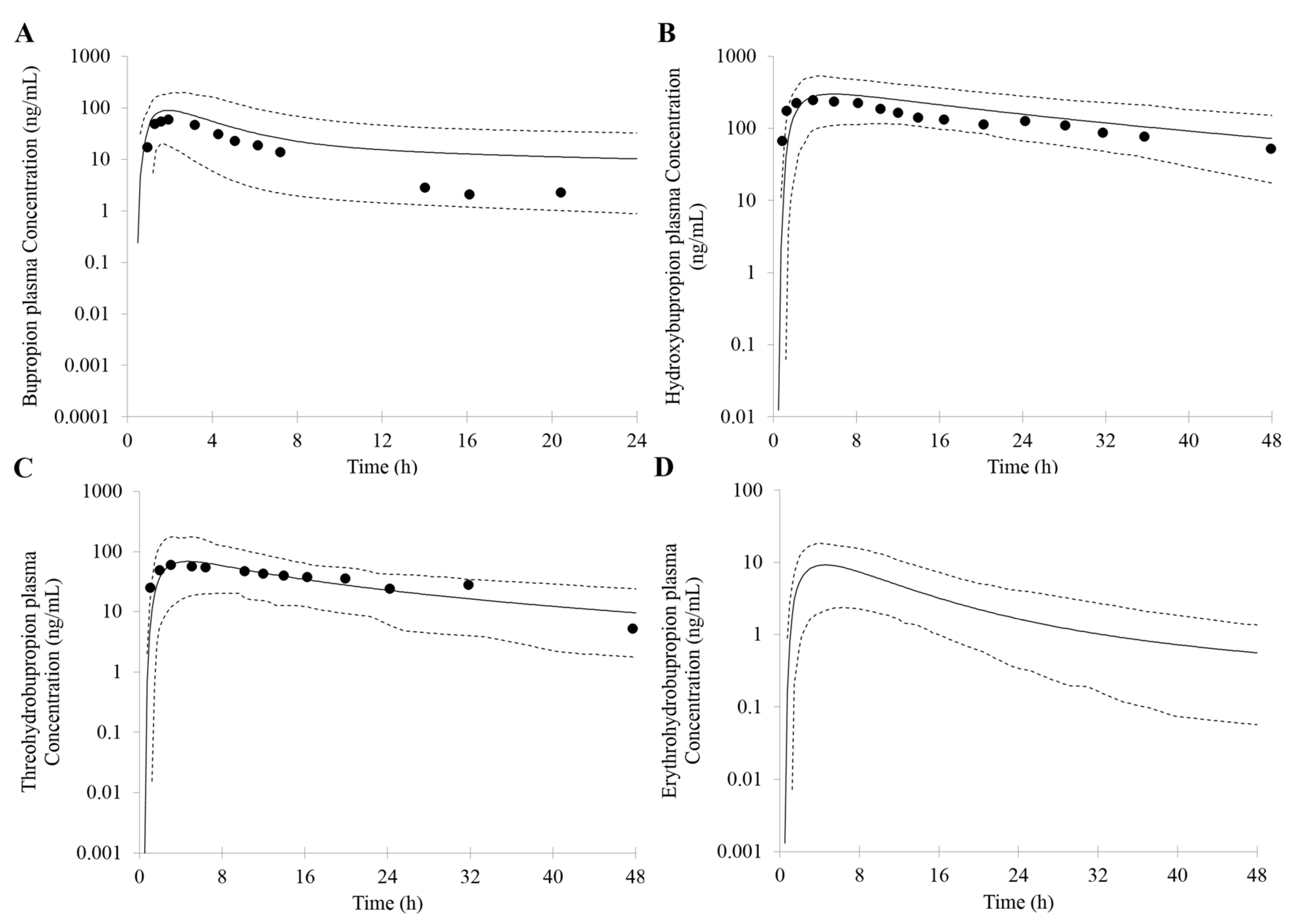
3.1. Prediction of Bupropion and Its Metabolites Pharmacokinetics
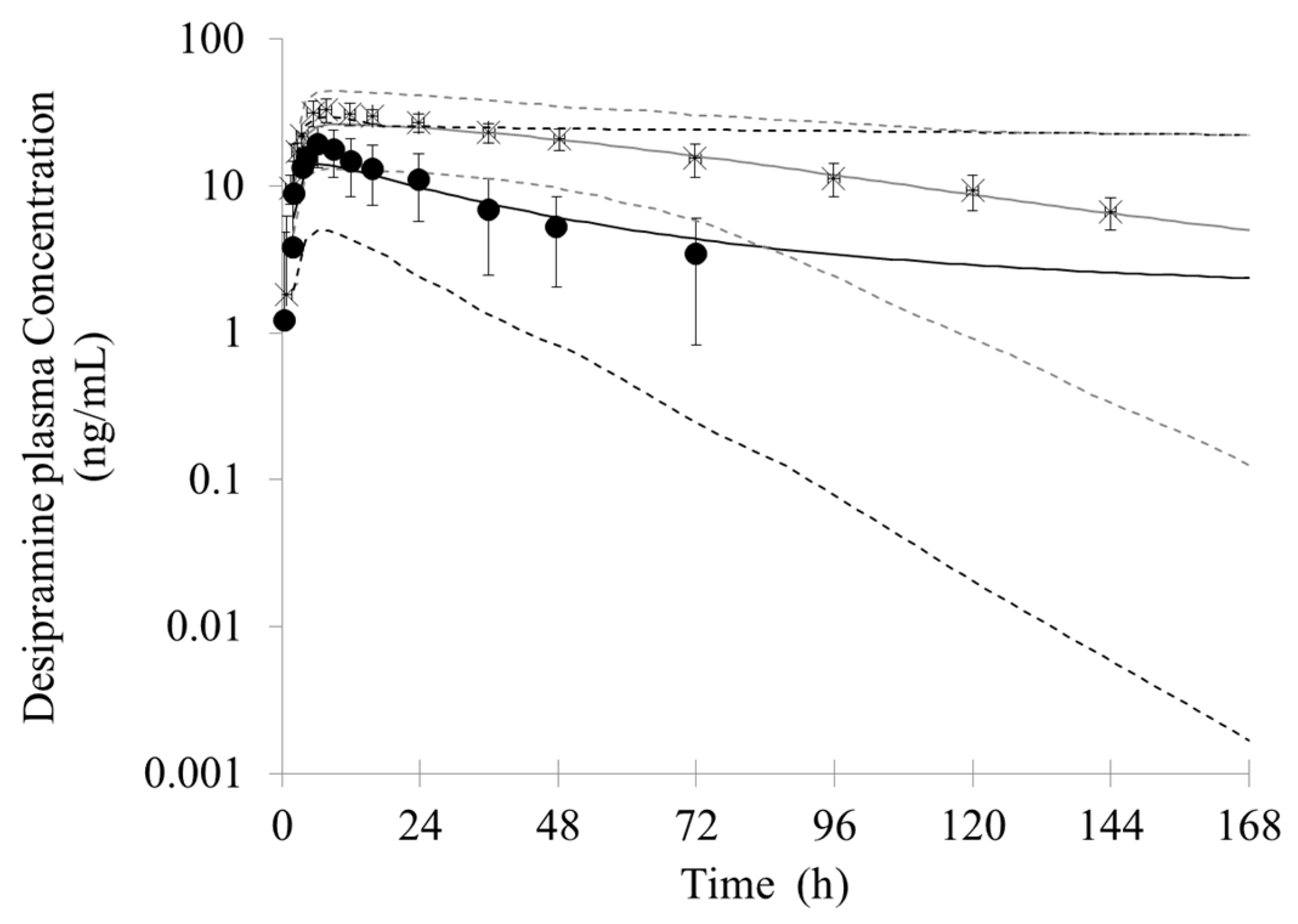
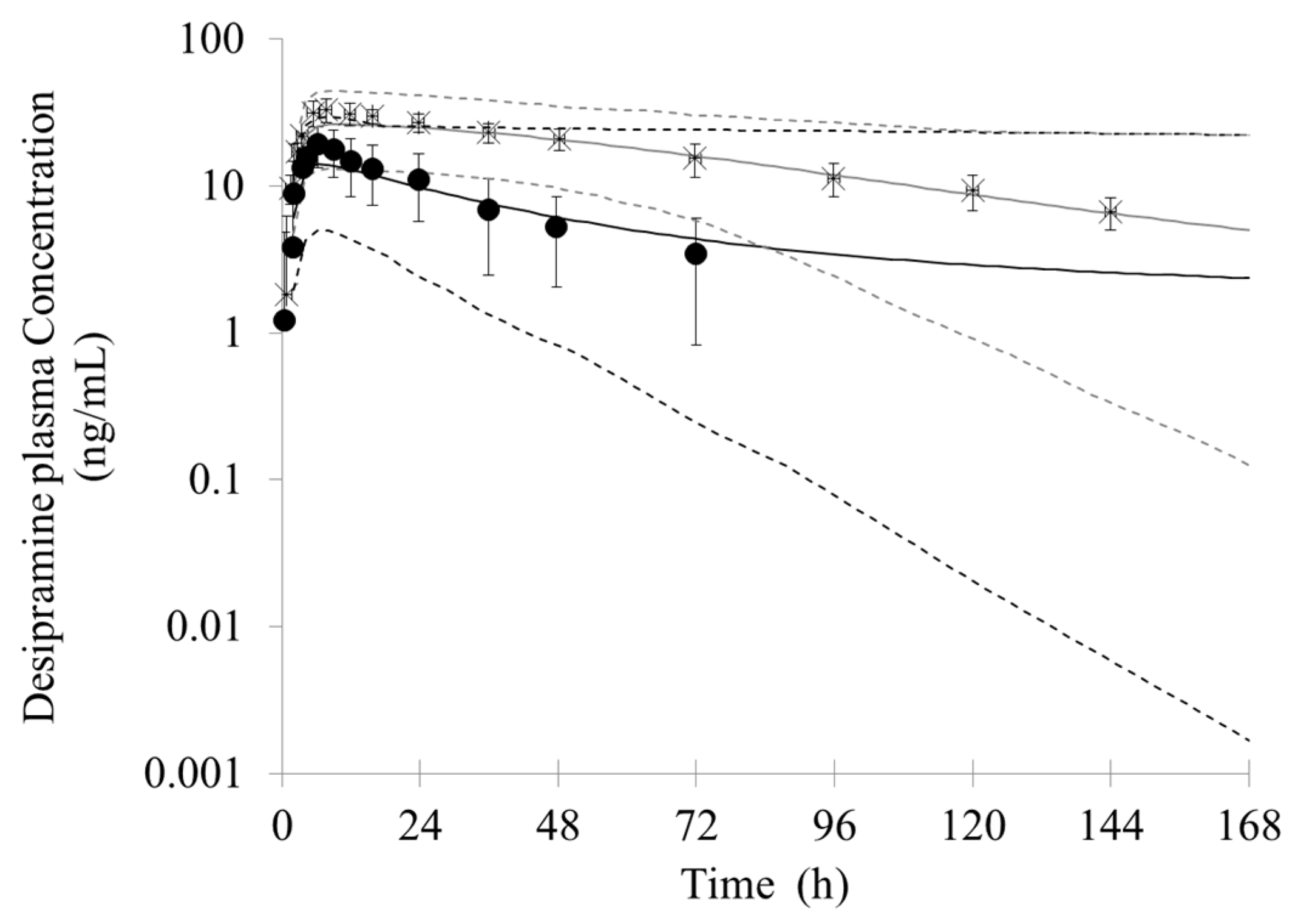
3.2. Prediction of the Bupropion-Desipramine DDI
3.3. Prediction of the Bupropion-Venlafaxine DDI
3.4. Prediction of DDI between Bupropion with Other Potential CYP2D6 Substrates
3.5. Prediction of Stereo-Selective Bupropion and Its Metabolites Pharmacokinetics and DDI
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Huang, S.M.; Lesko, L.J. Drug-drug, drug-dietary supplement, and drug-citrus fruit and other food interactions: What have we learned? J. Clin. Pharmacol. 2004, 44, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Krayenbühl, J.C.; Vozeh, S.; Kondo-Oestreicher, M.; Dayer, P. Drug-drug inleracüons or new active substances; mibefradil example. Eur. J. Clin. Pharmacol. 1999, 55, 559–565. [Google Scholar] [PubMed]
- Lee, S.Y.; Jang, H.; Lee, J.Y.; Kwon, K.I.; Oh, S.J.; Kim, S.K. Inhibition of cytochrome P450 by ethambutol in human liver microsomes. Toxicol. Lett. 2014, 229, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Wienkers, L.C.; Heath, T.G. Predicting in vivo drug interactions from in vitro drug discovery data. Nat. Rev. Drug Discov. 2005, 4, 825–833. [Google Scholar] [CrossRef] [PubMed]
- Prueksaritanont, T.; Chu, X.; Gibson, C.; Cui, D.; Yee, K.L.; Ballard, J.; Cabalu, T.; Hochman, J. Drug-drug interaction studies: Regulatory guidance and an industry perspective. AAPS J. 2013, 15, 629–645. [Google Scholar] [CrossRef] [PubMed]
- Callegari, E.; Kalgutkar, A.S.; Leung, L.; Obach, R.S.; Plowchalk, D.R.; Tse, S. Drug metabolites as cytochrome p450 inhibitors: A retrospective analysis and proposed algorithm for evaluation of the pharmacokinetic interaction potential of metabolites in drug discovery and development. Drug Metab. Dispos. 2013, 41, 2047–2055. [Google Scholar] [CrossRef] [PubMed]
- Isoherranen, N.; Hachad, H.; Yeung, C.K.; Levy, R.H. Qualitative analysis of the role of metabolites in inhibitory drug-drug interactions: Literature evaluation based on the metabolism and transport drug interaction database. Chem. Res. Toxicol. 2009, 22, 294–298. [Google Scholar] [CrossRef] [PubMed]
- Yeung, C.K.; Fujioka, Y.; Hachad, H.; Levy, R.H.; Isoherranen, N. Are circulating metabolites important in drug-drug interactions? Quantitative analysis of risk prediction and inhibitory potency. Clin. Pharmacol. Ther. 2011, 89, 105–113. [Google Scholar] [CrossRef] [PubMed]
- US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER). Drug Interaction Studies—Study Design, Data Analysis, Implications for Dosing, and Labeling Recommendations Draft Guidance. Available online: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm292362.pdf (accessed on 31 October 2017).
- European Medicines Agency, Committee for Human Medicinal Products (CHMP). Guideline on the Investigation of Drug Interactions. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606.pdf (accessed on 31 October 2017).
- Huang, S.M.; Rowland, M. The role of physiologically based pharmacokinetic modeling in regulatory review. Clin. Pharmacol. Ther. 2012, 91, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Fenneteau, F.; Poulin, P.; Nekka, F. Physiologically based predictions of the impact of inhibition of intestinal and hepatic metabolism on human pharmacokinetics of CYP3A substrates. J. Pharm. Sci. 2010, 99, 486–514. [Google Scholar] [CrossRef] [PubMed]
- Jamei, M.; Marciniak, S.; Feng, K.; Barnett, A.; Tucker, G.T.; Rostami-Hodjegan, A. The Simcyp population-based ADME simulator. Expert Opin. Drug Metab. Toxicol. 2009, 5, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Shitara, Y.; Sato, H.; Yoshisue, K.; Hirano, M.; Ikeda, T.; Sugiyama, Y. The quantitative prediction of CYP mediated drug interaction by physiologically based pharmacokineticmodeling. Pharm. Res. 2008, 25, 1891–1901. [Google Scholar] [CrossRef] [PubMed]
- Vossen, M.; Sevestre, M.; Niederalt, C.; Jang, I.J.; Willmann, S.; Edginton, A.N. Dynamically simulating the interaction of midazolam and the CYP3A4 inhibitor itraconazole using individual coupled wholebody physiologically-based pharmacokinetic (WB-PBPK) models. Theor. Biol. Med. Model. 2007, 4, 13. [Google Scholar] [CrossRef] [PubMed]
- Fahmi, O.A.; Maurer, T.S.; Kish, M.; Cardenas, E.; Boldt, S.; Nettleton, D. A combined model for predicting CYP3A4 clinical net drug-drug interaction based on CYP3A4 inhibition, inactivation, and induction determined in vitro. Drug Metab. Dispos. 2008, 36, 1698–1708. [Google Scholar] [CrossRef] [PubMed]
- Rostami-Hodjegan, A.; Tucker, G.T. ‘In silico’ simulations to assess the ‘in vivo’ consequences of ‘in vitro’ metabolic drug-drug interactions. Drug Discov. Today Technol. 2004, 1, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Rowland Yeo, K.; Jamei, M.; Yang, J.; Tucker, G.T.; Rostami-Hodjegan, A. Physiologically basedmechanistic modeling to predict complex drug-drug interactions involving simultaneous competitive and time-dependent enzyme inhibition by parent compound and its metabolite in both liver and gut-the effect of diltiazem on the time-course of exposure to triazolam. Eur. J. Pharm. Sci. 2010, 39, 298–309. [Google Scholar] [PubMed]
- Zhang, X.; Quinney, S.K.; Gorski, J.C.; Jones, D.R.; Hall, S.D. Semiphysiologically based pharmacokinetic models for the inhibition of midazolam clearance by diltiazem and its major metabolite. Drug Metab. Dispos. 2009, 37, 1587–1597. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.M. PBPK as a tool in regulatory review. Biopharm. Drug Dispos. 2012, 33, 51–52. [Google Scholar] [CrossRef] [PubMed]
- Leong, R.; Vieira, M.L.; Zhao, P.; Mulugeta, Y.; Lee, C.S.; Huang, S.M.; Burckart, G.J. Regulatory experience with physiologically based pharmacokinetic modeling for pediatric drug trials. Clin. Pharmacol. Ther. 2012, 91, 926–931. [Google Scholar] [CrossRef] [PubMed]
- Sinha, V.; Zhao, P.; Huang, S.M.; Zineh, I. Physiologically based pharmacokinetic modeling: From regulatory science to regulatory policy. Clin. Pharmacol. Ther. 2014, 95, 478–480. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Rowland, M.; Huang, S.M. Best practice in the use of physiologically based pharmacokinetic modeling and simulation to address clinical pharmacology regulatory questions. Clin. Pharmacol. Ther. 2012, 92, 17–20. [Google Scholar] [CrossRef] [PubMed]
- Faucette, S.R.; Hawke, R.L.; Lecluyse, E.L.; Shord, S.S.; Yan, B.; Laethem, R.M.; Lindley, C.M. Validation of bupropion hydroxylation as a selective marker of human cytochrome P450 2B6 catalytic activity. Drug Metab. Dispos. 2000, 28, 1222–1230. [Google Scholar] [PubMed]
- Golden, R.N.; De Vane, C.L.; Laizure, S.C.; Rudorfer, M.V.; Sherer, M.A.; Potter, W.Z. Bupropion in depression. II. The role of metabolites in clinical outcome. Arch. Gen. Psychiatry 1988, 45, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, D.H. Metabolism and kinetics of bupropion. J. Clin. Psychiatry 1983, 44, 79–81. [Google Scholar] [PubMed]
- Hesse, L.M.; Nevkatakrishnan, K.; Court, M.H.; von Moltke, L.L.; Duan, S.X.; Shader, R.I.; Greenblatt, D.J. 2B6 mediates the in vitro hydroxylation of bupropion: Potential drug interactions with other antidipressants. Drug Metab. Dispos. 2000, 28, 1176–1183. [Google Scholar] [PubMed]
- Reese, M.J.; Wurm, R.M.; Muir, K.T.; Generaux, G.T.; St John-Williams, L.; McConn, D.J. An in vitro mechanistic study to elucidate the desipramine/bupropion clinical drug-drug interaction. Drug Metab. Dispos. 2008, 36, 1198–1201. [Google Scholar] [CrossRef] [PubMed]
- Jefferson, J.W.; Pradko, J.F.; Muir, K.T. Bupropion for major depressive disorder: Pharmacokinetic and formulation considerations. Clin. Ther. 2005, 27, 1685–1695. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, T.; Rowland, M. Physiologically based pharmacokinetic modelling 2: Predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J. Pharm. Sci. 2006, 95, 1238–1257. [Google Scholar] [CrossRef] [PubMed]
- Findlay, J.W.; Van Wyck Fleet, J.; Smith, P.G.; Butz, R.F.; Hinton, M.L.; Blum, M.R.; Schroeder, D.H. Pharmacokinetics of bupropion, a novel antidepressant agent, following oral administration to healthy subjects. Eur. J. Clin. Pharmacol. 1981, 21, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Kirchheiner, J.; Klein, C.; Meineke, I.; Sasse, J.; Zanger, U.M.; Mürdter, T.E.; Roots, I.; Brockmöller, J. Bupropion and 4-OH-bupropion pharmacokinetics in relation to genetic polymorphisms in CYP2B6. Pharmacogenetics 2003, 13, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Connarn, J.N.; Zhang, X.; Babiskin, A.; Sun, D. Metabolism of bupropion by carbonyl reductases in liver and intestine. Drug Metab. Dispos. 2015, 43, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Ketter, T.A.; Jenkins, J.B.; Schroeder, D.H.; Pazzaglia, P.J.; Marangell, L.B.; George, M.S.; Callahan, A.M.; Hinton, M.L.; Chao, J.; Post, R.M. Carbamazepine but not valproate induces bupropion metabolism. J. Clin. Psychopharmacol. 1995, 15, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Poulin, P.; Theil, F.P. Prediction of pharmacokinetics prior to in vivo studies. 1. Mechanism-based prediction of volume of distribution. J. Pharm. Sci. 2002, 91, 129–156. [Google Scholar] [CrossRef] [PubMed]
- Ereshefsky, L.; Dugan, D. Review of the pharmacokinetics, pharmacogenetics, and drug interaction potential of antidepressants: Focus on venlafaxine. Depress. Anxiety 2000, 12 (Suppl. 1), 30–44. [Google Scholar] [CrossRef]
- Magalhães, P.; Alves, G.; LLerena, A.; Falcão, A. Clinical drug-drug interaction: Focus on venlafaxine. Drug Metab. Pers. Ther. 2015, 30, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Taft, D.R.; Iyer, G.R.; Behar, L.; DiGregorio, R.V. Application of a first-pass effect model to characterize the pharmacokinetic disposition of venlafaxine after oraladministration to human subjects. Drug Metab. Dispos. 1997, 25, 1215–1218. [Google Scholar] [PubMed]
- Fogelman, S.M.; Schmider, J.; Venkatakrishnan, K.; von Moltke, L.L.; Harmatz, J.S.; Shader, R.I.; Greenblatt, D.J. O-and N-demethylation of venlafaxine in vitro by human liver microsomes and by microsomes from cDNA-transfected cells: Effect of metabolic inhibitors and SSRI antidepressants. Neuropsychopharmacology 1999, 20, 480–490. [Google Scholar] [CrossRef]
- Siccardi, M.; Marzolini, C.; Seden, K.; Almond, L.; Kirov, A.; Khoo, S.; Owen, A.; Back, D. Prediction of drug-drug interactions between various antidepressants and efavirenz or boosted protease inhibitors using a physiologically based pharmacokinetic modelling approach. Clin. Pharmacokinet. 2013, 52, 583–592. [Google Scholar] [CrossRef] [PubMed]
- De Buck, S.S.; Sinha, V.K.; Fenu, L.A.; Gilissen, R.A.; Mackie, C.E.; Nijsen, M.J. The prediction of drug metabolism, tissue distribution, and bioavailability of 50 structurally diverse compounds in rat using mechanism-based absorption, distribution, and metabolism prediction tools. Drug Metab. Dispos. 2007, 35, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Jones, H.M.; Parrott, N.; Jorga, K.; Lavé, T. A novel strategy for physiologically based predictions of human pharmacokinetics. Clin. Pharmacokinet. 2006, 45, 511–542. [Google Scholar] [CrossRef] [PubMed]
- Parrott, N.; Paquereau, N.; Coassolo, P.; Lave, T. An evaluation of the utility of physiologically based models of pharmacokinetics in early drug discovery. J. Pharm. Sci. 2005, 94, 2327–2343. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, S.; Skaptason, J.; Romero, D.; Vekich, S.; Jones, H.M.; Tan, W.; Wilner, K.D.; Koudriakova, T. Prediction of oral pharmacokinetics of cMet kinase inhibitors in humans: Physiologically based pharmacokinetic model versus traditional one compartment model. Drug Metab. Dispos. 2011, 39, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Sager, J.E.; Price, L.S.; Isoherranen, N. Stereoselective metabolism of bupropion to OHbupropion, threohydrobupropion, erythrohydrobupropion, and 4’-OH-bupropion in vitro. Drug Metab. Dispos. 2016, 44, 1709–1719. [Google Scholar] [CrossRef] [PubMed]
- Dash, R.P.; Rais, R.; Srinivas, N.R. Chirality and neuropsychiatric drugs: An update on stereoselective disposition and clinical pharmacokinetics of bupropion. Xenobiotica 2017, 13, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Hsyu, P.H.; Singh, A.; Giargiari, T.D.; Dunn, J.A.; Ascher, J.A.; Johnston, J.A. Pharmacokinetics of bupropion and its metabolites in cigarette smokers versus nonsmokers. J. Clin. Pharmacol. 1997, 37, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Hesse, L.M.; Greenblatt, D.J.; von Moltke, L.L.; Court, M.H. Ritonavir has minimal impact on the pharmacokinetic disposition of a single dose of bupropion administered to human volunteers. J. Clin. Pharmacol. 2006, 46, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Posner, J.; Bye, A.; Dean, K.; Peck, A.W.; Whiteman, P.D. The disposition of bupropion and its metabolites in healthy male volunteers after single and multiple doses. Eur. J. Clin. Pharmacol. 1985, 29, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, S.H.; McCann, S.M.; Masellis, M.; McIntyre, R.S.; Raskin, J.; McKay, G.; Baker, G.B. Combining bupropion SR with venlafaxine, paroxetine, or fluoxetine: A preliminary report on pharmacokinetic, therapeutic, and sexual dysfunction effects. J. Clin. Psychiatry 2002, 63, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Kim, H.D.; Na, H.S.; Lee, S.Y.; Seo, D.W.; Choi, J.Y.; Ha, J.H.; Shin, H.J.; Kim, Y.H.; Chung, M.W. The influences of CYP2D6 genotypes and drug interactions on the pharmacokinetics of venlafaxine: Exploring predictive biomarkers for treatment outcomes. Psychopharmacology 2015, 232, 1899–1909. [Google Scholar] [CrossRef] [PubMed]
- Kotlyar, M.; Brauer, L.H.; Tracy, T.S.; Hatsukami, D.K.; Harris, J.; Bronars, C.A.; Adson, D.E. Inhibition of CYP2D6 activity by bupropion. J. Clin. Psychopharmacol. 2005, 25, 226–229. [Google Scholar] [CrossRef] [PubMed]
- McCollum, D.L.; Greene, J.L.; McGuire, D.K. Severe sinus bradycardia after initiation of bupropion therapy: A probable drug-drug interaction with metoprolol. Cardiovasc. Drugs Ther. 2004, 18, 329–330. [Google Scholar] [CrossRef] [PubMed]
- Masters, A.R.; Gufford, B.T.; Lu, J.B.; Metzger, I.F.; Jones, D.R.; Desta, Z. Chiral plasma pharmacokinetics and urinary excretion of bupropion and metabolites in healthy volunteers. J. Pharmacol. Exp. Ther. 2016, 358, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Sager, J.E.; Tripathy, S.; Price, L.S.; Nath, A.; Chang, J.; Stephenson-Famy, A.; Isoherranen, N. In vitro to in vivo extrapolation of the complex drug-drug interaction of bupropion and its metabolites with CYP2D6; simultaneous reversible inhibition and CYP2D6 downregulation. Biochem. Pharmacol. 2017, 123, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Chenel, M.; Bouzom, F.; Cazade, F.; Ogungbenro, K.; Aarons, L.; Mentré, F. Drug-drug interaction predictions with PBPK models and optimal multiresponse sampling time designs: Application to midazolam and a phase I compound. Part 2: Clinical trial results. J. Pharmacokinet. Pharmacodyn. 2008, 35, 661–681. [Google Scholar] [CrossRef] [PubMed]
- Jones, H.M.; Dickins, M.; Youdim, K.; Gosset, J.R.; Attkins, N.J.; Hay, T.L.; Gurrell, I.K.; Logan, Y.R.; Bungay, P.J.; Jones, B.C.; et al. Application of PBPK modelling in drug discovery and development at Pfizer. Xenobiotica 2012, 42, 94–106. [Google Scholar] [CrossRef] [PubMed]
- Perdaems, N.; Blasco, H.; Vinson, C.; Chenel, M.; Whalley, S.; Cazade, F.; Bouzom, F. Predictions of metabolic drug-drug interactions using physiologically based modelling: Two cytochrome P450 3A4 substrates coadministered with ketoconazole or verapamil. Clin. Pharmacokinet. 2010, 49, 239–258. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Mao, J.; Hop, C.E. Physiologically based pharmacokinetic modeling to predict drug-drug interactions involving inhibitory metabolite: A case study of amiodarone. Drug Metab. Dispos. 2015, 43, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Kudo, T.; Hisaka, A.; Sugiyama, Y.; Ito, K. Analysis of the repaglinide concentration increase produced by gemfibrozil and itraconazole based on the inhibition of the hepatic uptake transporter and metabolic enzymes. Drug Metab. Dispos. 2013, 41, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Varma, M.V.S.; Lai, Y.; Kimoto, E.; Goosen, T.C.; El-Kattan, A.F.; Kumar, V. Mechanistic modeling to predict the transporter- and enzyme-mediated drug-drug interactions of repaglinide. Pharm. Res. 2013, 30, 1188–1199. [Google Scholar] [CrossRef] [PubMed]
- Loboz, K.K.; Gross, A.S.; Ray, J.; McLachlan, A.J. HPLC assay for bupropion and its major metabolites in human plasma. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2005, 823, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, T.; Shintani, S.; Hara, A. Multiplicity of mammalian reductases for xenobiotic carbonyl compounds. Drug Metab. Pharmacokinet. 2006, 21, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Daviss, W.B.; Perel, J.M.; Rudolph, G.R.; Axelson, D.A.; Gilchrist, R.; Nuss, S.; Birmaher, B.; Brent, D.A. Steady-state pharmacokinetics of bupropion SR in juvenile patients. J. Am. Acad. Child. Adolesc. Psychiatry 2005, 44, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Loboz, K.K.; Gross, A.S.; Williams, K.M.; Liauw, W.S.; Day, R.O.; Blievernicht, J.K.; Zanger, U.M.; McLachlan, A.J. Cytochrome P450 2B6 activity as measured by bupropion hydroxylation: Effect of induction by rifampin and ethnicity. Clin. Pharmacol. Ther. 2006, 80, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Palovaara, S.; Pelkonen, O.; Uusitalo, J.; Lundgren, S.; Laine, K. Inhibition of cytochrome P450 2B6 activity by hormone replacement therapy and oral contraceptive as measured by bupropion hydroxylation. Clin. Pharmacol. Ther. 2003, 74, 326–333. [Google Scholar] [CrossRef]
- Stewart, J.J.; Berkel, H.J.; Parish, R.C.; Simar, M.R.; Syed, A.; Bocchini, J.A., Jr.; Wilson, J.T.; Manno, J.E. Single-dose pharmacokinetics of bupropion in adolescents: Effects of smoking status and gender. J. Clin. Pharmacol. 2001, 41, 770–778. [Google Scholar] [CrossRef] [PubMed]
- Worrall, S.P.; Almond, M.K.; Dhillon, S. Pharmacokinetics of bupropion and its metabolites in haemodialysis patients who smoke. A single dose study. Nephron. Clin. Pract. 2004, 97, c83–c89. [Google Scholar] [CrossRef] [PubMed]
- Bondarev, M.L.; Bondareva, T.S.; Young, R.; Glennon, R.A. Behavioral and biochemical investigations of bupropion metabolites. Eur. J. Pharmacol. 2003, 474, 85–93. [Google Scholar] [CrossRef]
- Damaj, M.I.; Carroll, F.I.; Eaton, J.B.; Navarro, H.A.; Blough, B.E.; Mirza, S.; Lukas, R.J.; Martin, B.R. Enantioselective effects of hydroxy metabolites of bupropion on behavior and on function of monoamine transporters and nicotinic receptors. Mol. Pharmacol. 2004, 66, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Damaj, M.I.; Grabus, S.D.; Navarro, H.A.; Vann, R.E.; Warner, J.A.; King, L.S.; Wiley, J.L.; Blough, B.E.; Lukas, R.J.; Carroll, F.I. Effects of hydroxymetabolites of bupropion on nicotine dependence behavior in mice. J. Pharmacol. Exp. Ther. 2010, 334, 1087–1095. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.Z.; Cox, L.S.; Nollen, N.; Faseru, B.; Okuyemi, K.S.; Ahluwalia, J.S.; Benowitz, N.L.; Tyndale, R.F. CYP2B6 and bupropion’s smoking-cessation pharmacology: The role of hydroxybupropion. Clin. Pharmacol. Ther. 2012, 92, 771–777. [Google Scholar] [CrossRef] [PubMed]
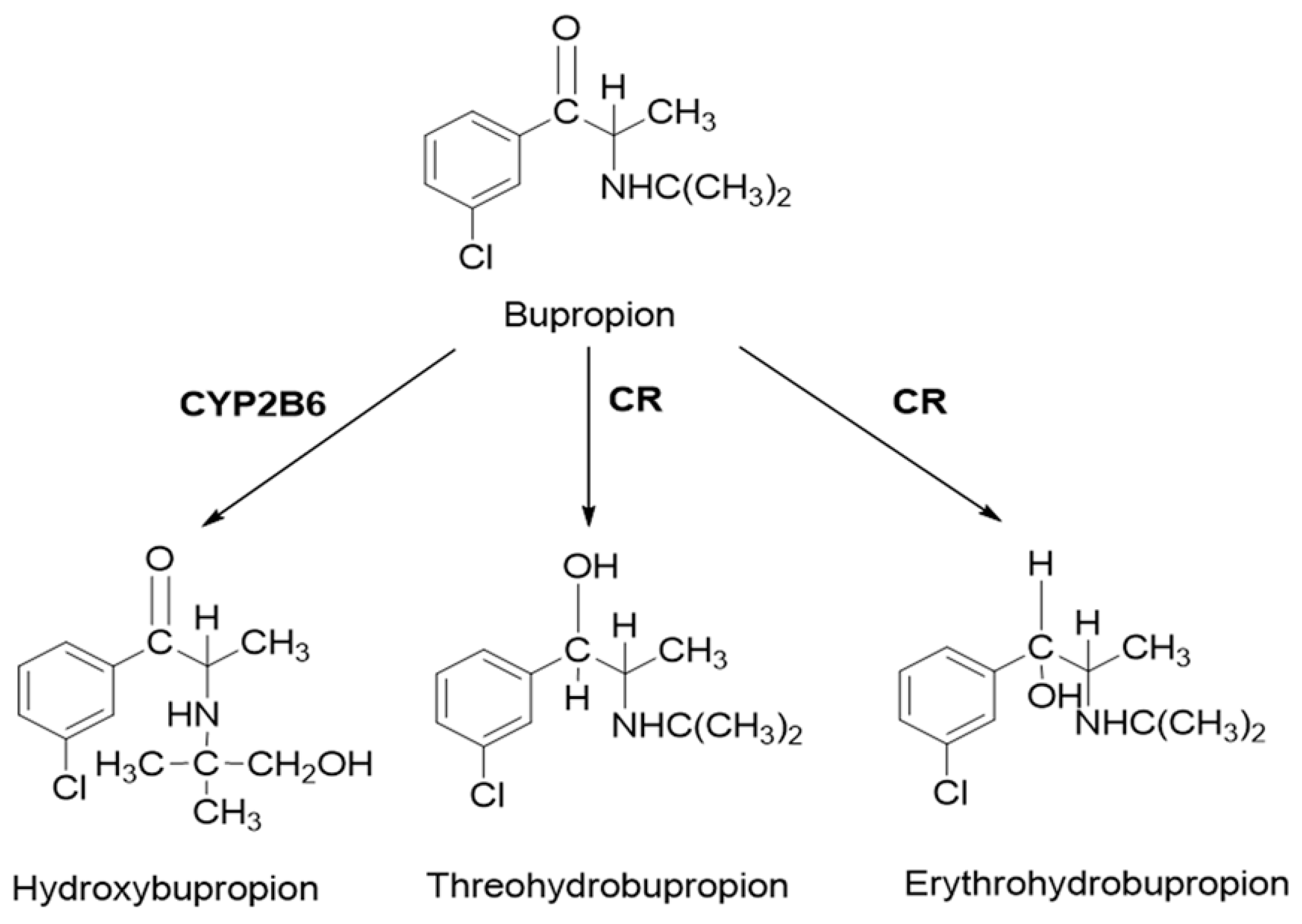


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Bupropion | |
|---|---|---|
| Value | References/Comments | |
| Mol weight (g/mol) | 239.74 | Drug bank |
| Log Po:w | 3.28 | Drug bank |
| pKa | 8.22 | Drug bank |
| B/P | 0.82 | [29] |
| fu,p | 0.16 | [28] |
| fa | 1 | [26] |
| ka (h−1) | 0.8 | [34] |
| Tlag (h) | 0.8 | [31] |
| Kp scalar | 5.4 | Simcyp best fit |
| Vss (L/kg) | 19 | [31] |
| Enzyme | CYP2B6 | Metabolite: hydroxybupropion |
| Vmax (pmol/min per milligram) | 3623 | [27] |
| Km (μM) | 89 | [27] |
| fu,mic | 0.16 | Assumed = fu,p |
| Enzyme | CYP2B6 | Metabolite: threohydrobupropion |
| Vmax (pmol/min per milligram) | 98.4 | [33] |
| Km (μM) | 186.3 | [33] |
| fu,mic | 0.003 | Simcyp best fit, correct expression of carbonyl reductase |
| Enzyme | CYP2B6 | Metabolite: erythrohydrobupropion |
| Vmax (pmol/min per milligram) | 2.6 | [33] |
| Km (μM) | 41.4 | [33] |
| fu,mic | 0.003 | Simcyp best fit, correct expression of carbonyl reductase |
| Parameter | Hydroxybupropion | Threohydrobupropion | Erythrohydrobupropion | |||
|---|---|---|---|---|---|---|
| Value | References/Comments | Value | References/Comments | Value | References/Comments | |
| Mol weight (g/mol) | 255.74 | ACD-ilab | 241.757 | ACD-ilab | 241.757 | ACD-ilab |
| Log Po:w | 2.03 | ACD-ilab | 2.88 | ACD-ilab | 2.88 | ACD-ilab |
| pKa | 7.4 | ACD-ilab | 7.4 | ACD-ilab | 9.6 | ACD-ilab |
| B/P | 0.82 | Assigned using bupropion value | 0.82 | Assigned using bupropion value | 0.82 | Assigned using bupropion value |
| fu,p | 0.23 | [28] | 0.58 | [28] | 0.58 | [28] |
| Vsac (L/kg) | 0.5 | Simcyp best fit | 5.83 | Simcyp best fit | N/A | |
| Vss (L/kg) | 2.15 | Predicted with Rogers method | 9.11 | Predicted with Rogers method | 1.47 | Predicted with Rogers method |
| Kp scalar | 1 | Simcyp default value | 1 | Simcyp default value | 2 | Simcyp best fit |
| CLpo (L/h) | 5.76 | Simcyp best fit | 21.15 | Simcyp best fit | 21.69 | Simcyp best fit |
| Parameter | Venlafaxine | |
|---|---|---|
| Value | References/Comments | |
| Mol weight (g/mol) | 277.402 | [40] |
| Log Po:w | 2.8 | [40] |
| pKa | 9.4 | [40] |
| B/P | 1.17 | [40] |
| fu,p | 0.73 | [40] |
| fa | 0.92 | [37] |
| ka (h−1) | 1.31 | [38] |
| Tlag (h) | 1.44 | Simcyp best fit |
| Kp scalar | 2.3 | Predicted with Poulin and Theil method |
| Vss (L/kg) | 7 | [38] |
| Enzyme | CYP2D6 | |
| CLint (µL/min/pmol of isoform) | 5.825 | Retrograde calculation in Simcyp to account for 80% Hep CL from CYP2D6 |
| CLint-additional (µL/min/mg protein) | 11.65 | Simcyp predicted |
| Parameter | Bupropion | Hydroxybupropion | Threohydrobupropion | Erythrohydrobupropion |
|---|---|---|---|---|
| Ki (μM) | 21 | 13 | 5.4 | 1.7 |
| Inhibitors | AUC Ratio | Cmax Ratio | Tmax Ratio |
|---|---|---|---|
| Bupropion + Desipramine (observed) | 5.2 | 1.9 | 2 |
| Bupropion (predicted) | 2.27 | 1.15 | 1.10 |
| Hydroxybupropion (predicted) | 4.58 | 1.76 | 1.84 |
| Threohydrobupropion (predicted) | 3.47 | 1.61 | 1.47 |
| Erythrohydrobupropion (predicted) | 2.83 | 1.45 | 1.47 |
| Bup + H-Bup + T-Bup + E-Bup (predicted) | 5.05 | 1.79 | 1.84 |
| Bupropion + Venlafaxine (observed) | N/A | 2.5 | N/A |
| Bupropion (predicted) | 1.30 | 1.27 | 1 |
| Hydroxybupropion (predicted) | 2.49 | 1.94 | 1 |
| Threohydrobupropion (predicted) | 2.14 | 1.80 | 1 |
| Erythrohydrobupropion (predicted) | 1.76 | 1.60 | 1 |
| Bup + H-Bup + T-Bup + E-Bup (predicted) | 3.03 | 2.24 | 1 |
| Substrate | AUC Ratio | Cmax Ratio |
|---|---|---|
| Bufuralol | 2.04 | 1.55 |
| Tolterodine | 2.91 | 2.17 |
| Metoprolol | 3.53 | 2.57 |
| Dextromethorphan | 4.06 | 3.05 |
| Parameter | Value | References/Comments | |
|---|---|---|---|
| R-BUP | |||
| Clint (μL/min per pmol) | |||
| CYP2B6 | 12 | Metabolite: RR-OHBUP | Retrograde calculation in Simcyp to account for 34% of total CL [46] |
| CYP2C19 | 5.36 | ||
| CYP3A4 | 0.58 | ||
| CYP2J2 | 27 | Metabolite: RR-TB | Retrograde calculation in Simcyp to account for 50% of total CL [46] |
| CYP2J2 | 4.24 | Metabolite: SR-EB | Retrograde calculation in Simcyp to account for 8% of total CL [46] |
| CYP2C19 | 4.24 | Metabolite: R-4′-OHBUP | Retrograde calculation in Simcyp to account for 8% of total CL [46] |
| S-BUP | |||
| Clint (μL/min per pmol) | |||
| CYP2B6 | 20.56 | Metabolite: SS-OHBUP | Retrograde calculation in Simcyp to account for 12% of total CL [46] |
| CYP2C19 | 12.61 | ||
| CYP3A4 | 1.37 | ||
| CYP2J2 | 236.16 | Metabolite: SS-TB | Retrograde calculation in Simcyp to account for 82% of total CL [46] |
| CYP2J2 | 11.52 | Metabolite: RS-EB | Retrograde calculation in Simcyp to account for 4% of total CL [46] |
| CYP2C19 | 5.76 | Metabolite: S- 4’-OHBUP | Retrograde calculation in Simcyp to account for 2% of total CL [46] |
| RR-OHBUP | |||
| CLpo (L/h) | 6.76 | Simcyp best fit | |
| SS-OHBUP | |||
| Vss (L/kg) | 10.5 | Predicted with Rogers method | |
| Kp scalar | 5 | Simcyp best fit | |
| CLpo (L/h) | 305.8 | Simcyp best fit | |
| RR-TB | |||
| Vss (L/kg) | 4.7 | Predicted with Poulin and Theil method | |
| Kp scalar | 1 | Simcyp default value | |
| CLpo (L/h) | 20 | Simcyp best fit | |
| SS-TB | |||
| Vss (L/kg) | 4.7 | Predicted with Poulin and Theil method | |
| Kp scalar | 1 | Simcyp default value | |
| CLpo (L/h) | 120 | Simcyp best fit | |
| SR-EB | |||
| Vss (L/kg) | 3.07 | Predicted with Poulin and Theil method | |
| Kp scalar | 1 | Simcyp default value | |
| CLpo (L/h) | 11.69 | Simcyp best fit | |
| RS-EB | |||
| Vss (L/kg) | 9.08 | Predicted with Poulin and Theil method | |
| Kp scalar | 3 | Simcyp best fit | |
| CLpo (L/h) | 52 | Simcyp best fit | |
| PK Parameter | AUC (nM·h) | Cmax (nM) | ||
|---|---|---|---|---|
| Predicted | Observed [54] | Predicted | Observed [54] | |
| R-BUP | 1343.68 | 1162 | 196.37 | 288 |
| S-BUP | 291.27 | 193 | 53.20 | 47 |
| RR-OHBUP | 37,777.63 | 37,421 | 1564.59 | 1240 |
| SS-OHBUP | 524.75 | 580 | 33.85 | 35.9 |
| RR-TB | 3228.59 | 3326 | 117.19 | 79.9 |
| SS-TB | 1813.4 | 1433 | 159.34 | 168 |
| SR-EB | 872.65 | 942 | 33.31 | 30.5 |
| RS-EB | 195.48 | 185 | 8.12 | 10.6 |
| Inhibitors | Ki | AUC Ratio | Cmax Ratio | Tmax Ratio |
|---|---|---|---|---|
| Bupropion + Desipramine (observed) | 5.2 | 1.9 | 2 | |
| R-BUP + RR-OHBUP + EB + TB (predicted) | 2.53 | 1.21 | 1.47 | |
| S-BUP + SS-OHBUP + EB + TB (predicted) | 1.93 | 1.03 | 1.10 | |
| R-BUP (predicted) | 12.5 | 1.83 | 0.96 | 1.10 |
| S-BUP (predicted) | 0.91 | 1.84 | 0.97 | 1.10 |
| RR-OHBUP (predicted) | 1.5 | 2.45 | 1.19 | 1.47 |
| SS-OHBUP (predicted) | 4.3 | 1.84 | 0.97 | 1.10 |
| Threohydrobupropion (predicted) | 3.97 | 1.88 | 0.99 | 1.10 |
| Erythrohydrobupropion (predicted) | 0.91 | 1.87 | 0.98 | 1.10 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xue, C.; Zhang, X.; Cai, W. Prediction of Drug-Drug Interactions with Bupropion and Its Metabolites as CYP2D6 Inhibitors Using a Physiologically-Based Pharmacokinetic Model. Pharmaceutics 2018, 10, 1. https://doi.org/10.3390/pharmaceutics10010001
Xue C, Zhang X, Cai W. Prediction of Drug-Drug Interactions with Bupropion and Its Metabolites as CYP2D6 Inhibitors Using a Physiologically-Based Pharmacokinetic Model. Pharmaceutics. 2018; 10(1):1. https://doi.org/10.3390/pharmaceutics10010001
Chicago/Turabian StyleXue, Caifu, Xunjie Zhang, and Weimin Cai. 2018. "Prediction of Drug-Drug Interactions with Bupropion and Its Metabolites as CYP2D6 Inhibitors Using a Physiologically-Based Pharmacokinetic Model" Pharmaceutics 10, no. 1: 1. https://doi.org/10.3390/pharmaceutics10010001
APA StyleXue, C., Zhang, X., & Cai, W. (2018). Prediction of Drug-Drug Interactions with Bupropion and Its Metabolites as CYP2D6 Inhibitors Using a Physiologically-Based Pharmacokinetic Model. Pharmaceutics, 10(1), 1. https://doi.org/10.3390/pharmaceutics10010001