Mitochondria-Targeted Drug Delivery in Cardiovascular Disease: A Long Road to Nano-Cardio Medicine



Abstract
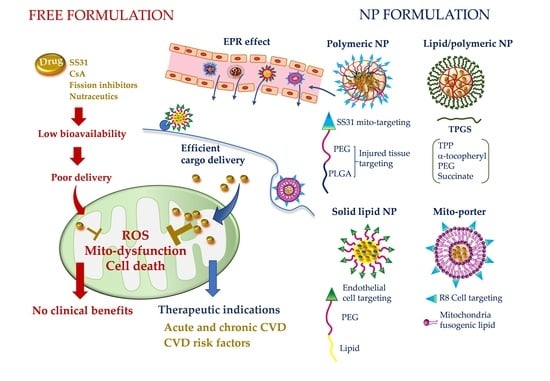
1. Introduction
2. Mitochondria-Targeted Antioxidants
2.1. Coenzyme Q10 (CoQ10)
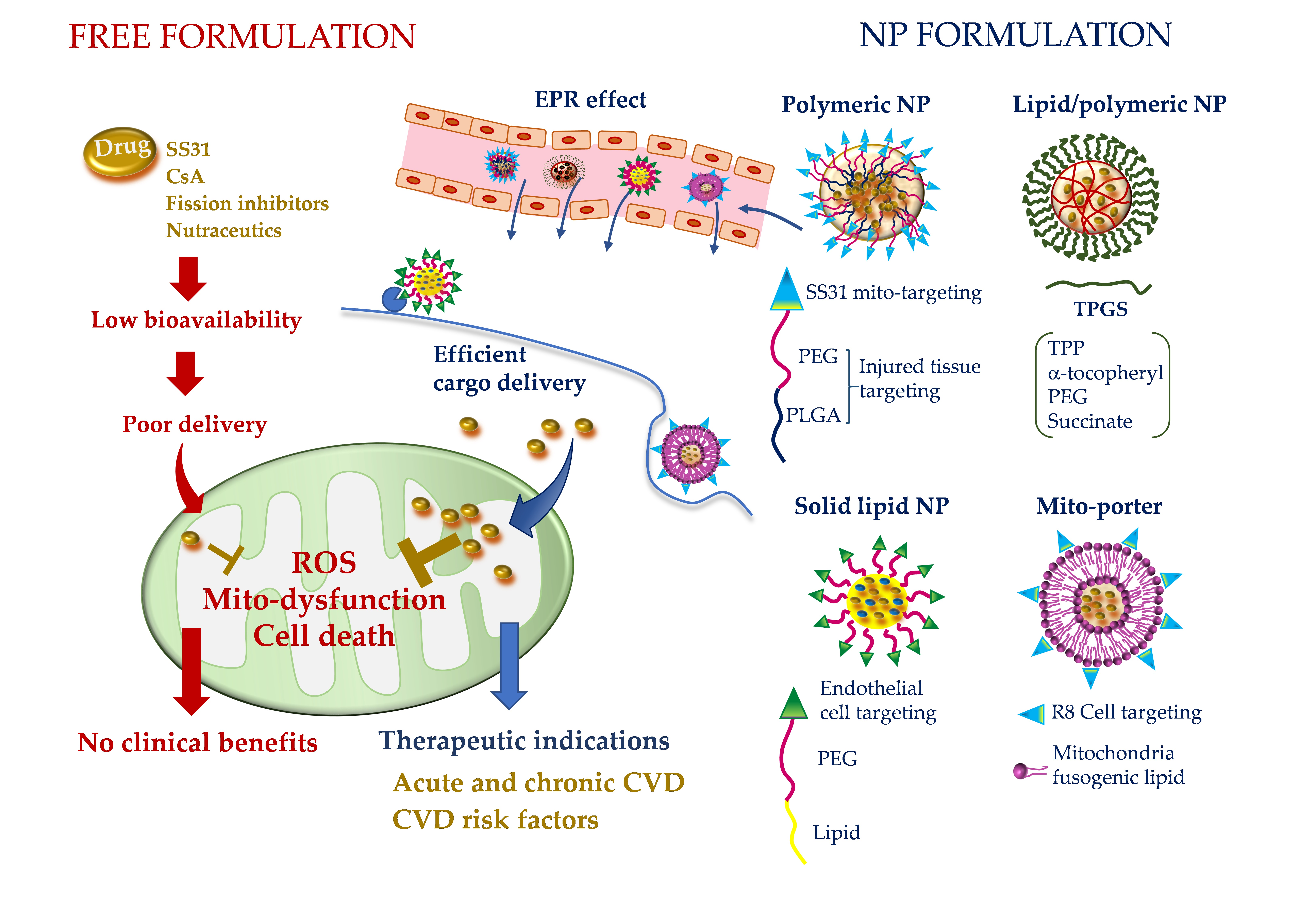
2.2. Mitoquinone (MitoQ)
2.3. 10-(6’-Plastoquinonyl)-Decyltriphenylphosphonium (SkQ1)
2.4. Mito-Tempo (MT)
2.5. Szeto–Schiller 31 (SS31)
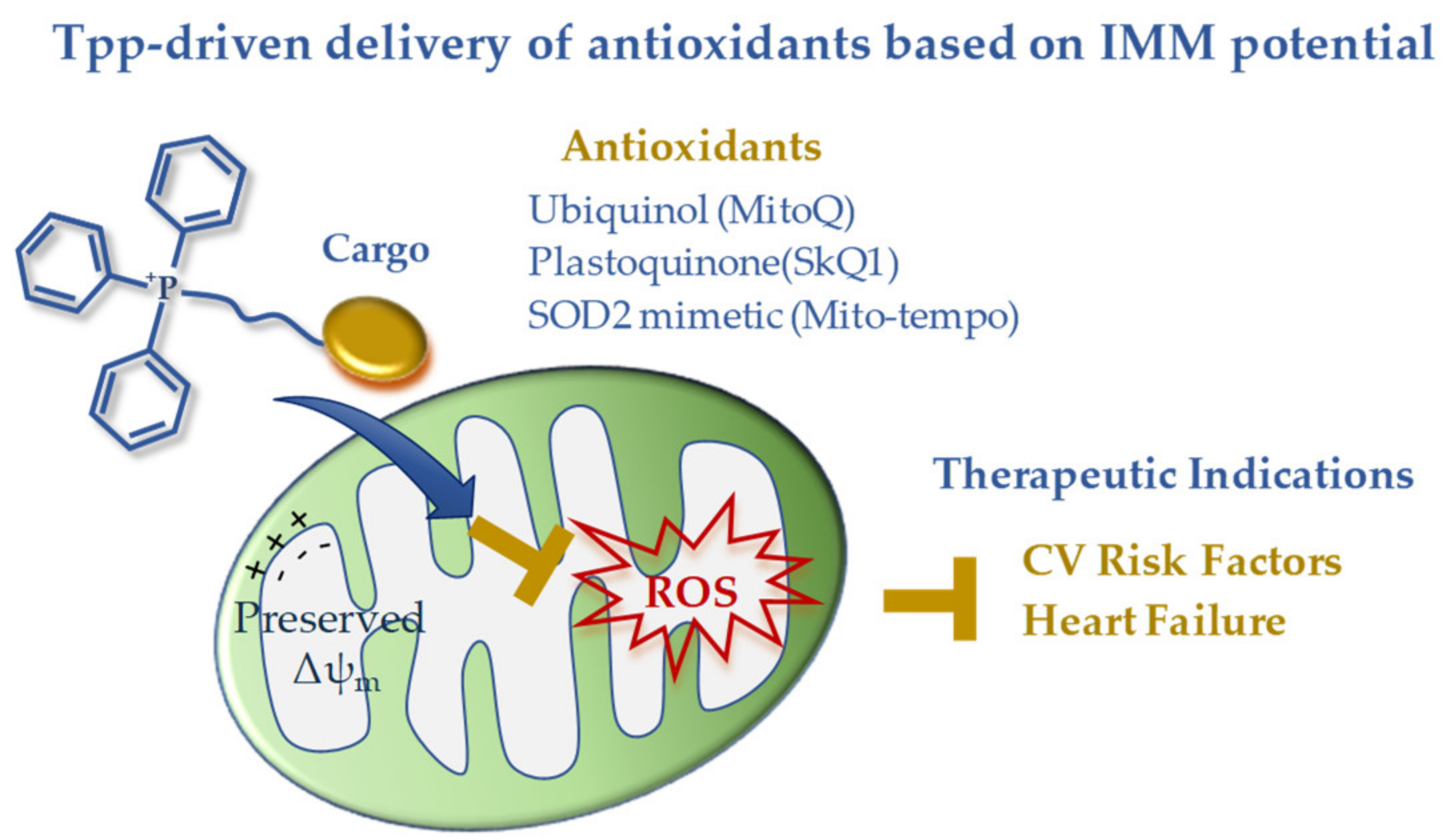
3. Inhibition of MPTP Opening
4. Mitochondria-Targeted Donors of Nitric Oxide (NO) and Hydrogen Sulfide (H2S)
4.1. MitoSNO
4.2. AP39
4.3. Isothiocyanate Derivatives
5. Inhibitors of Mitochondrial Fission
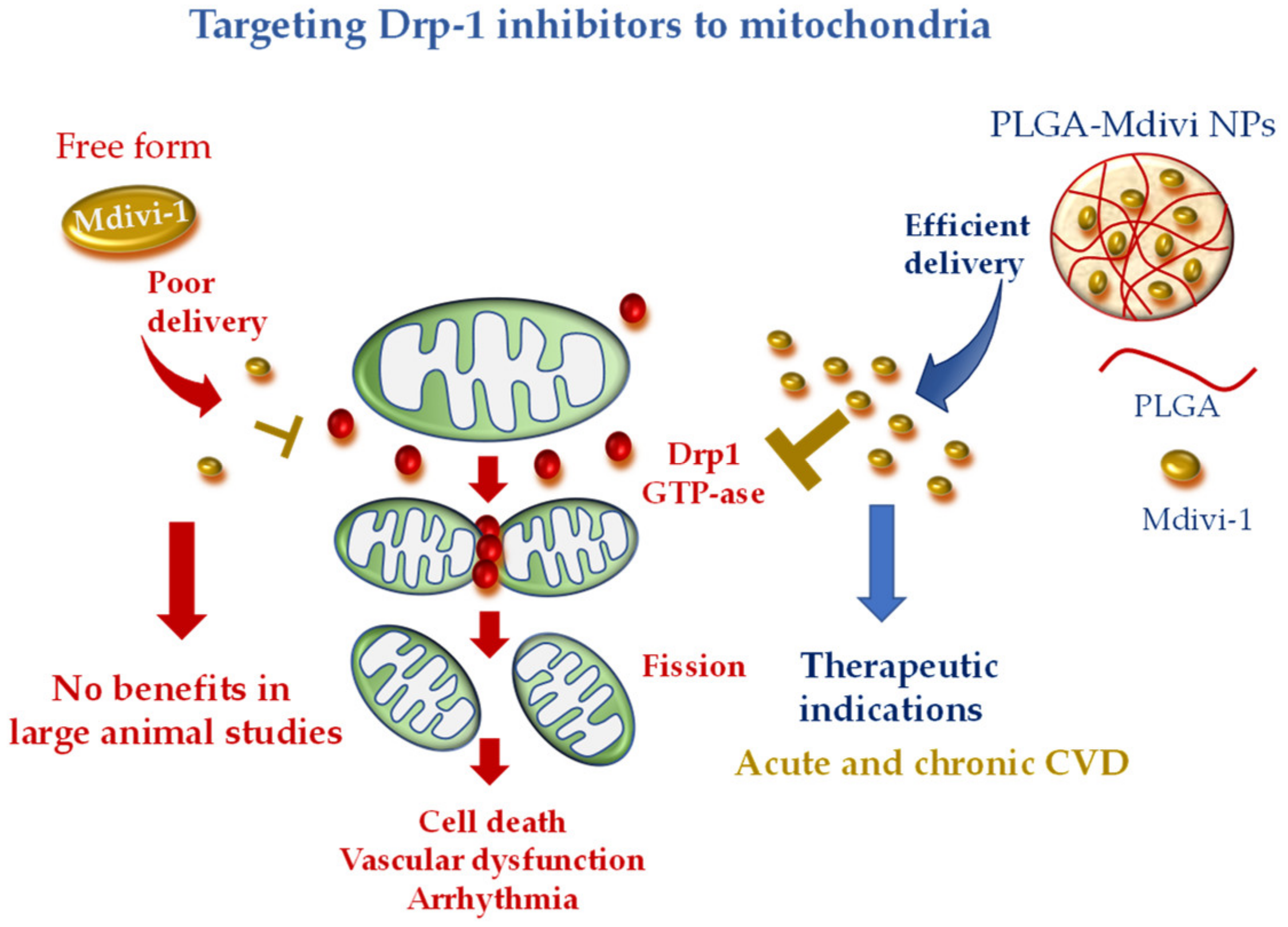
5.1. Mitochondrial Division Inhibitor 1 (Mdivi-1)
5.2. Drp1 inhibitor 1 (Driptor1) and Drp1 inhibitor 1a (Driptor1a)
6. Mitochondria-Targeting of Natural Compounds with Pleiotropic Effects
6.1. Resveratrol
6.2. Quercetin
6.3. Isosteviol
6.4. Tanshinone
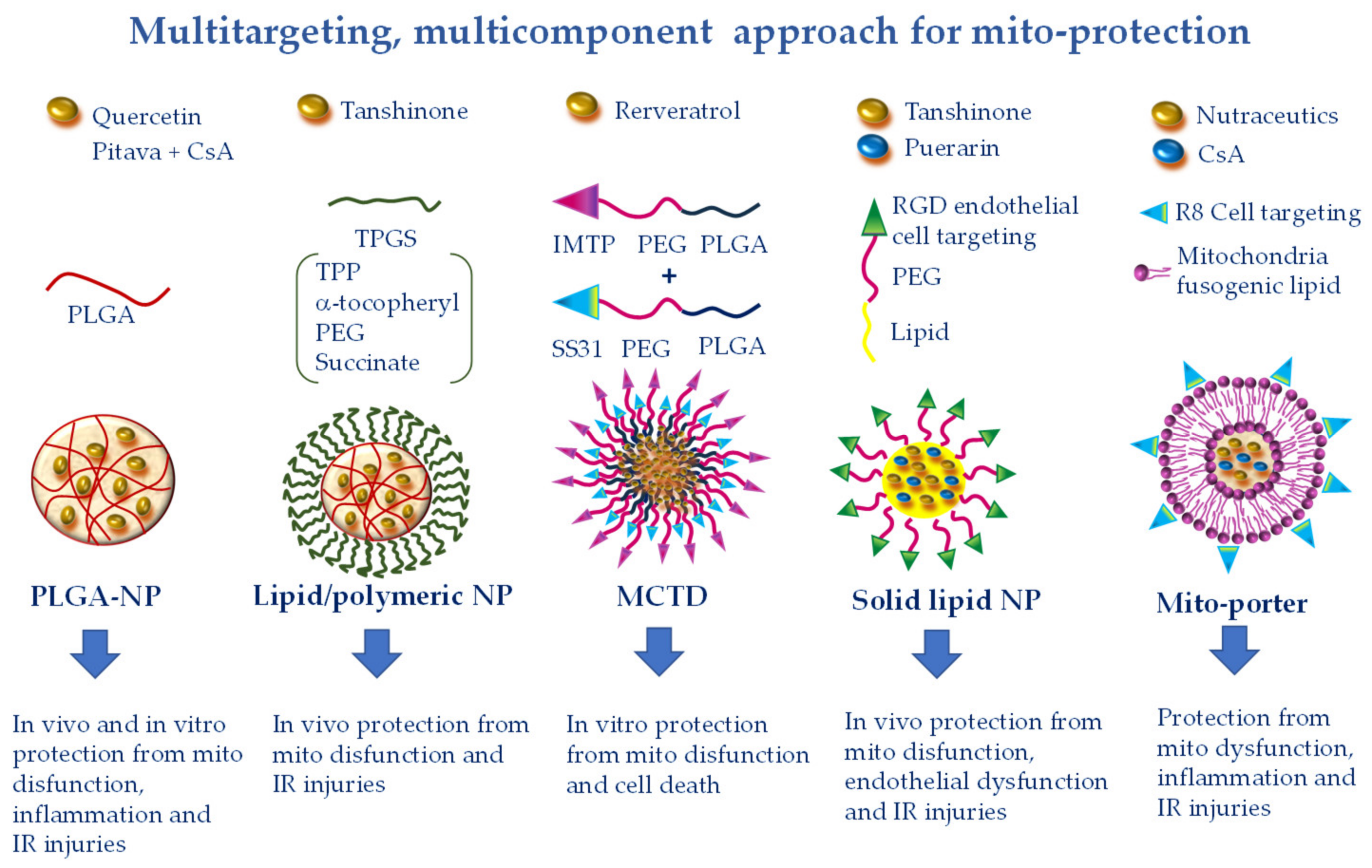
7. Simultaneous Drug Delivery for a More Efficient Combination Therapy
8. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Forini, F.; Nicolini, G.; Kusmic, C.; Iervasi, G. Protective Effects of Euthyroidism Restoration on Mitochondria Function and Quality Control in Cardiac Pathophysiology. Int. J. Mol. Sci. 2019, 20, 3377. [Google Scholar] [CrossRef]
- Forini, F.; Pitto, L.; Nicolini, G. Thyroid hormone, mitochondrial function and cardioprotection. In Thyroid and Heart. A comprehensive Translational Essay; Iervasi, G., Pingitore, A., Gerdes, A.M., Razvi, S., Eds.; Springer: Cham, Switzerland, 2020; pp. 109–126. [Google Scholar]
- Forini, F.; Nicolini, G.; Iervasi, G. Mitochondria as key targets of cardioprotection in cardiac ischemic disease: Role of thyroid hormone triiodothyronine. Int. J. Mol. Sci. 2015, 16, 6312–6336. [Google Scholar] [CrossRef]
- Dhingra, R.; Guberman, M.; Rabinovich-Nikitin, I.; Gerstein, J.; Margulets, V.; Gang, H.; Madden, N.; Thliveris, J.; Kirshenbaum, L.A. Impaired NF-κB signalling underlies cyclophilin D-mediated mitochondrial permeability transition pore opening in doxorubicin cardiomyopathy. Cardiovasc. Res. 2020, 116, 1161–1174. [Google Scholar] [CrossRef]
- Moon, S.H.; Liu, X.; Cedars, A.M.; Yang, K.; Kiebish, M.A.; Joseph, S.M.; Kelley, J.; Jenkins, C.M.; Gross, R.W. Heart failure-induced activation of phospholipase iPLA2γ generates hydroxyeicosatetraenoic acids opening the mitochondrial permeability transition pore. J. Biol. Chem. 2018, 293, 115–129. [Google Scholar] [CrossRef]
- Sesso, H.D.; Buring, J.E.; Christen, W.G.; Kurth, T.; Belanger, C.; MacFadyen, J.; Bubes, V.; Manson, J.E.; Glynn, R.J.; Gaziano, J.M. Vitamins E and C in the prevention of cardiovascular disease in men: The Physicians’ Health Study II randomized controlled trial. JAMA 2008, 300, 2123–2133. [Google Scholar] [CrossRef]
- Song, Y.; Cook, N.R.; Albert, C.M.; Van Denburgh, M.; Manson, J.E. Effects of vitamins C and E and beta-carotene on the risk of type 2 diabetes in women at high risk of cardiovascular disease: A randomized controlled trial. Am. J. Clin. Nutr. 2009, 90, 429–437. [Google Scholar] [CrossRef]
- Schmidt, H.H.; Stocker, R.; Vollbracht, C.; Paulsen, G.; Riley, D.; Daiber, A.; Cuadrado, A. Antioxidants in Translational Medicine. Antioxid. Redox Signal. 2015, 23, 1130–1143. [Google Scholar] [CrossRef]
- Adlam, V.J.; Harrison, J.C.; Porteous, C.M.; James, A.M.; Smith, R.A.; Murphy, M.P.; Sammut, I.A. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J. 2005, 19, 1088–1095. [Google Scholar] [CrossRef] [PubMed]
- Dare, A.J.; Logan, A.; Prime, T.A.; Rogatti, S.; Goddard, M.; Bolton, E.M.; Bradley, J.A.; Pettigrew, G.J.; Murphy, M.P.; Saeb-Parsy, K. The mitochondria-targeted anti-oxidant MitoQ decreases ischemia-reperfusion injury in a murine syngeneic heart transplant model. J. Heart Lung Transplant. 2015, 34, 1471–1480. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.; Huynh, N.N.; Hamilton, C.A.; Beattie, E.; Smith, R.A.; Cochemé, H.M.; Murphy, M.P.; Dominiczak, A.F. Mitochondria-targeted antioxidant MitoQ10 improves endothelial function and attenuates cardiac hypertrophy. Hypertension 2009, 54, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Goh, K.Y.; He, L.; Song, J.; Jinno, M.; Rogers, A.J.; Sethu, P.; Halade, G.V.; Rajasekaran, N.S.; Liu, X.; Prabhu, S.D.; et al. Mitoquinone ameliorates pressure overload-induced cardiac fibrosis and left ventricular dysfunction in mice. Redox Biol. 2019, 21, 101100. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Song, J.; Ernst, P.; Latimer, M.N.; Ha, C.M.; Goh, K.Y.; Ma, W.; Rajasekaran, N.S.; Zhang, J.; Liu, X.; et al. MitoQ regulates redox-related noncoding RNAs to preserve mitochondrial network integrity in pressure-overload heart failure. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H682–H695. [Google Scholar] [CrossRef] [PubMed]
- Rossman, M.J.; Santos-Parker, J.R.; Steward, C.A.C.; Bispham, N.Z.; Cuevas, L.M.; Rosenberg, H.L.; Woodward, K.A.; Chonchol, M.; Gioscia-Ryan, R.A.; Murphy, M.P.; et al. Chronic Supplementation with a Mitochondrial Antioxidant (MitoQ) Improves Vascular Function in Healthy Older Adults. Hypertension 2018, 71, 1056–1063. [Google Scholar] [CrossRef] [PubMed]
- Firsov, A.M.; Kotova, E.A.; Orlov, V.N.; Antonenko, Y.N.; Skulachev, V.P. A mitochondria-targeted antioxidant can inhibit peroxidase activity of cytochrome c by detachment of the protein from liposomes. FEBS Lett. 2016, 590, 2836–2843. [Google Scholar] [CrossRef] [PubMed]
- Antonenko, Y.N.; Avetisyan, A.V.; Bakeeva, L.E.; Chernyak, B.V.; Chertkov, V.A.; Domnina, L.V.; Ivanova, O.Y.; Izyumov, D.S.; Khailova, L.S.; Klishin, S.S.; et al. Mitochondria-targeted plastoquinone derivatives as tools to interrupt execution of the aging program. 1. Cationic plastoquinone derivatives: Synthesis and in vitro studies. Biochemistry 2008, 73, 1273–1287. [Google Scholar] [CrossRef] [PubMed]
- Bakeeva, L.E.; Barskov, I.V.; Egorov, M.V.; Isaev, N.K.; Kapelko, V.I.; Kazachenko, A.V.; Kirpatovsky, V.I.; Kozlovsky, S.V.; Lakomkin, V.L.; Levina, S.B.; et al. Mitochondria-targeted plastoquinone derivatives as tools to interrupt execution of the aging program. 2. Treatment of some ROS- and age-related diseases (heart arrhythmia, heart infarctions, kidney ischemia, and stroke). Biochemistry 2008, 73, 1288–1299. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.B.; Meng, Y.H.; Chang, S.; Zhang, R.Y.; Shi, C. High fructose causes cardiac hypertrophy via mitochondrial signaling pathway. Am. J. Transl. Res. 2016, 8, 4869–4880. [Google Scholar]
- Agarkov, A.A.; Popova, T.N.; Boltysheva, Y.G. Influence of 10-(6-plastoquinonyl) decyltriphenylphosphonium on free-radical homeostasis in the heart and blood serum of rats with streptozotocin-induced hyperglycemia. World J. Diabetes 2019, 10, 546–559. [Google Scholar] [CrossRef]
- Shabalina, I.G.; Vyssokikh, M.Y.; Gibanova, N.; Csikasz, R.I.; Edgar, D.; Hallden-Waldemarson, A.; Rozhdestvenskaya, Z.; Bakeeva, L.E.; Vays, V.B.; Pustovidko, A.V.; et al. Improved health-span and lifespan in mtDNA mutator mice treated with the mitochondrially targeted antioxidant SkQ1. Aging 2017, 9, 315–339. [Google Scholar] [CrossRef]
- Pung, Y.F.; Rocic, P.; Murphy, M.P.; Smith, R.A.; Hafemeister, J.; Ohanyan, V.; Guarini, G.; Yin, L.; Chilian, W.M. Resolution of mitochondrial oxidative stress rescues coronary collateral growth in Zucker obese fatty rats. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 325–334. [Google Scholar] [CrossRef]
- Luo, M.; Guan, X.; Luczak, E.D.; Lang, D.; Kutschke, W.; Gao, Z.; Yang, J.; Glynn, P.; Sossalla, S.; Swaminathan, P.D.; et al. Diabetes increases mortality after myocardial infarction by oxidizing CaMKII. J. Clin. Investig. 2013, 123, 1262–1274. [Google Scholar] [CrossRef] [PubMed]
- Ni, R.; Cao, T.; Xiong, S.; Ma, J.; Fan, G.C.; Lacefield, J.C.; Lu, Y.; Le Tissier, S.; Peng, T. Therapeutic inhibition of mitochondrial reactive oxygen species with mito-TEMPO reduces diabetic cardiomyopathy. Free Radic. Biol. Med. 2016, 90, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Jeong, E.M.; Chung, J.; Liu, H.; Go, Y.; Gladstein, S.; Farzaneh-Far, A.; Lewandowski, E.D.; Dudley, S.C., Jr. Role of Mitochondrial Oxidative Stress in Glucose Tolerance, Insulin Resistance, and Cardiac Diastolic Dysfunction. J. Am. Heart Assoc. 2016, 5, e003046. [Google Scholar] [CrossRef] [PubMed]
- Dikalova, A.E.; Bikineyeva, A.T.; Budzyn, K.; Nazarewicz, R.R.; McCann, L.; Lewis, W.; Harrison, D.G.; Dikalov, S.I. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ. Res. 2010, 107, 106–116. [Google Scholar] [CrossRef]
- Sovari, A.A.; Rutledge, C.A.; Jeong, E.M.; Dolmatova, E.; Arasu, D.; Liu, H.; Vahdani, N.; Gu, L.; Zandieh, S.; Xiao, L.; et al. Mitochondria oxidative stress, connexin43 remodeling, and sudden arrhythmic death. Circ. Arrhythmia Electrophysiol. 2013, 6, 623–631. [Google Scholar] [CrossRef]
- Dikalov, S.I.; Nazarewicz, R.R.; Bikineyeva, A.; Hilenski, L.; Lassègue, B.; Griendling, K.K.; Harrison, D.G.; Dikalova, A.E. Nox2-induced production of mitochondrial superoxide in angiotensin II-mediated endothelial oxidative stress and hypertension. Antioxid. Redox Signal. 2014, 20, 281–294. [Google Scholar] [CrossRef]
- Cao, T.; Fan, S.; Zheng, D.; Wang, G.; Yu, Y.; Chen, R.; Song, L.S.; Fan, G.C.; Zhang, Z.; Peng, T. Increased calpain-1 in mitochondria induces dilated heart failure in mice: Role of mitochondrial superoxide anion. Basic Res. Cardiol. 2019, 114, 17. [Google Scholar] [CrossRef]
- Hoshino, A.; Okawa, Y.; Ariyoshi, M.; Kaimoto, S.; Uchihashi, M.; Fukai, K.; Iwai-Kanai, E.; Matoba, S. Oxidative post-translational modifications develop LONP1 dysfunction in pressure overload heart failure. Circ. Heart Fail. 2014, 7, 500–509. [Google Scholar] [CrossRef]
- Dey, S.; DeMazumder, D.; Sidor, A.; Foster, D.B.; O’Rourke, B. Mitochondrial ROS Drive Sudden Cardiac Death and Chronic Proteome Remodeling in Heart Failure. Circ Res. 2018, 123, 356–371. [Google Scholar] [CrossRef]
- Kizhakekuttu, T.J.; Wang, J.; Dharmashankar, K.; Ying, R.; Gutterman, D.D.; Vita, J.A.; Widlansky, M.E. Adverse alterations in mitochondrial function contribute to type 2 diabetes mellitus-related endothelial dysfunction in humans. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2531–2539. [Google Scholar] [CrossRef]
- Prime, T.A.; Blaikie, F.H.; Evans, C.; Nadtochiy, S.M.; James, A.M.; Dahm, C.C.; Vitturi, D.A.; Patel, R.P.; Hiley, C.R.; Abakumova, I.; et al. A Mitochondria-Targeted S-Nitrosothiol Modulates Respiration, Nitrosates Thiols, and Protects against Ischemia-Reperfusion Injury. Proc. Natl. Acad. Sci. USA 2009, 106, 10764–10769. [Google Scholar] [CrossRef] [PubMed]
- Chouchani, E.T.; Hurd, T.R.; Nadtochiy, S.M.; Brookes, P.S.; Fearnley, I.M.; Lilley, K.S.; Smith, R.A.; Murphy, M.P. Identification of S-nitrosated mitochondrial proteins by S-nitrosothiol difference in gel electrophoresis (SNO-DIGE): Implications for the regulation of mitochondrial function by reversible S-nitrosation. Biochem. J. 2010, 430, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Chouchani, E.T.; Methner, C.; Nadtochiy, S.M.; Logan, A.; Pell, V.R.; Ding, S.; James, A.M.; Cochemé, H.M.; Reinhold, J.; Lilley, K.S.; et al. Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat. Med. 2013, 19, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Methner, C.; Chouchani, E.T.; Buonincontri, G.; Pell, V.R.; Sawiak, S.J.; Murphy, M.P.; Krieg, T. Mitochondria selective S-nitrosation by mitochondria-targeted S-nitrosothiol protects against post-infarct heart failure in mouse hearts. Eur. J. Heart Fail. 2014, 16, 712–717. [Google Scholar] [CrossRef]
- Gerő, D.; Torregrossa, R.; Perry, A.; Waters, A.; Le-Trionnaire, S.; Whatmore, J.L.; Wood, M.; Whiteman, M. The novel mitochondria-targeted hydrogen sulfide (H2S) donors AP123 and AP39 protect against hyperglycemic injury in microvascular endothelial cells in vitro. Pharmacol. Res. 2016, 113, 186–198. [Google Scholar] [CrossRef]
- Chatzianastasiou, A.; Bibli, S.I.; Andreadou, I.; Efentakis, P.; Kaludercic, N.; Wood, M.E.; Whiteman, M.; Di Lisa, F.; Daiber, A.; Manolopoulos, V.G.; et al. Cardioprotection by H2S Donors: Nitric Oxide-Dependent and Independent Mechanisms. J. Pharmacol. Exp. Ther. 2016, 358, 431–440. [Google Scholar] [CrossRef]
- Ikeda, K.; Marutani, E.; Hirai, S.; Wood, M.E.; Whiteman, M.; Ichinose, F. Mitochondria-targeted hydrogen sulfide donor AP39 improves neurological outcomes after cardiac arrest in mice. Nitric Oxide 2015, 49, 90–96. [Google Scholar] [CrossRef]
- Karwi, Q.G.; Bornbaum, J.; Boengler, K.; Torregrossa, R.; Whiteman, M.; Wood, M.E.; Schulz, R.; Baxter, G.F. AP39, a mitochondria-targeting hydrogen sulfide (H2S) donor, protects against myocardial reperfusion injury independently of salvage kinase signalling. Br. J. Pharmacol. 2017, 174, 287–301. [Google Scholar] [CrossRef]
- Zhu, C.; Su, Y.; Juriasingani, S.; Zheng, H.; Veramkovich, V.; Jiang, J.; Sener, A.; Whiteman, M.; Lacefield, J.; Nagpal, D.; et al. Supplementing preservation solution with mitochondria-targeted H2S donor AP39 protects cardiac grafts from prolonged cold ischemia-reperfusion injury in heart transplantation. Am. J. Transplant. 2019, 19, 3139–3148. [Google Scholar] [CrossRef]
- Testai, L.; Strobykina, I.; Semenov, V.V.; Semenova, M.; Pozzo, E.D.; Martelli, A.; Citi, V.; Martini, C.; Breschi, M.C.; Kataev, V.E.; et al. Mitochondriotropic and Cardioprotective Effects of Triphenylphosphonium-Conjugated Derivatives of the Diterpenoid Isosteviol. Int. J. Mol. Sci. 2017, 18, 2060. [Google Scholar] [CrossRef]
- Liu, D.; Jin, F.; Shu, G.; Xu, X.; Qi, J.; Kang, X.; Yu, H.; Lu, K.; Jiang, S.; Han, F.; et al. Enhanced efficiency of mitochondria-targeted peptide SS-31 for acute kidney injury by pH-responsive and AKI-kidney targeted nanopolyplexes. Biomaterials 2019, 211, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, G.; Matoba, T.; Nakano, Y.; Nagaoka, K.; Ishikita, A.; Nakano, K.; Funamoto, D.; Sunagawa, K.; Egashira, K. Nanoparticle-Mediated Targeting of Cyclosporine A Enhances Cardioprotection Against Ischemia-Reperfusion Injury Through Inhibition of Mitochondrial Permeability Transition Pore Opening. Sci. Rep. 2016, 6, 20467. [Google Scholar] [CrossRef]
- Zhang, C.X.; Cheng, Y.; Liu, D.Z.; Liu, M.; Cui, H.; Zhang, B.L.; Mei, Q.B.; Zhou, S.Y. Mitochondria-targeted cyclosporin A delivery system to treat myocardial ischemia reperfusion injury of rats. J. Nanobiotechnology 2019, 17, 18. [Google Scholar] [CrossRef] [PubMed]
- Hoque, A.; Sivakumaran, P.; Bond, S.T.; Ling, N.X.Y.; Kong, A.M.; Scott, J.W.; Bandara, N.; Hernández, D.; Liu, G.S.; Wong, R.C.B.; et al. Mitochondrial fission protein Drp1 inhibition promotes cardiac mesodermal differentiation of human pluripotent stem cells. Cell Death Discov. 2018, 4, 39. [Google Scholar] [CrossRef]
- Lozano, O.; Lázaro-Alfaro, A.; Silva-Platas, C.; Oropeza-Almazán, Y.; Torres-Quintanilla, A.; Bernal-Ramírez, J.; Alves-Figueiredo, H.; García-Rivas, G. Nanoencapsulated Quercetin Improves Cardioprotection during Hypoxia-Reoxygenation Injury through Preservation of Mitochondrial Function. Oxidative Med. Cell. Longev. 2019, 2019, 7683051. [Google Scholar] [CrossRef] [PubMed]
- Okahara, A.; Koga, J.I.; Matoba, T.; Fujiwara, M.; Tokutome, M.; Ikeda, G.; Nakano, K.; Tachibana, M.; Ago, T.; Kitazono, T.; et al. Simultaneous targeting of mitochondria and monocytes enhances neuroprotection against ischemia-reperfusion injury. Sci. Rep. 2020, 10, 14435. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Li, J.; Hu, S.; Wu, F.; Zhang, X. Triphenylphosphonium and D-α-tocopheryl polyethylene glycol 1000 succinate-modified, tanshinone IIA-loaded lipid-polymeric nanocarriers for the targeted therapy of myocardial infarction. Int. J. Nanomed. 2018, 13, 4045–4057. [Google Scholar] [CrossRef]
- Cheng, Y.; Liu, D.Z.; Zhang, C.X.; Cui, H.; Liu, M.; Zhang, B.L.; Mei, Q.B.; Lu, Z.F.; Zhou, S.Y. Mitochondria-targeted antioxidant delivery for precise treatment of myocardial ischemia-reperfusion injury through a multistage continuous targeted strategy. Nanomedicine 2019, 16, 236–249. [Google Scholar] [CrossRef]
- Kawamura, E.; Maruyama, M.; Abe, J.; Sudo, A.; Takeda, A.; Takada, S.; Yokota, T.; Kinugawa, S.; Harashima, H.; Yamada, Y. Validation of Gene Therapy for Mutant Mitochondria by Delivering Mitochondrial RNA Using a MITO-Porter. Mol. Ther. Nucleic Acids 2020, 20, 687–698. [Google Scholar] [CrossRef]
- Furukawa, R.; Yamada, Y.; Kawamura, E.; Harashima, H. Mitochondrial delivery of antisense RNA by MITO-Porter results in mitochondrial RNA knockdown, and has a functional impact on mitochondria. Biomaterials 2015, 57, 107–115. [Google Scholar] [CrossRef]
- Ishikawa, T.; Somiya, K.; Munechika, R.; Harashima, H.; Yamada, Y. Mitochondrial transgene expression via an artificial mitochondrial DNA vector in cells from a patient with a mitochondrial disease. J. Control Release 2018, 274, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Ramachandra, C.J.A.; Hernandez-Resendiz, S.; Crespo-Avilan, G.E.; Lin, Y.H.; Hausenloy, D.J. Mitochondria in acute myocardial infarction and cardioprotection. EBioMedicine 2020, 57, 102884. [Google Scholar] [CrossRef] [PubMed]
- Szeto, H.H. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br. J. Pharmacol. 2014, 171, 2029–2050. [Google Scholar] [CrossRef] [PubMed]
- Suk, J.S.; Xu, Q.; Kim, N.; Hanes, J.; Ensign, L.M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 2016, 99, 28–51. [Google Scholar] [CrossRef]
- Yamada, Y.; Akita, H.; Kamiya, H.; Kogure, K.; Yamamoto, T.; Shinohara, Y.; Yamashita, K.; Kobayashi, H.; Kikuchi, H.; Harashima, H. MITO-Porter: A liposome-based carrier system for delivery of macromolecules into mitochondria via membrane fusion. Biochim. Biophys. Acta 2008, 1778, 423–432. [Google Scholar] [CrossRef]
- Kelso, G.F.; Porteous, C.M.; Coulter, C.V.; Hughes, G.; Porteous, W.K.; Ledgerwood, E.C.; Smith, R.A.; Murphy, M.P. Selective targeting of a redox-active ubiquinone to mitochondria within cells: Antioxidant and antiapoptotic properties. J. Biol. Chem. 2001, 276, 4588–4596. [Google Scholar] [CrossRef]
- Ayer, A.; Macdonald, P.; Stocker, R. CoQ₁₀ Function and Role in Heart Failure and Ischemic Heart Disease. Annu. Rev. Nutr. 2015, 35, 175–213. [Google Scholar] [CrossRef]
- Mortensen, S.A.; Rosenfeldt, F.; Kumar, A.; Dolliner, P.; Filipiak, K.J.; Pella, D.; Alehagen, U.; Steurer, G.; Littarru, G.P. Q-SYMBIO Study Investigators. The effect of coenzyme Q10 on morbidity and mortality in chronic heart failure: Results from Q-SYMBIO: A randomized double-blind trial. JACC Heart Fail. 2014, 2014, 641–649. [Google Scholar] [CrossRef]
- Gholami, M.; Rezvanfar, M.R.; Delavar, M.; Abdollahi, M.; Khosrowbeygi, A. Effects of Coenzyme Q10 Supplementation on Serum Values of Gamma-glutamyl transferase, Pseudocholinesterase, Bilirubin, Ferritin, and High-Sensitivity C-Reactive Protein in Women with Type 2 Diabetes. Exp. Clin. Endocrinol. Diabetes 2019, 127, 311–319. [Google Scholar] [CrossRef]
- Zielonka, J.; Joseph, J.; Sikora, A.; Hardy, M.; Ouari, O.; Vasquez-Vivar, J.; Cheng, G.; Lopez, M.; Kalyanaraman, B. Mitochondria-Targeted Triphenylphosphonium-Based Compounds: Syntheses, Mechanisms of Action, and Therapeutic and Diagnostic Applications. Chem. Rev. 2017, 117, 10043–10120. [Google Scholar] [CrossRef]
- Neuzil, J.; Widén, C.; Gellert, N.; Swettenham, E.; Zobalova, R.; Dong, L.F.; Wang, X.F.; Lidebjer, C.; Dalen, H.; Headrick, J.P.; et al. Mitochondria transmit apoptosis signalling in cardiomyocyte-like cells and isolated hearts exposed to experimental ischemia-reperfusion injury. Redox Rep. 2007, 12, 148–162. [Google Scholar] [CrossRef] [PubMed]
- Zernii, E.Y.; Gancharova, O.S.; Baksheeva, V.E.; Golovastova, M.O.; Kabanova, E.I.; Savchenko, M.S.; Tiulina, V.V.; Sotnikova, L.F.; Zamyatnin, A.A., Jr.; Philippov, P.P.; et al. Mitochondria-Targeted Antioxidant SkQ1 Prevents Anesthesia-Induced Dry Eye Syndrome. Oxid Med. Cell Longev. 2017, 2017, 9281519. [Google Scholar] [CrossRef]
- Trnka, J.; Blaikie, F.H.; Smith, R.A.; Murphy, M.P. A mitochondria-targeted nitroxide is reduced to its hydroxylamine by ubiquinol in mitochondria. Free Radic. Biol. Med. 2008, 44, 1406–1419. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Luo, G.; Giannelli, S.; Szeto, H.H. Mitochondria-targeted peptide prevents mitochondrial depolarization and apoptosis induced by tert-butyl hydroperoxide in neuronal cell lines. Biochem. Pharmacol. 2005, 70, 1796–1806. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, M.; Spencer, J.P.; Szeto, H.H.; Armstrong, J.S. Do mitochondriotropic antioxidants prevent chlorinative stress-induced mitochondrial and cellular injury? Antioxid. Redox Signal. 2008, 10, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Birk, A.V.; Chao, W.M.; Bracken, C.; Warren, J.D.; Szeto, H.H. Targeting mitochondrial cardiolipin and the cytochrome c/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br. J. Pharmacol. 2014, 171, 2017–2028. [Google Scholar] [CrossRef] [PubMed]
- Chavez, J.D.; Tang, X.; Campbell, M.D.; Reyes, G.; Kramer, P.A.; Stuppard, R.; Keller, A.; Zhang, H.; Rabinovitch, P.S.; Marcinek, D.J.; et al. Mitochondrial protein interaction landscape of SS-31. Proc. Natl. Acad. Sci. USA 2020, 117, 15363–15373. [Google Scholar] [CrossRef]
- Kloner, R.A.; Hale, S.L.; Dai, W.; Gorman, R.C.; Shuto, T.; Koomalsingh, K.J.; Gorman, J.H., III; Sloan, R.C.; Frasier, C.R.; Watson, C.A.; et al. Reduction of ischemia/reperfusion injury with bendavia, a mitochondria-targeting cytoprotective Peptide. J. Am. Heart Assoc. 2012, 1, e001644. [Google Scholar] [CrossRef]
- Brown, D.A.; Hale, S.L.; Baines, C.P.; del Rio, C.L.; Hamlin, R.L.; Yueyama, Y.; Kijtawornrat, A.; Yeh, S.T.; Frasier, C.R.; Stewart, L.M.; et al. Reduction of early reperfusion injury with the mitochondria-targeting peptide bendavia. J. Cardiovasc. Pharmacol. Ther. 2014, 19, 121–132. [Google Scholar] [CrossRef]
- Cho, J.; Won, K.; Wu, D.; Soong, Y.; Liu, S.; Szeto, H.H.; Hong, M.K. Potent mitochondria-targeted peptides reduce myocardial infarction in rats. Coron. Artery Dis. 2007, 18, 215–220. [Google Scholar] [CrossRef]
- Dai, D.F.; Chen, T.; Szeto, H.; Nieves-Cintrón, M.; Kutyavin, V.; Santana, L.F.; Rabinovitch, P.S. Mitochondrial targeted antioxidant peptide ameliorates hypertensive cardiomyopathy. J. Am. Coll. Cardiol. 2011, 58, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.F.; Hsieh, E.J.; Chen, T.; Menendez, L.G.; Basisty, N.B.; Tsai, L.; Beyer, R.P.; Crispin, D.A.; Shulman, N.J.; Szeto, H.H.; et al. Global proteomics and pathway analysis of pressure-overload-induced heart failure and its attenuation by mitochondrial-targeted peptides. Circ. Heart Fail. 2013, 6, 1067–1076. [Google Scholar] [CrossRef] [PubMed]
- Sabbah, H.N.; Gupta, R.C.; Kohli, S.; Wang, M.; Hachem, S.; Zhang, K. Chronic Therapy with Elamipretide (MTP-131), a Novel Mitochondria-Targeting Peptide, Improves Left Ventricular and Mitochondrial Function in Dogs with Advanced Heart Failure. Circ. Heart Fail. 2016, 9, e002206. [Google Scholar] [CrossRef] [PubMed]
- Yuan, F.; Woollard, J.R.; Jordan, K.L.; Lerman, A.; Lerman, L.O.; Eirin, A. Mitochondrial targeted peptides preserve mitochondrial organization and decrease reversible myocardial changes in early swine metabolic syndrome. Cardiovasc. Res. 2018, 114, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Gibson, C.M.; Giugliano, R.P.; Kloner, R.A.; Bode, C.; Tendera, M.; Jánosi, A.; Merkely, B.; Godlewski, J.; Halaby, R.; Korjian, S.; et al. EMBRACE STEMI study: A Phase 2a trial to evaluate the safety, tolerability, and efficacy of intravenous MTP-131 on reperfusion injury in patients undergoing primary percutaneous coronary intervention. Eur. Heart J. 2016, 37, 1296–1303. [Google Scholar] [CrossRef] [PubMed]
- Daubert, M.A.; Yow, E.; Dunn, G.; Marchev, S.; Barnhart, H.; Douglas, P.S.; O’Connor, C.; Goldstein, S.; Udelson, J.E.; Sabbah, H.N. Novel Mitochondria-Targeting Peptide in Heart Failure Treatment: A Randomized, Placebo-Controlled Trial of Elamipretide. Circ. Heart Fail. 2017, 10, e004389. [Google Scholar] [CrossRef]
- Butler, J.; Khan, M.S.; Anker, S.D.; Fonarow, G.C.; Kim, R.J.; Nodari, S.; O’Connor, C.M.; Pieske, B.; Pieske-Kraigher, E.; Sabbah, H.N.; et al. Effects of Elamipretide on Left Ventricular Function in Patients with Heart Failure with Reduced Ejection Fraction: The PROGRESS-HF Phase 2 Trial. J. Card. Fail. 2020, 26, 429–437. [Google Scholar] [CrossRef]
- Chatfield, K.C.; Sparagna, G.C.; Chau, S.; Phillips, E.K.; Ambardekar, A.V.; Aftab, M.; Mitchell, M.B.; Sucharov, C.C.; Miyamoto, S.D.; Stauffer, B.L. Elamipretide Improves Mitochondrial Function in the Failing Human Heart. JACC Basic Transl. Sci. 2019, 4, 147–157. [Google Scholar] [CrossRef]
- Agnihotri, S.A.; Mallikarjuna, N.N.; Aminabhavi, T.M. Recent advances on chitosan-based micro- and nanoparticles in drug delivery. J. Control Release 2004, 100, 5–28. [Google Scholar] [CrossRef]
- Huebener, P.; Abou-Khamis, T.; Zymek, P.; Bujak, M.; Ying, X.; Chatila, K.; Haudek, S.; Thakker, G.; Frangogiannis, N.G. CD44 is critically involved in infarct healing by regulating the inflammatory and fibrotic response. J. Immunol. 2008, 180, 2625–2633. [Google Scholar] [CrossRef]
- Dalal, S.; Zha, Q.; Daniels, C.R.; Steagall, R.J.; Joyner, W.L.; Gadeau, A.P.; Singh, M.; Singh, K. Osteopontin stimulates apoptosis in adult cardiac myocytes via the involvement of CD44 receptors, mitochondrial death pathway, and endoplasmic reticulum stress. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H1182–H1191. [Google Scholar] [CrossRef] [PubMed]
- Suleiman, M.; Abdulrahman, N.; Yalcin, H.; Mraiche, F. The role of CD44, hyaluronan and NHE1 in cardiac remodeling. Life Sci. 2018, 209, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Javadov, S.; Karmazyn, M. Mitochondrial permeability transition pore opening as an endpoint to initiate cell death and as a putative target for cardioprotection. Cell Physiol. Biochem. 2007, 20, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Boston-Griffiths, E.A.; Yellon, D.M. Cyclosporin A and cardioprotection: From investigative tool to therapeutic agent. Br. J. Pharmacol. 2012, 165, 1235–1245. [Google Scholar] [CrossRef]
- Mewton, N.; Cung, T.T.; Morel, O.; Cayla, G.; Bonnefoy-Cudraz, E.; Rioufol, G.; Angoulvant, D.; Guerin, P.; Elbaz, M.; Delarche, N.; et al. CIRCUS Study Investigators: Rationale and design of the Cyclosporine to ImpRove Clinical oUtcome in ST-elevation myocardial infarction patients (the CIRCUS trial). Am. Heart J. 2015, 169, 758–766. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Davidson, A.M. Inhibition of Ca2(+)-induced large-amplitude swelling of liver and heart mitochondria by cyclosporin is probably caused by the inhibitor binding to mitochondrial-matrix peptidyl-prolyl cis-trans isomerase and preventing it interacting with the adenine nucleotide translocase. Biochem. J. 1990, 268, 153–160. [Google Scholar] [CrossRef]
- Chiari, P.; Angoulvant, D.; Mewton, N.; Desebbe, O.; Obadia, J.F.; Robin, J.; Farhat, F.; Jegaden, O.; Bastien, O.; Lehot, J.J.; et al. Cyclosporine protects the heart during aortic valve surgery. Anesthesiology 2014, 121, 232–238. [Google Scholar] [CrossRef]
- Piot, C.; Croisille, P.; Staat, P.; Thibault, H.; Rioufol, G.; Mewton, N.; Elbelghiti, R.; Cung, T.T.; Bonnefoy, E.; Angoulvant, D.; et al. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N. Engl. J. Med. 2008, 359, 473–481. [Google Scholar] [CrossRef]
- Cung, T.T.; Morel, O.; Cayla, G.; Rioufol, G.; Garcia-Dorado, D.; Angoulvant, D.; Bonnefoy-Cudraz, E.; Guérin, P.; Elbaz, M.; Delarche, N.; et al. Cyclosporine before PCI in Patients with Acute Myocardial Infarction. N. Engl. J. Med. 2015, 373, 1021–1031. [Google Scholar] [CrossRef]
- Ottani, F.; Latini, R.; Staszewsky, L.; La Vecchia, L.; Locuratolo, N.; Sicuro, M.; Masson, S.; Barlera, S.; Milani, V.; Lombardi, M.; et al. Cyclosporine A in Reperfused Myocardial Infarction: The Multicenter, Controlled, Open-Label CYCLE Trial. J. Am. Coll. Cardiol. 2016, 67, 365–374. [Google Scholar] [CrossRef]
- Cormack, S.; Mohammed, A.; Panahi, P.; Das, R.; Steel, A.J.; Chadwick, T.; Bryant, A.; Egred, M.; Stellos, K.; Spyridopoulos, I.; et al. Effect of ciclosporin on safety, lymphocyte kinetics and left ventricular remodelling in acute myocardial infarction. Br. J. Clin. Pharmacol. 2020, 86, 1387–1397. [Google Scholar] [CrossRef] [PubMed]
- Pandita, D.; Kumar, S.; Lather, V. Hybrid poly(lactic-co-glycolic acid) nanoparticles: Design and delivery prospectives. Drug Discov. Today 2015, 20, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Andreadou, I.; Iliodromitis, E.K.; Rassaf, T.; Schulz, R.; Papapetropoulos, A.; Ferdinandy, P. The role of gasotransmitters NO, H2S and CO in myocardial ischaemia/reperfusion injury and cardioprotection by preconditioning, postconditioning and remote conditioning. Br. J. Pharmacol. 2015, 172, 1587–1606. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.M.; Maddock, H.L.; Yellon, D.M. The cardioprotective and mitochondrial depolarising properties of exogenous nitric oxide in mouse heart. Cardiovasc. Res. 2003, 57, 405–415. [Google Scholar] [CrossRef][Green Version]
- Penna, C.; Perrelli, M.G.; Tullio, F.; Angotti, C.; Camporeale, A.; Poli, V.; Pagliaro, P. Diazoxide postconditioning induces mitochondrial protein S-nitrosylation and a redox-sensitive mitochondrial phosphorylation/translocation of RISK elements: No role for SAFE. Basic Res. Cardiol. 2013, 108, 371. [Google Scholar] [CrossRef]
- Shimizu, Y.; Polavarapu, R.; Eskla, K.L.; Nicholson, C.K.; Koczor, C.A.; Wang, R.; Lewis, W.; Shiva, S.; Lefer, D.J.; Calvert, J.W. Hydrogen sulfide regulates cardiac mitochondrial biogenesis via the activation of AMPK. J. Mol. Cell Cardiol. 2018, 116, 29–40. [Google Scholar] [CrossRef]
- Sun, J.; Aponte, A.M.; Menazza, S.; Gucek, M.; Steenbergen, C.; Murphy, E. Additive cardioprotection by pharmacological postconditioning with hydrogen sulfide and nitric oxide donors in mouse heart: S-sulfhydration vs. S-nitrosylation. Cardiovasc. Res. 2016, 110, 96–106. [Google Scholar] [CrossRef]
- Martelli, A.; Testai, L.; Citi, V.; Marino, A.; Bellagambi, F.G.; Ghimenti, S.; Breschi, M.C.; Calderone, V. Pharmacological characterization of the vascular effects of aryl isothiocyanates: Is hydrogen sulfide the real player? Vascul. Pharmacol. 2014, 60, 32–41. [Google Scholar] [CrossRef]
- Testai, L.; Marino, A.; Piano, I.; Brancaleone, V.; Tomita, K.; Di Cesare Mannelli, L.; Martelli, A.; Citi, V.; Breschi, M.C.; Levi, R.; et al. The novel H2S-donor 4-carboxyphenyl isothiocyanate promotes cardioprotective effects against ischemia/reperfusion injury through activation of mitoKATP channels and reduction of oxidative stress. Pharmacol. Res. 2016, 113, 290–299. [Google Scholar] [CrossRef]
- Xu, M.; Yigang, W.; Ahmar, A.; Muhammad, A. Mitochondrial KATP channel activation reduces anoxic injury by restoring mitochondrial membrane potential. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H1295–H1303. [Google Scholar] [CrossRef]
- Paggio, A.; Checchetto, V.; Campo, A.; Menabò, R.; Di Marco, G.; Di Lisa, F.; Szabo, I.; Rizzuto, R.; De Stefani, D. Identification of an ATP-sensitive potassium channel in mitochondria. Nature 2019, 572, 609–613. [Google Scholar] [CrossRef] [PubMed]
- Citi, V.; Corvino, A.; Fiorino, F.; Frecentese, F.; Magli, E.; Perissutti, E.; Santagada, V.; Brogi, S.; Flori, L.; Gorica, E.; et al. Structure-activity relationships study of isothiocyanates for H2S releasing properties: 3-Pyridyl-isothiocyanate as a new promising cardioprotective agent. J. Adv. Res. 2020. [Google Scholar] [CrossRef]
- Wu, D.; Hu, Q.; Tan, B.; Rose, P.; Zhu, D.; Zhu, Y.Z. Amelioration of mitochondrial dysfunction in heart failure through S-sulfhydration of Ca2+/calmodulin-dependent protein kinase II. Redox Biol. 2018, 19, 250–262. [Google Scholar] [CrossRef] [PubMed]
- Jhun, B.S.; O-Uchi, J.; Adaniya, S.M.; Cypress, M.W.; Yoon, Y. Adrenergic Regulation of Drp1-Driven Mitochondrial Fission in Cardiac Physio-Pathology. Antioxidants 2018, 7, 195. [Google Scholar] [CrossRef] [PubMed]
- Maneechote, C.; Palee, S.; Chattipakorn, S.C.; Chattipakorn, N. Roles of mitochondrial dynamics modulators in cardiac ischaemia/reperfusion injury. J. Cell Mol. Med. 2017, 21, 2643–2653. [Google Scholar] [CrossRef]
- Cassidy-Stone, A.; Chipuk, J.E.; Ingerman, E.; Song, C.; Yoo, C.; Kuwana, T.; Kurth, M.J.; Shaw, J.T.; Hinshaw, J.E.; Green, D.R.; et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev. Cell 2008, 14, 193–204. [Google Scholar] [CrossRef]
- Ong, S.B.; Subrayan, S.; Lim, S.Y.; Yellon, D.M.; Davidson, S.M.; Hausenloy, D.J. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation 2010, 121, 2012–2022. [Google Scholar] [CrossRef]
- Sharp, W.W.; Fang, Y.H.; Han, M.; Zhang, H.J.; Hong, Z.; Banathy, A.; Morrow, E.; Ryan, J.J.; Archer, S.L. Dynamin-related protein 1 (Drp1)-mediated diastolic dysfunction in myocardial ischemia-reperfusion injury: Therapeutic benefits of Drp1 inhibition to reduce mitochondrial fission. FASEB J. 2014, 28, 316–326. [Google Scholar] [CrossRef]
- Givvimani, S.; Munjal, C.; Tyagi, N.; Sen, U.; Metreveli, N.; Tyagi, S.C. Mitochondrial division/mitophagy inhibitor (Mdivi) ameliorates pressure overload induced heart failure. PLoS ONE. 2012, 7, e32388. [Google Scholar] [CrossRef]
- Qi, J.; Wang, F.; Yang, P.; Wang, X.; Xu, R.; Chen, J.; Yuan, Y.; Lu, Z.; Duan, J. Mitochondrial Fission Is Required for Angiotensin II-Induced Cardiomyocyte Apoptosis Mediated by a Sirt1-p53 Signaling Pathway. Front. Pharmacol. 2018, 9, 176. [Google Scholar] [CrossRef]
- Tanner, M.J.; Wang, J.; Ying, R.; Suboc, T.B.; Malik, M.; Couillard, A.; Branum, A.; Puppala, V.; Widlansky, M.E. Dynamin-related protein 1 mediates low glucose-induced endothelial dysfunction in human arterioles. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H515–H527. [Google Scholar] [CrossRef] [PubMed]
- Ishikita, A.; Matoba, T.; Ikeda, G.; Koga, J.; Mao, Y.; Nakano, K.; Takeuchi, O.; Sadoshima, J.; Egashira, K. Nanoparticle-Mediated Delivery of Mitochondrial Division Inhibitor 1 to the Myocardium Protects the Heart From Ischemia-Reperfusion Injury Through Inhibition of Mitochondria Outer Membrane Permeabilization: A New Therapeutic Modality for Acute Myocardial Infarction. J. Am. Heart Assoc. 2016, 5, e003872. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Dasgupta, A.; Chen, K.H.; Neuber-Hess, M.; Patel, J.; Hurst, T.E.; Mewburn, J.D.; Lima, P.D.A.; Alizadeh, E.; Martin, A.; et al. Identification of novel dynamin-related protein 1 (Drp1) GTPase inhibitors: Therapeutic potential of Drpitor1 and Drpitor1a in cancer and cardiac ischemia-reperfusion injury. FASEB J. 2020, 34, 1447–1464. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.M.; Ferdinandy, P.; Andreadou, I.; Bøtker, H.E.; Heusch, G.; Ibáñez, B.; Ovize, M.; Schulz, R.; Yellon, D.M.; Hausenloy, D.J.; et al. Multitarget Strategies to Reduce Myocardial Ischemia/Reperfusion Injury: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 73, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.; Zhang, T.; Zhang, W.; Zhao, Z.; Sun, J. Natural Drugs as a Treatment Strategy for Cardiovascular Disease through the Regulation of Oxidative Stress. Oxidative Med. Cell. Longev. 2020, 2020, 5430407. [Google Scholar] [CrossRef] [PubMed]
- Wiciński, M.; Socha, M.; Walczak, M.; Wódkiewicz, E.; Malinowski, B.; Rewerski, S.; Górski, K.; Pawlak-Osińska, K. Beneficial Effects of Resveratrol Administration-Focus on Potential Biochemical Mechanisms in Cardiovascular Conditions. Nutrients 2018, 10, 1813. [Google Scholar] [CrossRef]
- Arinno, A.; Apaijai, N.; Chattipakorn, S.C.; Chattipakorn, N. The roles of resveratrol on cardiac mitochondrial function in cardiac diseases. Eur. J. Nutr. 2020. [Google Scholar] [CrossRef]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef]
- Li, H.; Xia, N.; Hasselwander, S.; Daiber, A. Resveratrol and Vascular Function. Int. J. Mol. Sci. 2019, 20, 2155. [Google Scholar] [CrossRef]
- Bagul, P.K.; Katare, P.B.; Bugga, P.; Dinda, A.K.; Banerjee, S.K. SIRT-3 Modulation by Resveratrol Improves Mitochondrial Oxidative Phosphorylation in Diabetic Heart through Deacetylation of TFAM. Cells 2018, 7, 235. [Google Scholar] [CrossRef]
- Ramírez-Garza, S.L.; Laveriano-Santos, E.P.; Marhuenda-Muñoz, M.; Storniolo, C.E.; Tresserra-Rimbau, A.; Vallverdú-Queralt, A.; Lamuela-Raventós, R.M. Health Effects of Resveratrol: Results from Human Intervention Trials. Nutrients 2018, 10, 1892. [Google Scholar] [CrossRef] [PubMed]
- De Ligt, M.; Bergman, M.; Fuentes, R.M.; Essers, H.; Moonen-Kornips, E.; Havekes, B.; Schrauwen-Hinderling, V.B.; Schrauwen, P. No effect of resveratrol supplementation after 6 months on insulin sensitivity in overweight adults: A randomized trial. Am. J. Clin. Nutr. 2020, 112, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yao, J.; Han, C.; Yang, J.; Chaudhry, M.T.; Wang, S.; Liu, H.; Yin, Y. Quercetin, Inflammation and Immunity. Nutrients 2016, 8, 167. [Google Scholar] [CrossRef] [PubMed]
- Çelik, N.; Vurmaz, A.; Kahraman, A. Protective effect of quercetin on homocysteine-induced oxidative stress. Nutrition 2017, 33, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Ali, T.; Rehman, S.U.; Khan, M.S.; Alam, S.I.; Ikram, M.; Muhammad, T.; Saeed, K.; Badshah, H.; Kim, M.O. Neuroprotective Effect of Quercetin Against the Detrimental Effects of LPS in the Adult Mouse Brain. Front. Pharmacol. 2018, 9, 1383. [Google Scholar] [CrossRef]
- Qiu, L.; Luo, Y.; Chen, X. Quercetin attenuates mitochondrial dysfunction and biogenesis via upregulated AMPK/SIRT1 signaling pathway in OA rats. Biomed. Pharmacother. 2018, 103, 1585–1591. [Google Scholar] [CrossRef]
- Serban, M.C.; Sahebkar, A.; Zanchetti, A.; Mikhailidis, D.P.; Howard, G.; Antal, D.; Andrica, F.; Ahmed, A.; Aronow, W.S.; Muntner, P.; et al. Lipid and Blood Pressure Meta-analysis Collaboration (LBPMC) Group. Effects of Quercetin on Blood Pressure: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. J. Am. Heart Assoc. 2016, 5, e002713. [Google Scholar] [CrossRef]
- Chatsudthipong, V.; Muanprasat, C. Stevioside and related compounds: Therapeutic benefits beyond sweetness. Pharmacol. Ther. 2009, 121, 41–54. [Google Scholar] [CrossRef]
- Liu, F.; Su, H.; Liu, B.; Mei, Y.; Ke, Q.; Sun, X.; Tan, W. STVNa Attenuates Isoproterenol-Induced Cardiac Hypertrophy Response through the HDAC4 and Prdx2/ROS/Trx1 Pathways. Int. J. Mol. Sci. 2020, 21, 682. [Google Scholar] [CrossRef]
- Mei, Y.; Liu, B.; Su, H.; Zhang, H.; Liu, F.; Ke, Q.; Sun, X.; Tan, W. Isosteviol sodium protects the cardiomyocyte response associated with the SIRT1/PGC-1α pathway. J. Cell. Mol. Med. 2020, 24, 10866–10875. [Google Scholar] [CrossRef]
- Xu, D.; Li, Y.; Wang, J.; Davey, A.K.; Zhang, S.; Evans, A.M. The cardioprotective effect of isosteviol on rats with heart ischemia-reperfusion injury. Life Sci. 2007, 80, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Zhang, S.; Foster, D.J.; Wang, J. The effects of isosteviol against myocardium injury induced by ischaemia-reperfusion in the isolated guinea pig heart. Clin. Exp. Pharmacol. Physiol. 2007, 34, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zou, J.; Cao, D.; Ma, X. Pharmacological basis of tanshinone and new insights into tanshinone as a multitarget natural product for multifaceted diseases. Biomed. Pharmacother. 2020, 130, 110599. [Google Scholar] [CrossRef]
- Tan, S.; Zou, C.; Zhang, W.; Yin, M.; Gao, X.; Tang, Q. Recent developments in d-α-tocopheryl polyethylene glycol-succinate-based nanomedicine for cancer therapy. Drug Deliv. 2017, 24, 1831–1842. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Li, L.; Li, L.; Ni, J.; Guo, R.; Mao, J.; Fan, G. Effects of the combination of tanshinone IIA and puerarin on cardiac function and inflammatory response in myocardial ischemia mice. J. Mol. Cell. Cardiol. 2019, 137, 59–70. [Google Scholar] [CrossRef]
- Gao, Y.; Wang, X.; He, C. An isoflavonoid-enriched extract from Pueraria lobata (kudzu) root protects human umbilical vein endothelial cells against oxidative stress induced apoptosis. J. Ethnopharmacol. 2016, 193, 524–530. [Google Scholar] [CrossRef]
- Dong, Z.; Guo, J.; Xing, X.; Zhang, X.; Du, Y.; Lu, Q. RGD modified and PEGylated lipid nanoparticles loaded with puerarin: Formulation, characterization and protective effects on acute myocardial ischemia model. Biomed. Pharmacother. 2017, 89, 297–304. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Carrier | Delivery Strategy | Cargo | Cardioprotective Effect |
|---|---|---|---|
| TTP small molecules (lipophilic cation) | Negative IMM potential | Antioxidants, H2S and NO donors, isosteviol | ROS scavenging [9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31] PTM [32,33,34,35,36,37,38,39,40] mitoKATP activation [41] |
| PH-sensitive polymeric NPs (chitosan, HA) | CD44-dependent cell targeting, SS31(Mito-targeting) | SS31 | Cardiolipin binding [42] |
| Polymeric NPs (PLGA; PEG; PEG-PLGA) | EPR effect (injured tissue-targeting), RGD (endothelial cell targeting), SS31 (mito-targeting) | CsA, fission inhibitors, nutraceutics | Cyclophylin D binding [43,44], inhibition of MPTPO [45], pleiotropic [46,47] |
| Lipid/polymeric NP (TPGS-PLGA) | EPR (injured tissue targeting), TTP (mito-targeting) | Tanshinone | Pleiotropic [48] |
| Lipid/polymeric NPs (MCTD) | EPR effect (injured tissue targeting), IMTP (cell targeting), SS31 (mito-targeting) | Resveratrol | Pleiotropic [49] |
| Liposome (mito-porter) | R8 (cell targeting), fusogenic lipid (mito-targeting) | Multiple mitoprotective agents | Pleiotropic [50,51,52,53] |
| Carrier | Function | Cardioprotective Mechanism | Therapeutic Indications |
|---|---|---|---|
| TTP-conjugation MitoSNO | NO donor | Reversible mitochondria protein nitrosilation | Cardiac IR Post ischemic HF |
| TTP-conjugation AP39 | H2S donor | MTPT opening inhibition | Cardioplegic solution Cardiac transplantation HF |
| 4CPI | H2S donor | MitoK-ATP activation | Cardioplegic solution Cardiac transplantation HF |
| 3PI | H2S donor | MitoK-ATP activation | Cardioplegic solution Cardiac transplantation HF |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Forini, F.; Canale, P.; Nicolini, G.; Iervasi, G. Mitochondria-Targeted Drug Delivery in Cardiovascular Disease: A Long Road to Nano-Cardio Medicine. Pharmaceutics 2020, 12, 1122. https://doi.org/10.3390/pharmaceutics12111122
Forini F, Canale P, Nicolini G, Iervasi G. Mitochondria-Targeted Drug Delivery in Cardiovascular Disease: A Long Road to Nano-Cardio Medicine. Pharmaceutics. 2020; 12(11):1122. https://doi.org/10.3390/pharmaceutics12111122
Chicago/Turabian StyleForini, Francesca, Paola Canale, Giuseppina Nicolini, and Giorgio Iervasi. 2020. "Mitochondria-Targeted Drug Delivery in Cardiovascular Disease: A Long Road to Nano-Cardio Medicine" Pharmaceutics 12, no. 11: 1122. https://doi.org/10.3390/pharmaceutics12111122
APA StyleForini, F., Canale, P., Nicolini, G., & Iervasi, G. (2020). Mitochondria-Targeted Drug Delivery in Cardiovascular Disease: A Long Road to Nano-Cardio Medicine. Pharmaceutics, 12(11), 1122. https://doi.org/10.3390/pharmaceutics12111122