
RNAi-Based Approaches for Pancreatic Cancer Therapy

 and
and
Abstract
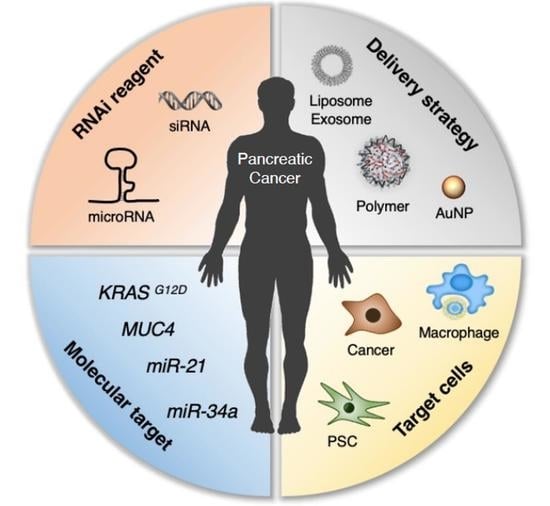
:
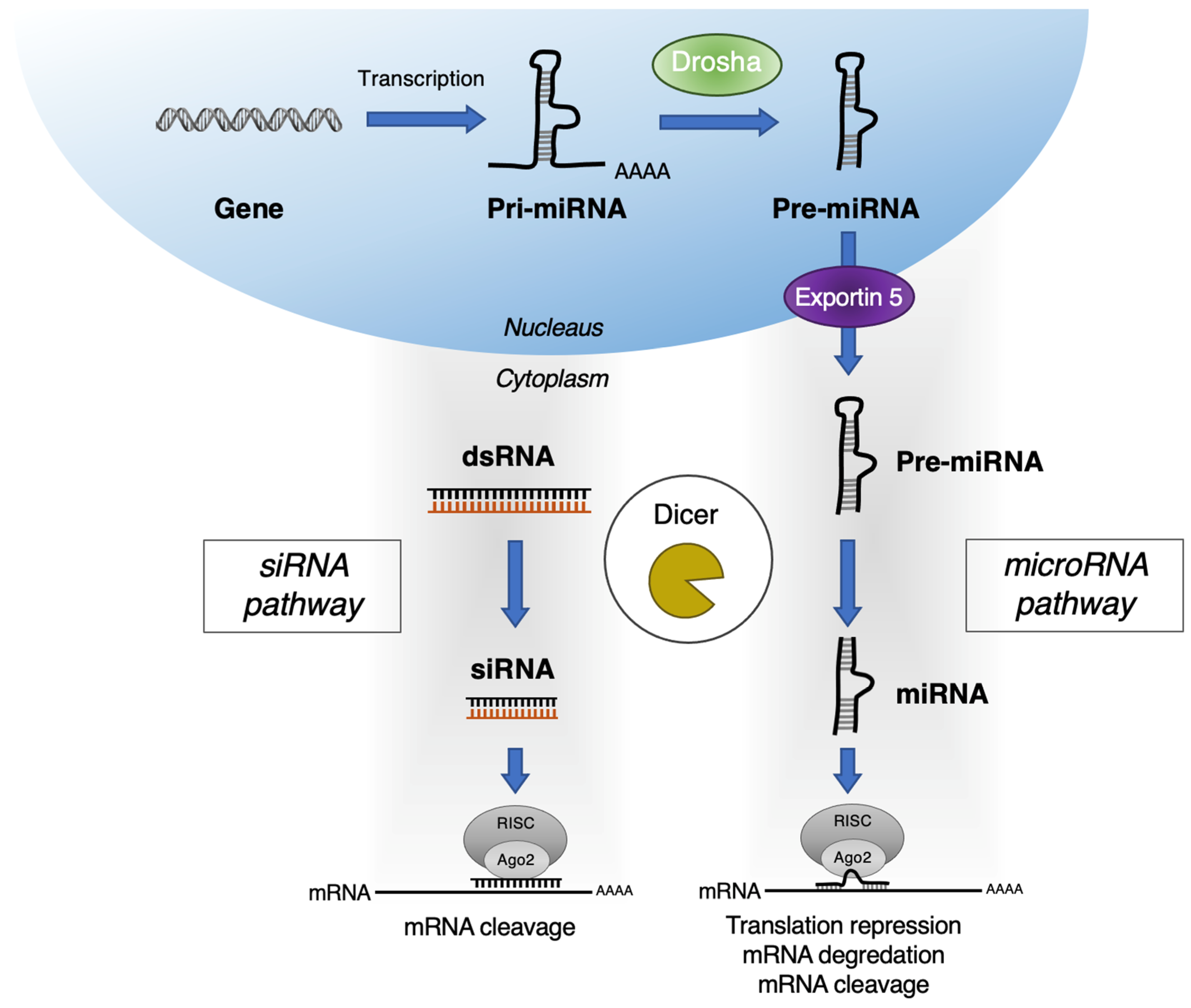
1. Introduction
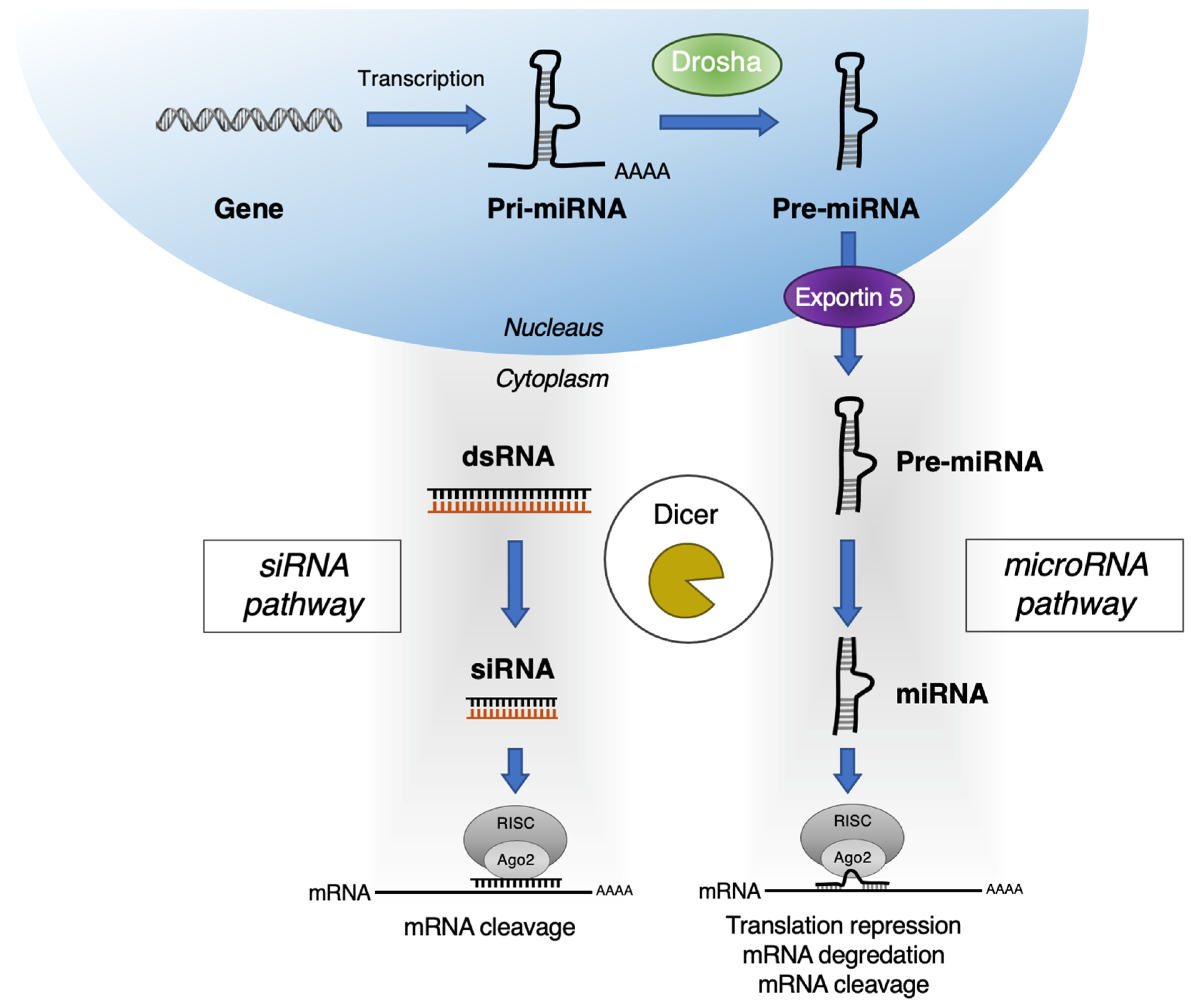
2. RNAi Targets for Pancreatic Cancer Therapy
2.1. Pancreatic Tumor Targets

{kind=link}
{kind=link}
{kind=link}
| Category | Delivery Vehicle | siRNA Target | Tumor Model | Drug Route | Combination Therapy | Reference |
|---|---|---|---|---|---|---|
| Lipid-based Nanoparticles | Lipid nanoparticle (LNPK15) | KRAS | MIA PaCa-2 s.c. | I.V. | [50] | |
| Lipoplex | KRAS | PANC1 s.c. | I.V. | [51] | ||
| Lipoplex (Atu027) | PKN3 | DanG orthotopic | I.V. | [52] | ||
| Liposome | KRAS | PANC-1 s.c. | I.V. | Gemcitabine | [53] | |
| Polymer-based Nanoparticles | Gold nanocluster siRNA (GNC-siRNA) | Nerve growth factor | PANC-1 s.c., orthotopic and PDX | I.T. | [54] | |
| Superparamagnetic iron oxide nanoparticles (siPLK1-StAv-SPIONs) | PLK1 | 6606PDA orthotopic [55] | I.V. | [56] | ||
| Star polymeric nanoparticles different lengths of cationic PDMAEMA side-arms and varied amounts of POEGMA | βIII-tubulin | MiaPaCa-2 and HPAF-II orthotopic | I.T. | [57,58] | ||
| Poly(ethylene glycol) and charge-conversional polymer (PEG-CCP) | VEGF | L1-Luc/TAg transgenic [59] | I.V. | [60,61] | ||
| Local Drug EluteR, LODER (PLGA) | KRAS | PANC1-Luc or Capan1-Luc s.c., synograft, and orthotopic | I.T. | [62] | ||
| PLGA/poloxamer | EPAS1 | BxPC3 s.c. | I.T. | [63] | ||
| Cholesterol-modified polymeric CXCR4 antagonist (PCX) nanoparticles | NCOA3 | CD18/HPAF orthotopic | I.V. | [64] | ||
| Cholesterol-modified polymeric CXCR4 antagonist (PCX) nanoparticles | KRAS | KPC8060 orthotopic | I.V. and I.P. | [65] | ||
| BCPV | KRAS | MiaPaCa-2 s.c. | Peritumoral | [66] | ||
| Folic acid (FA)-modified PEG-chitosan oligosaccharide lactate (COL) nanoparticles | ARHGEF4, CCDC88A, LAMTOR2, mTOR, NUP85, and WASF2 | S2-013 orthotopic | I.V. | [67] | ||
| PEGylated iRGD-guided tumor-penetrating nanocomplexes (TPN) | KRAS | KP D8-175 orthotopic from Pdx1-Cre; Krasþ/LSL-G12D; Trp53fl/ (KPC) mouse | I.V. | [68] | ||
| poly(ethylene glycol)-block-poly-L-lysine (PEG-PLL) | KRAS | PANC-1 (mutant KrasGGT_GAT), BXPC-3 (KrasWT) s.c. | I.V. | Arsenic therapy | [69] | |
| Peptide-conjugated PSPG (PSPGP) | TR3 | PANC-1 s.c. | I.V. | Paclitaxel | [70] | |
| Magnetic nanocarrier | PD-L1 | PANC-02 syngeneic | I.V. | Gemcitabine | [71] | |
| Extracellular vesicle | Exosome | KRAS | PANC-1 or BxPC-3/1) PANC-1, BxPC-3, or KPC689 orthotopic tumor mice model 2) KTC (Ptf1acre/+;LSL-KrasG12D/+;Tgfbr2lox/lox) genetically engineered mouse | I.P. | [72] | |
| Extracellular vesicle | Galectin-9 | PANC-02 orthotopic | I.V. | Oxaliplatin | [73] |
2.2. Targets in the Pancreatic Tumor Stroma and Immunosuppressive Microenvironment
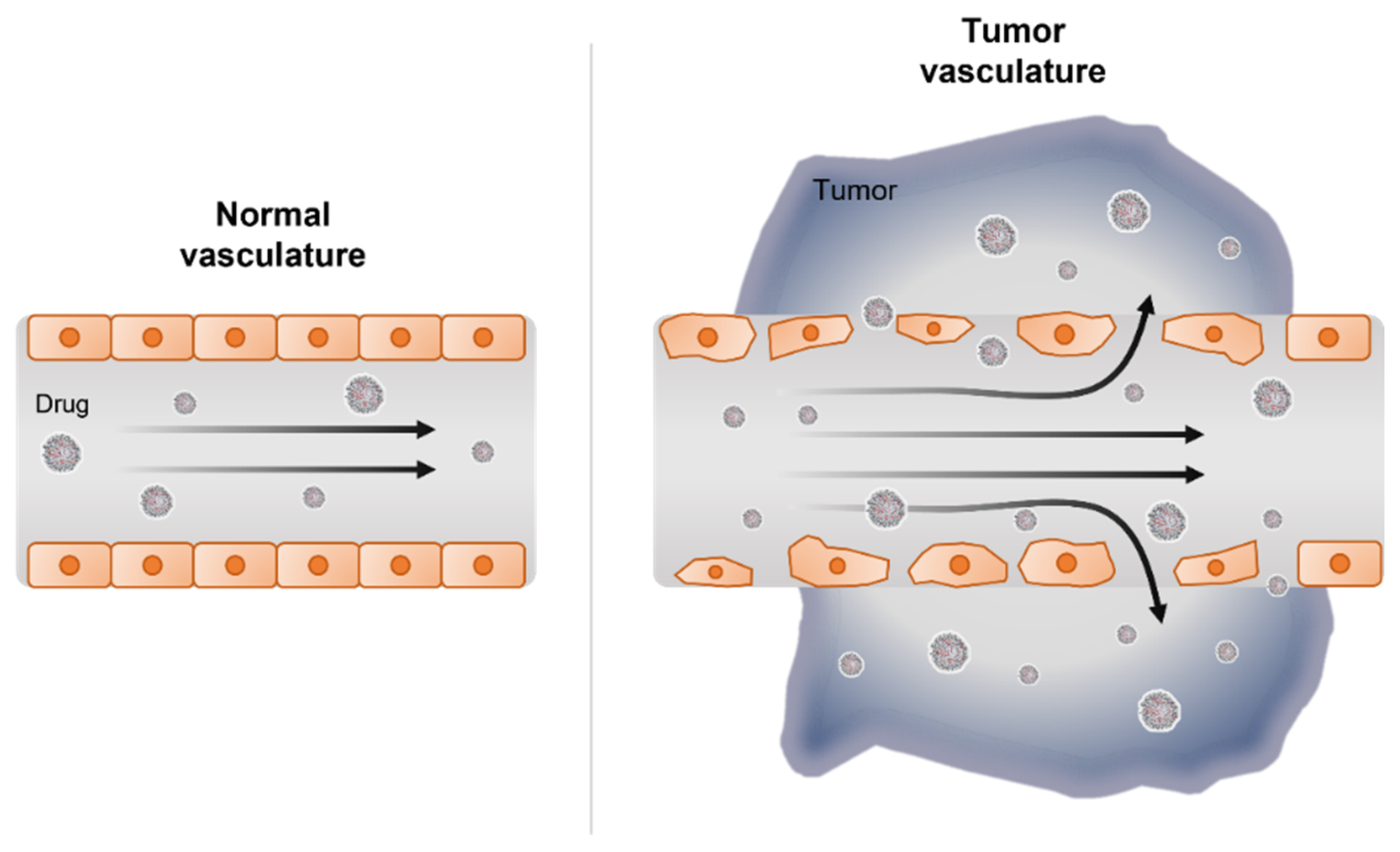
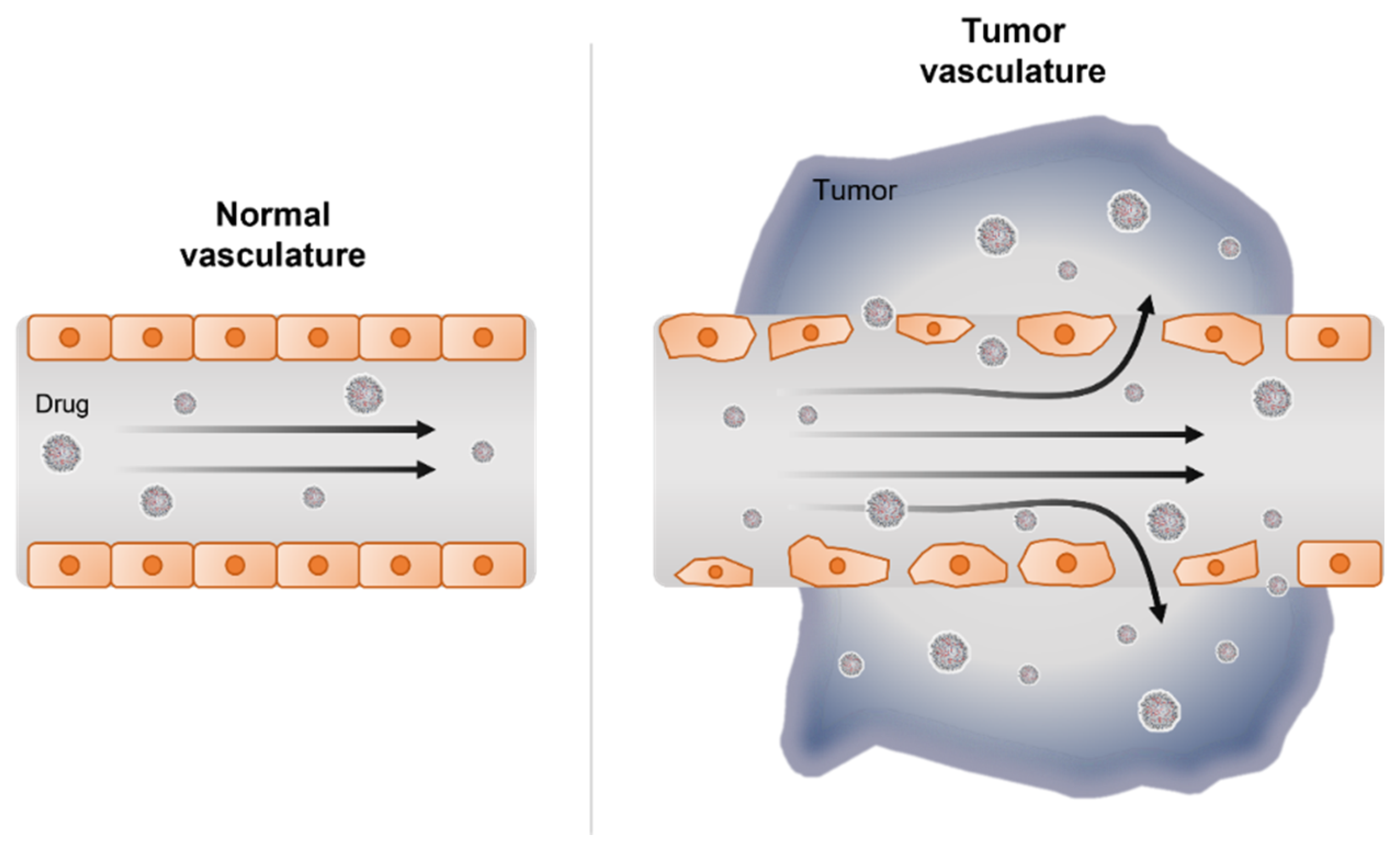
3. RNAi Delivery Strategies for Pancreatic Cancer Therapy
3.1. Nanocarrier-Mediated RNAi Therapy
3.1.1. Lipid-Based Nanoparticle Delivery
3.1.2. Metal Nanoparticle USE for Delivery
3.1.3. Polymer-Based Nanoparticle Delivery
3.1.4. Extracellular Vesicle-Mediated Delivery
3.2. Combination Therapy
4. Challenges and Future Prospects
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamisawa, T.; Wood, L.D.; Itoi, T.; Takaori, K. Pancreatic cancer. Lancet 2016, 388, 73–85. [Google Scholar] [CrossRef]
- Roth, M.T.; Cardin, D.B.; Berlin, J.D. Recent advances in the treatment of pancreatic cancer. F1000 Res. 2020, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2, 16022. [Google Scholar] [CrossRef]
- Ho, W.J.; Jaffee, E.M.; Zheng, L. The tumour microenvironment in pancreatic cancer—Clinical challenges and opportunities. Nat. Rev. Clin. Oncol. 2020, 17, 527–540. [Google Scholar] [CrossRef]
- Clark, C.E.; Hingorani, S.R.; Mick, R.; Combs, C.; Tuveson, D.A.; Vonderheide, R.H. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007, 67, 9518–9527. [Google Scholar] [CrossRef] [Green Version]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef] [Green Version]
- Stromnes, I.M.; Brockenbrough, J.S.; Izeradjene, K.; Carlson, M.A.; Cuevas, C.; Simmons, R.M.; Greenberg, P.D.; Hingorani, S.R. Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut 2014, 63, 1769–1781. [Google Scholar] [CrossRef] [Green Version]
- Provenzano, P.P.; Hingorani, S.R. Hyaluronan, fluid pressure, and stromal resistance in pancreas cancer. Br. J. Cancer 2013, 108, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Daniel, S.K.; Sullivan, K.M.; Labadie, K.P.; Pillarisetty, V.G. Hypoxia as a barrier to immunotherapy in pancreatic adenocarcinoma. Clin. Transl. Med. 2019, 8, 10. [Google Scholar] [CrossRef]
- Doktorova, H.; Hrabeta, J.; Khalil, M.A.; Eckschlager, T. Hypoxia-induced chemoresistance in cancer cells: The role of not only HIF-1. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc. Czech Repub. 2015, 159, 166–177. [Google Scholar] [CrossRef] [Green Version]
- Bachem, M.G.; Schneider, E.; Gross, H.; Weidenbach, H.; Schmid, R.M.; Menke, A.; Siech, M.; Beger, H.; Grunert, A.; Adler, G. Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology 1998, 115, 421–432. [Google Scholar] [CrossRef]
- Xu, Z.; Vonlaufen, A.; Phillips, P.A.; Fiala-Beer, E.; Zhang, X.; Yang, L.; Biankin, A.V.; Goldstein, D.; Pirola, R.C.; Wilson, J.S.; et al. Role of pancreatic stellate cells in pancreatic cancer metastasis. Am. J. Pathol. 2010, 177, 2585–2596. [Google Scholar] [CrossRef]
- Fu, Y.; Liu, S.; Zeng, S.; Shen, H. The critical roles of activated stellate cells-mediated paracrine signaling, metabolism and onco-immunology in pancreatic ductal adenocarcinoma. Mol. Cancer 2018, 17, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusiak, A.A.; Szopa, M.D.; Jakubowska, M.A.; Ferdek, P.E. Signaling in the Physiology and Pathophysiology of Pancreatic Stellate Cells—A Brief Review of Recent Advances. Front. Physiol. 2020, 11, 78. [Google Scholar] [CrossRef]
- Waters, A.M.; Der, C.J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a031435. [Google Scholar] [CrossRef]
- Hu, H.F.; Ye, Z.; Qin, Y.; Xu, X.W.; Yu, X.J.; Zhuo, Q.F.; Ji, S.R. Mutations in key driver genes of pancreatic cancer: Molecularly targeted therapies and other clinical implications. Acta Pharmacol. Sin. 2021. [Google Scholar] [CrossRef] [PubMed]
- Kristen, A.V.; Ajroud-Driss, S.; Conceicao, I.; Gorevic, P.; Kyriakides, T.; Obici, L. Patisiran, an RNAi therapeutic for the treatment of hereditary transthyretin-mediated amyloidosis. Neurodegener. Dis. Manag. 2019, 9, 5–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Setten, R.L.; Rossi, J.J.; Han, S.P. The current state and future directions of RNAi-based therapeutics. Nat. Rev. Drug Discov. 2019, 18, 421–446. [Google Scholar] [CrossRef]
- Hu, B.; Weng, Y.; Xia, X.H.; Liang, X.J.; Huang, Y. Clinical advances of siRNA therapeutics. J. Gene Med. 2019, 21, e3097. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, C.; Sharma, A.R.; Sharma, G.; Doss, C.G.P.; Lee, S.S. Therapeutic miRNA and siRNA: Moving from Bench to Clinic as Next Generation Medicine. Mol. Ther. Nucleic Acids 2017, 8, 132–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saw, P.E.; Song, E.W. siRNA therapeutics: A clinical reality. Sci. China Life Sci. 2020, 63, 485–500. [Google Scholar] [CrossRef]
- Singh, A.; Trivedi, P.; Jain, N.K. Advances in siRNA delivery in cancer therapy. Artif. Cells Nanomed. Biotechnol. 2018, 46, 274–283. [Google Scholar] [CrossRef]
- Tatiparti, K.; Sau, S.; Kashaw, S.K.; Iyer, A.K. siRNA Delivery Strategies: A Comprehensive Review of Recent Developments. Nanomaterials 2017, 7, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rana, T.M. Illuminating the silence: Understanding the structure and function of small RNAs. Nat. Rev. Mol. Cell Biol. 2007, 8, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Sajid, M.I.; Moazzam, M.; Kato, S.; Yeseom Cho, K.; Tiwari, R.K. Overcoming Barriers for siRNA Therapeutics: From Bench to Bedside. Pharmaceuticals 2020, 13, 294. [Google Scholar] [CrossRef]
- Gebert, L.F.R.; MacRae, I.J. Regulation of microRNA function in animals. Nat. Rev. Mol. Cell Biol. 2019, 20, 21–37. [Google Scholar] [CrossRef]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal Transduct. Target Ther. 2016, 1, 15004. [Google Scholar] [CrossRef] [Green Version]
- Gurbuz, N.; Ozpolat, B. MicroRNA-based Targeted Therapeutics in Pancreatic Cancer. Anticancer Res. 2019, 39, 529–532. [Google Scholar] [CrossRef] [Green Version]
- Bjornmalm, M.; Thurecht, K.J.; Michael, M.; Scott, A.M.; Caruso, F. Bridging Bio-Nano Science and Cancer Nanomedicine. ACS Nano 2017, 11, 9594–9613. [Google Scholar] [CrossRef]
- Hruban, R.H.; Adsay, N.V.; Albores-Saavedra, J.; Compton, C.; Garrett, E.S.; Goodman, S.N.; Kern, S.E.; Klimstra, D.S.; Kloppel, G.; Longnecker, D.S.; et al. Pancreatic intraepithelial neoplasia: A new nomenclature and classification system for pancreatic duct lesions. Am. J. Surg. Pathol. 2001, 25, 579–586. [Google Scholar] [CrossRef]
- Hou, P.; Kapoor, A.; Zhang, Q.; Li, J.; Wu, C.J.; Li, J.; Lan, Z.; Tang, M.; Ma, X.; Ackroyd, J.J.; et al. Tumor Microenvironment Remodeling Enables Bypass of Oncogenic KRAS Dependency in Pancreatic Cancer. Cancer Discov. 2020, 10, 1058–1077. [Google Scholar] [CrossRef]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Kokkinos, J.; Ignacio, R.M.C.; Sharbeen, G.; Boyer, C.; Gonzales-Aloy, E.; Goldstein, D.; Australian Pancreatic Cancer Genome Initiative, A.; McCarroll, J.A.; Phillips, P.A. Targeting the undruggable in pancreatic cancer using nano-based gene silencing drugs. Biomaterials 2020, 240, 119742. [Google Scholar] [CrossRef]
- Dang, C.V.; Reddy, E.P.; Shokat, K.M.; Soucek, L. Drugging the ‘undruggable’ cancer targets. Nat. Rev. Cancer 2017, 17, 502–508. [Google Scholar] [CrossRef]
- Morris, J.P.t.; Wang, S.C.; Hebrok, M. KRAS, Hedgehog, Wnt and the twisted developmental biology of pancreatic ductal adenocarcinoma. Nat. Rev. Cancer 2010, 10, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Kanda, M.; Matthaei, H.; Wu, J.; Hong, S.M.; Yu, J.; Borges, M.; Hruban, R.H.; Maitra, A.; Kinzler, K.; Vogelstein, B.; et al. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology 2012, 142, 730–733 e739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Distler, M.; Aust, D.; Weitz, J.; Pilarsky, C.; Grutzmann, R. Precursor lesions for sporadic pancreatic cancer: PanIN, IPMN, and MCN. Biomed. Res. Int. 2014, 2014, 474905. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Komar, C.A.; Bengsch, F.; Graham, K.; Beatty, G.L. Genetically Engineered Mouse Models of Pancreatic Cancer: The KPC Model (LSL-Kras(G12D/+);LSL-Trp53(R172H/+);Pdx-1-Cre), Its Variants, and Their Application in Immuno-oncology Drug Discovery. Curr. Protoc. Pharmacol. 2016, 73, 143911–143920. [Google Scholar] [CrossRef] [Green Version]
- Gopinathan, A.; Morton, J.P.; Jodrell, D.I.; Sansom, O.J. GEMMs as preclinical models for testing pancreatic cancer therapies. Dis. Model Mech. 2015, 8, 1185–1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardeesy, N.; Aguirre, A.J.; Chu, G.C.; Cheng, K.H.; Lopez, L.V.; Hezel, A.F.; Feng, B.; Brennan, C.; Weissleder, R.; Mahmood, U.; et al. Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc. Natl. Acad. Sci. USA 2006, 103, 5947–5952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguirre, A.J.; Bardeesy, N.; Sinha, M.; Lopez, L.; Tuveson, D.A.; Horner, J.; Redston, M.S.; DePinho, R.A. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003, 17, 3112–3126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRAS(G12C) Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, M.; Wang, H.; Fisher, W.E.; Lin, P.H.; Yao, Q.; Chen, C. Profiling of 95 microRNAs in pancreatic cancer cell lines and surgical specimens by real-time PCR analysis. World J. Surg. 2009, 33, 698–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, M.Z.; Kong, X.; Weng, M.Z.; Cheng, K.; Gong, W.; Quan, Z.W.; Peng, C.H. Candidate microRNA biomarkers of pancreatic ductal adenocarcinoma: Meta-analysis, experimental validation and clinical significance. J. Exp. Clin. Cancer Res. 2013, 32, 71. [Google Scholar] [CrossRef] [Green Version]
- Passadouro, M.; Faneca, H. Managing Pancreatic Adenocarcinoma: A Special Focus in MicroRNA Gene Therapy. Int. J. Mol. Sci. 2016, 17, 718. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.; Fesler, A.; Wang, H.; Ju, J. microRNA based prognostic biomarkers in pancreatic Cancer. Biomark. Res. 2018, 6, 18. [Google Scholar] [CrossRef]
- Sasayama, Y.; Hasegawa, M.; Taguchi, E.; Kubota, K.; Kuboyama, T.; Naoi, T.; Yabuuchi, H.; Shimai, N.; Asano, M.; Tokunaga, A.; et al. In vivo activation of PEGylated long circulating lipid nanoparticle to achieve efficient siRNA delivery and target gene knock down in solid tumors. J. Control. Release 2019, 311-312, 245–256. [Google Scholar] [CrossRef]
- Rao, D.D.; Luo, X.; Wang, Z.; Jay, C.M.; Brunicardi, F.C.; Maltese, W.; Manning, L.; Senzer, N.; Nemunaitis, J. KRAS mutant allele-specific expression knockdown in pancreatic cancer model with systemically delivered bi-shRNA KRAS lipoplex. PLoS ONE 2018, 13, e0193644. [Google Scholar] [CrossRef]
- Aleku, M.; Schulz, P.; Keil, O.; Santel, A.; Schaeper, U.; Dieckhoff, B.; Janke, O.; Endruschat, J.; Durieux, B.; Roder, N.; et al. Atu027, a liposomal small interfering RNA formulation targeting protein kinase N3, inhibits cancer progression. Cancer Res. 2008, 68, 9788–9798. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Zhang, Z. Nanoformulation of Apolipoprotein E3-Tagged Liposomal Nanoparticles for the co-Delivery of KRAS-siRNA and Gemcitabine for Pancreatic Cancer Treatment. Pharm. Res. 2020, 37, 247. [Google Scholar] [CrossRef]
- Lei, Y.; Tang, L.; Xie, Y.; Xianyu, Y.; Zhang, L.; Wang, P.; Hamada, Y.; Jiang, K.; Zheng, W.; Jiang, X. Gold nanoclusters-assisted delivery of NGF siRNA for effective treatment of pancreatic cancer. Nat. Commun. 2017, 8, 15130. [Google Scholar] [CrossRef]
- Partecke, I.L.; Kaeding, A.; Sendler, M.; Albers, N.; Kuhn, J.P.; Speerforck, S.; Roese, S.; Seubert, F.; Diedrich, S.; Kuehn, S.; et al. In vivo imaging of pancreatic tumours and liver metastases using 7 Tesla MRI in a murine orthotopic pancreatic cancer model and a liver metastases model. BMC Cancer 2011, 11, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahajan, U.M.; Teller, S.; Sendler, M.; Palankar, R.; van den Brandt, C.; Schwaiger, T.; Kuhn, J.P.; Ribback, S.; Glockl, G.; Evert, M.; et al. Tumour-specific delivery of siRNA-coupled superparamagnetic iron oxide nanoparticles, targeted against PLK1, stops progression of pancreatic cancer. Gut 2016, 65, 1838–1849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teo, J.; McCarroll, J.A.; Boyer, C.; Youkhana, J.; Sagnella, S.M.; Duong, H.T.; Liu, J.; Sharbeen, G.; Goldstein, D.; Davis, T.P.; et al. A Rationally Optimized Nanoparticle System for the Delivery of RNA Interference Therapeutics into Pancreatic Tumors in Vivo. Biomacromolecules 2016, 17, 2337–2351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarroll, J.A.; Sharbeen, G.; Kavallaris, M.; Phillips, P.A. The Use of Star Polymer Nanoparticles for the Delivery of siRNA to Mouse Orthotopic Pancreatic Tumor Models. Methods Mol. Biol. 2019, 1974, 329–353. [Google Scholar] [CrossRef]
- Zhang, N.; Lyons, S.; Lim, E.; Lassota, P. A spontaneous acinar cell carcinoma model for monitoring progression of pancreatic lesions and response to treatment through noninvasive bioluminescence imaging. Clin. Cancer Res. 2009, 15, 4915–4924. [Google Scholar] [CrossRef] [Green Version]
- Pittella, F.; Cabral, H.; Maeda, Y.; Mi, P.; Watanabe, S.; Takemoto, H.; Kim, H.J.; Nishiyama, N.; Miyata, K.; Kataoka, K. Systemic siRNA delivery to a spontaneous pancreatic tumor model in transgenic mice by PEGylated calcium phosphate hybrid micelles. J. Control. Release 2014, 178, 18–24. [Google Scholar] [CrossRef]
- Pittella, F.; Miyata, K.; Maeda, Y.; Suma, T.; Watanabe, S.; Chen, Q.; Christie, R.J.; Osada, K.; Nishiyama, N.; Kataoka, K. Pancreatic cancer therapy by systemic administration of VEGF siRNA contained in calcium phosphate/charge-conversional polymer hybrid nanoparticles. J. Control. Release 2012, 161, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Zorde Khvalevsky, E.; Gabai, R.; Rachmut, I.H.; Horwitz, E.; Brunschwig, Z.; Orbach, A.; Shemi, A.; Golan, T.; Domb, A.J.; Yavin, E.; et al. Mutant KRAS is a druggable target for pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20723–20728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bangoura, G.; Yang, L.Y.; Huang, G.W.; Wang, W. Expression of HIF-2alpha/EPAS1 in hepatocellular carcinoma. World J. Gastroenterol. 2004, 10, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kumar, S.; Rachagani, S.; Sajja, B.R.; Xie, Y.; Hang, Y.; Jain, M.; Li, J.; Boska, M.D.; Batra, S.K.; et al. Polyplex-mediated inhibition of chemokine receptor CXCR4 and chromatin-remodeling enzyme NCOA3 impedes pancreatic cancer progression and metastasis. Biomaterials 2016, 101, 108–120. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Hang, Y.; Wang, Y.; Sleightholm, R.; Prajapati, D.R.; Bader, J.; Yu, A.; Tang, W.; Jaramillo, L.; Li, J.; et al. Stromal Modulation and Treatment of Metastatic Pancreatic Cancer with Local Intraperitoneal Triple miRNA/siRNA Nanotherapy. ACS Nano 2020, 14, 255–271. [Google Scholar] [CrossRef]
- Lin, G.; Chen, C.K.; Yin, F.; Yang, C.; Tian, J.; Chen, T.; Xu, G.; He, C.; Lin, M.C.; Wang, J.; et al. Biodegradable nanoparticles as siRNA carriers for in vivo gene silencing and pancreatic cancer therapy. J. Mater. Chem. B 2017, 5, 3327–3337. [Google Scholar] [CrossRef]
- Taniuchi, K.; Yawata, T.; Tsuboi, M.; Ueba, T.; Saibara, T. Efficient delivery of small interfering RNAs targeting particular mRNAs into pancreatic cancer cells inhibits invasiveness and metastasis of pancreatic tumors. Oncotarget 2019, 10, 2869–2886. [Google Scholar] [CrossRef] [Green Version]
- Lo, J.H.; Hao, L.; Muzumdar, M.D.; Raghavan, S.; Kwon, E.J.; Pulver, E.M.; Hsu, F.; Aguirre, A.J.; Wolpin, B.M.; Fuchs, C.S.; et al. iRGD-guided Tumor-penetrating Nanocomplexes for Therapeutic siRNA Delivery to Pancreatic Cancer. Mol. Cancer. Ther. 2018, 17, 2377–2388. [Google Scholar] [CrossRef] [Green Version]
- Zeng, L.; Li, J.; Wang, Y.; Qian, C.; Chen, Y.; Zhang, Q.; Wu, W.; Lin, Z.; Liang, J.; Shuai, X.; et al. Combination of siRNA-directed Kras oncogene silencing and arsenic-induced apoptosis using a nanomedicine strategy for the effective treatment of pancreatic cancer. Nanomedicine 2014, 10, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, H.; Wang, K.; Hu, Q.; Yao, Q.; Shen, Y.; Yu, G.; Tang, G. Targeted Co-delivery of PTX and TR3 siRNA by PTP Peptide Modified Dendrimer for the Treatment of Pancreatic Cancer. Small 2017, 13, 10–1002. [Google Scholar] [CrossRef] [PubMed]
- Yoo, B.; Jordan, V.C.; Sheedy, P.; Billig, A.M.; Ross, A.; Pantazopoulos, P.; Medarova, Z. RNAi-Mediated PD-L1 Inhibition for Pancreatic Cancer Immunotherapy. Sci. Rep. 2019, 9, 4712. [Google Scholar] [CrossRef]
- Kamerkar, S.; LeBleu, V.S.; Sugimoto, H.; Yang, S.; Ruivo, C.F.; Melo, S.A.; Lee, J.J.; Kalluri, R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 2017, 546, 498–503. [Google Scholar] [CrossRef]
- Zhou, W.; Zhou, Y.; Chen, X.; Ning, T.; Chen, H.; Guo, Q.; Zhang, Y.; Liu, P.; Zhang, Y.; Li, C.; et al. Pancreatic cancer-targeting exosomes for enhancing immunotherapy and reprogramming tumor microenvironment. Biomaterials 2021, 268, 120546. [Google Scholar] [CrossRef]
- Feig, C.; Gopinathan, A.; Neesse, A.; Chan, D.S.; Cook, N.; Tuveson, D.A. The pancreas cancer microenvironment. Clin. Cancer Res. 2012, 18, 4266–4276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, L.; Xue, J.; Jaffee, E.M.; Habtezion, A. Role of immune cells and immune-based therapies in pancreatitis and pancreatic ductal adenocarcinoma. Gastroenterology 2013, 144, 1230–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lafaro, K.J.; Melstrom, L.G. The Paradoxical Web of Pancreatic Cancer Tumor Microenvironment. Am. J. Pathol. 2019, 189, 44–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnittert, J.; Bansal, R.; Prakash, J. Targeting Pancreatic Stellate Cells in Cancer. Trends Cancer 2019, 5, 128–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najafi, M.; Mortezaee, K.; Majidpoor, J. Stromal reprogramming: A target for tumor therapy. Life Sci. 2019, 239, 117049. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Yoo, W.; Lee, J.; Kim, H.; Lee, H.; Kim, Y.S.; Kim, D.U.; Oh, J. Formation of vitamin A lipid droplets in pancreatic stellate cells requires albumin. Gut 2009, 58, 1382–1390. [Google Scholar] [CrossRef] [PubMed]
- Neesse, A.; Algul, H.; Tuveson, D.A.; Gress, T.M. Stromal biology and therapy in pancreatic cancer: A changing paradigm. Gut 2015, 64, 1476–1484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masamune, A.; Kikuta, K.; Watanabe, T.; Satoh, K.; Hirota, M.; Shimosegawa, T. Hypoxia stimulates pancreatic stellate cells to induce fibrosis and angiogenesis in pancreatic cancer. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, G709–G717. [Google Scholar] [CrossRef] [Green Version]
- Kollmann, T.R.; Levy, O.; Montgomery, R.R.; Goriely, S. Innate immune function by Toll-like receptors: Distinct responses in newborns and the elderly. Immunity 2012, 37, 771–783. [Google Scholar] [CrossRef] [Green Version]
- Masamune, A.; Kikuta, K.; Watanabe, T.; Satoh, K.; Satoh, A.; Shimosegawa, T. Pancreatic stellate cells express Toll-like receptors. J. Gastroenterol. 2008, 43, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yue, D.; Cheng, L.; Huang, A.; Tong, N.; Cheng, P. Vitamin A-coupled liposomes carrying TLR4-silencing shRNA induce apoptosis of pancreatic stellate cells and resolution of pancreatic fibrosis. J. Mol. Med. 2018, 96, 445–458. [Google Scholar] [CrossRef]
- Ishiwatari, H.; Sato, Y.; Murase, K.; Yoneda, A.; Fujita, R.; Nishita, H.; Birukawa, N.K.; Hayashi, T.; Sato, T.; Miyanishi, K.; et al. Treatment of pancreatic fibrosis with siRNA against a collagen-specific chaperone in vitamin A-coupled liposomes. Gut 2013, 62, 1328–1339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, X.; Li, Y.; Xu, Y.; Zhao, X.; Zhang, Y.; Yang, X.; Wang, Y.; Zhao, R.; Anderson, G.J.; Zhao, Y.; et al. Reversal of pancreatic desmoplasia by re-educating stellate cells with a tumour microenvironment-activated nanosystem. Nat. Commun. 2018, 9, 3390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeNardo, D.G.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef]
- Argyle, D.; Kitamura, T. Targeting Macrophage-Recruiting Chemokines as a Novel Therapeutic Strategy to Prevent the Progression of Solid Tumors. Front. Immunol. 2018, 9, 2629. [Google Scholar] [CrossRef]
- Qian, B.Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef] [Green Version]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [Green Version]
- Qian, Y.; Qiao, S.; Dai, Y.; Xu, G.; Dai, B.; Lu, L.; Yu, X.; Luo, Q.; Zhang, Z. Molecular-Targeted Immunotherapeutic Strategy for Melanoma via Dual-Targeting Nanoparticles Delivering Small Interfering RNA to Tumor-Associated Macrophages. ACS Nano 2017, 11, 9536–9549. [Google Scholar] [CrossRef]
- Li, M.; Li, M.; Yang, Y.; Liu, Y.; Xie, H.; Yu, Q.; Tian, L.; Tang, X.; Ren, K.; Li, J.; et al. Remodeling tumor immune microenvironment via targeted blockade of PI3K-gamma and CSF-1/CSF-1R pathways in tumor associated macrophages for pancreatic cancer therapy. J. Control. Release 2020, 321, 23–35. [Google Scholar] [CrossRef]
- Arora, S.; Swaminathan, S.K.; Kirtane, A.; Srivastava, S.K.; Bhardwaj, A.; Singh, S.; Panyam, J.; Singh, A.P. Synthesis, characterization, and evaluation of poly (D,L-lactide-co-glycolide)-based nanoformulation of miRNA-150: Potential implications for pancreatic cancer therapy. Int. J. Nanomedicine 2014, 9, 2933–2942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, G.K.; Khan, M.A.; Bhardwaj, A.; Srivastava, S.K.; Zubair, H.; Patton, M.C.; Singh, S.; Khushman, M.; Singh, A.P. Exosomes confer chemoresistance to pancreatic cancer cells by promoting ROS detoxification and miR-155-mediated suppression of key gemcitabine-metabolising enzyme, DCK. Br. J. Cancer 2017, 116, 609–619. [Google Scholar] [CrossRef] [Green Version]
- Schnittert, J.; Kuninty, P.R.; Bystry, T.F.; Brock, R.; Storm, G.; Prakash, J. Anti-microRNA targeting using peptide-based nanocomplexes to inhibit differentiation of human pancreatic stellate cells. Nanomedicine 2017, 12, 1369–1384. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhao, L.; Ischenko, I.; Bao, Q.; Schwarz, B.; Niess, H.; Wang, Y.; Renner, A.; Mysliwietz, J.; Jauch, K.W.; et al. Antisense inhibition of microRNA-21 and microRNA-221 in tumor-initiating stem-like cells modulates tumorigenesis, metastasis, and chemotherapy resistance in pancreatic cancer. Target Oncol. 2015, 10, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, Y.; Li, J.; Zhang, Z.; Huang, C.; Lian, G.; Yang, K.; Chen, S.; Lin, Y.; Wang, L.; et al. Co-delivery of microRNA-21 antisense oligonucleotides and gemcitabine using nanomedicine for pancreatic cancer therapy. Cancer Sci. 2017, 108, 1493–1503. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zhou, Y.; Zhi, X.; Ma, T.; Liu, H.; Chen, B.W.; Zheng, X.; Xie, S.; Zhao, B.; Feng, X.; et al. Delivery of miR-212 by chimeric peptide-condensed supramolecular nanoparticles enhances the sensitivity of pancreatic ductal adenocarcinoma to doxorubicin. Biomaterials 2019, 192, 590–600. [Google Scholar] [CrossRef]
- Uz, M.; Kalaga, M.; Pothuraju, R.; Ju, J.; Junker, W.M.; Batra, S.K.; Mallapragada, S.; Rachagani, S. Dual delivery nanoscale device for miR-345 and gemcitabine co-delivery to treat pancreatic cancer. J. Control. Release 2019, 294, 237–246. [Google Scholar] [CrossRef]
- Hu, Q.L.; Jiang, Q.Y.; Jin, X.; Shen, J.; Wang, K.; Li, Y.B.; Xu, F.J.; Tang, G.P.; Li, Z.H. Cationic microRNA-delivering nanovectors with bifunctional peptides for efficient treatment of PANC-1 xenograft model. Biomaterials 2013, 34, 2265–2276. [Google Scholar] [CrossRef]
- Pramanik, D.; Campbell, N.R.; Karikari, C.; Chivukula, R.; Kent, O.A.; Mendell, J.T.; Maitra, A. Restitution of tumor suppressor microRNAs using a systemic nanovector inhibits pancreatic cancer growth in mice. Mol. Cancer Ther. 2011, 10, 1470–1480. [Google Scholar] [CrossRef] [Green Version]
- Contado, C. Nanomaterials in consumer products: A challenging analytical problem. Front. Chem. 2015, 3, 48. [Google Scholar] [CrossRef] [Green Version]
- Tinkle, S.; McNeil, S.E.; Muhlebach, S.; Bawa, R.; Borchard, G.; Barenholz, Y.C.; Tamarkin, L.; Desai, N. Nanomedicines: Addressing the scientific and regulatory gap. Ann. N. Y. Acad. Sci. 2014, 1313, 35–56. [Google Scholar] [CrossRef]
- Behlke, M.A. Chemical modification of siRNAs for in vivo use. Oligonucleotides 2008, 18, 305–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenblum, D.; Joshi, N.; Tao, W.; Karp, J.M.; Peer, D. Progress and challenges towards targeted delivery of cancer therapeutics. Nat. Commun. 2018, 9, 1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greish, K. Enhanced permeability and retention (EPR) effect for anticancer nanomedicine drug targeting. Methods Mol. Biol. 2010, 624, 25–37. [Google Scholar] [CrossRef]
- Azzopardi, E.A.; Ferguson, E.L.; Thomas, D.W. The enhanced permeability retention effect: A new paradigm for drug targeting in infection. J. Antimicrob. Chemother. 2013, 68, 257–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 2017, 17, 20–37. [Google Scholar] [CrossRef]
- Attia, M.F.; Anton, N.; Wallyn, J.; Omran, Z.; Vandamme, T.F. An overview of active and passive targeting strategies to improve the nanocarriers efficiency to tumour sites. J. Pharm. Pharmacol. 2019, 71, 1185–1198. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, J.A.; Witzigmann, D.; Chen, S.; Cullis, P.R.; van der Meel, R. Lipid Nanoparticle Technology for Clinical Translation of siRNA Therapeutics. Acc. Chem. Res. 2019, 52, 2435–2444. [Google Scholar] [CrossRef]
- Yonezawa, S.; Koide, H.; Asai, T. Recent advances in siRNA delivery mediated by lipid-based nanoparticles. Adv. Drug Deliv. Rev. 2020, 154–155, 64–78. [Google Scholar] [CrossRef]
- Kim, Y.K. RNA Therapy: Current Status and Future Potential. Chonnam Med. J. 2020, 56, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Rao, D.D.; Maples, P.B.; Senzer, N.; Kumar, P.; Wang, Z.; Pappen, B.O.; Yu, Y.; Haddock, C.; Jay, C.; Phadke, A.P.; et al. Enhanced target gene knockdown by a bifunctional shRNA: A novel approach of RNA interference. Cancer Gene Ther. 2010, 17, 780–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santel, A.; Aleku, M.; Keil, O.; Endruschat, J.; Esche, V.; Durieux, B.; Loffler, K.; Fechtner, M.; Rohl, T.; Fisch, G.; et al. RNA interference in the mouse vascular endothelium by systemic administration of siRNA-lipoplexes for cancer therapy. Gene Ther. 2006, 13, 1360–1370. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.H.; Yap, T.A.; Yan, L.; Cunningham, D. Targeting the PI3K-AKT-mTOR signaling network in cancer. Chin. J. Cancer 2013, 32, 253–265. [Google Scholar] [CrossRef] [Green Version]
- Jopling, C.L.; Yi, M.; Lancaster, A.M.; Lemon, S.M.; Sarnow, P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science 2005, 309, 1577–1581. [Google Scholar] [CrossRef] [Green Version]
- Ohno, S.; Takanashi, M.; Sudo, K.; Ueda, S.; Ishikawa, A.; Matsuyama, N.; Fujita, K.; Mizutani, T.; Ohgi, T.; Ochiya, T.; et al. Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol. Ther. 2013, 21, 185–191. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Crawford, M.; Mao, Y.; Lee, R.J.; Davis, I.C.; Elton, T.S.; Lee, L.J.; Nana-Sinkam, S.P. Therapeutic Delivery of MicroRNA-29b by Cationic Lipoplexes for Lung Cancer. Mol. Ther. Nucleic Acids 2013, 2, e84. [Google Scholar] [CrossRef]
- Drury, R.E.; O’Connor, D.; Pollard, A.J. The Clinical Application of MicroRNAs in Infectious Disease. Front. Immunol. 2017, 8, 1182. [Google Scholar] [CrossRef]
- Beg, M.S.; Brenner, A.J.; Sachdev, J.; Borad, M.; Kang, Y.K.; Stoudemire, J.; Smith, S.; Bader, A.G.; Kim, S.; Hong, D.S. Phase I study of MRX34, a liposomal miR-34a mimic, administered twice weekly in patients with advanced solid tumors. Investig. New Drugs 2017, 35, 180–188. [Google Scholar] [CrossRef]
- Hanna, J.; Hossain, G.S.; Kocerha, J. The Potential for microRNA Therapeutics and Clinical Research. Front. Genet. 2019, 10, 478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Cheng, Z.; Wang, Y.; Han, T. The Risks of miRNA Therapeutics: In a Drug Target Perspective. Drug Des. Devel. Ther. 2021, 15, 721–733. [Google Scholar] [CrossRef]
- Sardar, R.; Funston, A.M.; Mulvaney, P.; Murray, R.W. Gold nanoparticles: Past, present, and future. Langmuir 2009, 25, 13840–13851. [Google Scholar] [CrossRef] [PubMed]
- Carabineiro, S.A.C. Applications of Gold Nanoparticles in Nanomedicine: Recent Advances in Vaccines. Molecules 2017, 22, 857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jobling, P.; Pundavela, J.; Oliveira, S.M.; Roselli, S.; Walker, M.M.; Hondermarck, H. Nerve-Cancer Cell Cross-talk: A Novel Promoter of Tumor Progression. Cancer Res. 2015, 75, 1777–1781. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Wu, H.; Shi, H.; Wang, M.; Huang, C.; Jia, N. A novel multifunctional biomimetic Au@BSA nanocarrier as a potential siRNA theranostic nanoplatform. J. Mater. Chem. B 2016, 4, 2519–2526. [Google Scholar] [CrossRef]
- Soetaert, F.; Korangath, P.; Serantes, D.; Fiering, S.; Ivkov, R. Cancer therapy with iron oxide nanoparticles: Agents of thermal and immune therapies. Adv. Drug Deliv. Rev. 2020, 163–164, 65–83. [Google Scholar] [CrossRef]
- Yin, F.; Hu, K.; Chen, Y.; Yu, M.; Wang, D.; Wang, Q.; Yong, K.T.; Lu, F.; Liang, Y.; Li, Z. SiRNA Delivery with PEGylated Graphene Oxide Nanosheets for Combined Photothermal and Genetherapy for Pancreatic Cancer. Theranostics 2017, 7, 1133–1148. [Google Scholar] [CrossRef] [Green Version]
- Anderson, T.; Hu, R.; Yang, C.; Yoon, H.S.; Yong, K.T. Pancreatic cancer gene therapy using an siRNA-functionalized single walled carbon nanotubes (SWNTs) nanoplex. Biomater. Sci. 2014, 2, 1244–1253. [Google Scholar] [CrossRef]
- Suk, J.S.; Xu, Q.; Kim, N.; Hanes, J.; Ensign, L.M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 2016, 99, 28–51. [Google Scholar] [CrossRef] [Green Version]
- Hofmann-Amtenbrink, M.; Grainger, D.W.; Hofmann, H. Nanoparticles in medicine: Current challenges facing inorganic nanoparticle toxicity assessments and standardizations. Nanomedicine 2015, 11, 1689–1694. [Google Scholar] [CrossRef]
- Sadat Tabatabaei Mirakabad, F.; Nejati-Koshki, K.; Akbarzadeh, A.; Yamchi, M.R.; Milani, M.; Zarghami, N.; Zeighamian, V.; Rahimzadeh, A.; Alimohammadi, S.; Hanifehpour, Y.; et al. PLGA-based nanoparticles as cancer drug delivery systems. Asian Pac. J. Cancer Prev. 2014, 15, 517–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, X.; Zhu, Q.; Sun, Y.; Li, L.; Zhu, Y.; Zhao, Z.; Zuo, J.; Fang, W.; Li, K. PLGA/poloxamer nanoparticles loaded with EPAS1 siRNA for the treatment of pancreatic cancer in vitro and in vivo. Int. J. Mol. Med. 2015, 35, 995–1002. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.K.; Law, W.C.; Aalinkeel, R.; Nair, B.; Kopwitthaya, A.; Mahajan, S.D.; Reynolds, J.L.; Zou, J.; Schwartz, S.A.; Prasad, P.N.; et al. Well-defined degradable cationic polylactide as nanocarrier for the delivery of siRNA to silence angiogenesis in prostate cancer. Adv. Healthc. Mater. 2012, 1, 751–761. [Google Scholar] [CrossRef] [PubMed]
- Garg, U.; Chauhan, S.; Nagaich, U.; Jain, N. Current Advances in Chitosan Nanoparticles Based Drug Delivery and Targeting. Adv. Pharm. Bull. 2019, 9, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Patel, M.; Yang, X.; Mitra, A.K. Recent advances in protein and Peptide drug delivery: A special emphasis on polymeric nanoparticles. Protein Pept. Lett. 2014, 21, 1102–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alipour, M.; Baneshi, M.; Hosseinkhani, S.; Mahmoudi, R.; Jabari Arabzadeh, A.; Akrami, M.; Mehrzad, J.; Bardania, H. Recent progress in biomedical applications of RGD-based ligand: From precise cancer theranostics to biomaterial engineering: A systematic review. J. Biomed. Mater. Res. A 2020, 108, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Pan, B.T.; Johnstone, R.M. Fate of the transferrin receptor during maturation of sheep reticulocytes in vitro: Selective externalization of the receptor. Cell 1983, 33, 967–978. [Google Scholar] [CrossRef]
- Thery, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Choi, Y.; Nam, G.H.; Kim, I.S. Functionalized exosome harboring bioactive molecules for cancer therapy. Cancer Lett. 2020, 489, 155–162. [Google Scholar] [CrossRef]
- Nam, G.H.; Choi, Y.; Kim, G.B.; Kim, S.; Kim, S.A.; Kim, I.S. Emerging Prospects of Exosomes for Cancer Treatment: From Conventional Therapy to Immunotherapy. Adv. Mater. 2020, 32, e2002440. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.B.; Nam, G.H.; Hong, Y.; Woo, J.; Cho, Y.; Kwon, I.C.; Yang, Y.; Kim, I.S. Xenogenization of tumor cells by fusogenic exosomes in tumor microenvironment ignites and propagates antitumor immunity. Sci. Adv. 2020, 6, eaaz2083. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Huang, L.; Mao, M.; Ding, J.; Wu, G.; Fan, W.; Yang, T.; Zhang, M.; Huang, Y.; Xie, H.Y. Viral Protein-Pseudotyped and siRNA-Electroporated Extracellular Vesicles for Cancer Immunotherapy. Adv. Funct. Mater. 2020, 30, 2006515. [Google Scholar] [CrossRef]
- Liu, C.; Su, C. Design strategies and application progress of therapeutic exosomes. Theranostics 2019, 9, 1015–1028. [Google Scholar] [CrossRef]
- O’Brien, K.; Lowry, M.C.; Corcoran, C.; Martinez, V.G.; Daly, M.; Rani, S.; Gallagher, W.M.; Radomski, M.W.; MacLeod, R.A.; O’Driscoll, L. miR-134 in extracellular vesicles reduces triple-negative breast cancer aggression and increases drug sensitivity. Oncotarget 2015, 6, 32774–32789. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Yang, C.; Shi, Y.; Zhao, L. Exosomes derived from siRNA against GRP78 modified bone-marrow-derived mesenchymal stem cells suppress Sorafenib resistance in hepatocellular carcinoma. J. Nanobiotechnol. 2018, 16, 103. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Chen, X.; Tian, B.; Liu, J.; Yang, L.; Zeng, L.; Chen, T.; Hong, A.; Wang, X. Nucleolin-targeted Extracellular Vesicles as a Versatile Platform for Biologics Delivery to Breast Cancer. Theranostics 2017, 7, 1360–1372. [Google Scholar] [CrossRef]
- Morad, G.; Carman, C.V.; Hagedorn, E.J.; Perlin, J.R.; Zon, L.I.; Mustafaoglu, N.; Park, T.E.; Ingber, D.E.; Daisy, C.C.; Moses, M.A. Tumor-Derived Extracellular Vesicles Breach the Intact Blood-Brain Barrier via Transcytosis. ACS Nano 2019, 13, 13853–13865. [Google Scholar] [CrossRef]
- Yang, T.; Fogarty, B.; LaForge, B.; Aziz, S.; Pham, T.; Lai, L.; Bai, S. Delivery of Small Interfering RNA to Inhibit Vascular Endothelial Growth Factor in Zebrafish Using Natural Brain Endothelia Cell-Secreted Exosome Nanovesicles for the Treatment of Brain Cancer. AAPS J. 2017, 19, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Katakowski, M.; Buller, B.; Zheng, X.; Lu, Y.; Rogers, T.; Osobamiro, O.; Shu, W.; Jiang, F.; Chopp, M. Exosomes from marrow stromal cells expressing miR-146b inhibit glioma growth. Cancer Lett. 2013, 335, 201–204. [Google Scholar] [CrossRef] [Green Version]
- Gourlay, J.; Morokoff, A.P.; Luwor, R.B.; Zhu, H.J.; Kaye, A.H.; Stylli, S.S. The emergent role of exosomes in glioma. J. Clin. Neurosci. 2017, 35, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Mendt, M.; Kamerkar, S.; Sugimoto, H.; McAndrews, K.M.; Wu, C.C.; Gagea, M.; Yang, S.; Blanko, E.V.R.; Peng, Q.; Ma, X.; et al. Generation and testing of clinical-grade exosomes for pancreatic cancer. JCI Insight 2018, 3, e99263. [Google Scholar] [CrossRef] [PubMed]
- Babu, A.; Munshi, A.; Ramesh, R. Combinatorial therapeutic approaches with RNAi and anticancer drugs using nanodrug delivery systems. Drug Dev. Ind. Pharm. 2017, 43, 1391–1401. [Google Scholar] [CrossRef]
- Creixell, M.; Peppas, N.A. Co-delivery of siRNA and therapeutic agents using nanocarriers to overcome cancer resistance. Nano Today 2012, 7, 367–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, S.; Pathak, K.; Vaidya, A. Molecular therapy using siRNA: Recent trends and advances of multi target inhibition of cancer growth. Int. J. Biol. Macromol. 2018, 116, 880–892. [Google Scholar] [CrossRef]
- Yu, Q.; Zhang, B.; Zhou, Y.; Ge, Q.; Chang, J.; Chen, Y.; Zhang, K.; Peng, D.; Chen, W. Co-delivery of gambogenic acid and VEGF-siRNA with anionic liposome and polyethylenimine complexes to HepG2 cells. J. Liposome Res. 2019, 29, 322–331. [Google Scholar] [CrossRef]
- Golan, T.; Khvalevsky, E.Z.; Hubert, A.; Gabai, R.M.; Hen, N.; Segal, A.; Domb, A.; Harari, G.; David, E.B.; Raskin, S.; et al. RNAi therapy targeting KRAS in combination with chemotherapy for locally advanced pancreatic cancer patients. Oncotarget 2015, 6, 24560–24570. [Google Scholar] [CrossRef] [Green Version]
- McCarroll, J.A.; Sharbeen, G.; Liu, J.; Youkhana, J.; Goldstein, D.; McCarthy, N.; Limbri, L.F.; Dischl, D.; Ceyhan, G.O.; Erkan, M.; et al. betaIII-tubulin: A novel mediator of chemoresistance and metastases in pancreatic cancer. Oncotarget 2015, 6, 2235–2249. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.X.; Wang, Y.; Blake, S.; Yu, M.; Mei, L.; Wang, H.; Shi, J. RNA Nanotechnology-Mediated Cancer Immunotherapy. Theranostics 2020, 10, 281–299. [Google Scholar] [CrossRef]
- Ma, J.; Dong, C.; Ji, C. MicroRNA and drug resistance. Cancer Gene Ther. 2010, 17, 523–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amrutkar, M.; Gladhaug, I.P. Pancreatic Cancer Chemoresistance to Gemcitabine. Cancers 2017, 9, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frixa, T.; Donzelli, S.; Blandino, G. Oncogenic MicroRNAs: Key Players in Malignant Transformation. Cancers 2015, 7, 2466–2485. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.H.; Tsao, C.J. Emerging role of microRNA-21 in cancer. Biomed. Rep. 2016, 5, 395–402. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Mondal, G.; Slavik, P.; Rachagani, S.; Batra, S.K.; Mahato, R.I. Codelivery of small molecule hedgehog inhibitor and miRNA for treating pancreatic cancer. Mol. Pharm. 2015, 12, 1289–1298. [Google Scholar] [CrossRef] [Green Version]
- Hidalgo, M.; Cascinu, S.; Kleeff, J.; Labianca, R.; Lohr, J.M.; Neoptolemos, J.; Real, F.X.; Van Laethem, J.L.; Heinemann, V. Addressing the challenges of pancreatic cancer: Future directions for improving outcomes. Pancreatology 2015, 15, 8–18. [Google Scholar] [CrossRef]
- Lewis, W.H. Pinocytosis by Malignant Cells. Am. J. Cancer 1937, 29, 666. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Sayed, N.; Ditsworth, D.; Thompson, C.B. Brick by brick: Metabolism and tumor cell growth. Curr. Opin. Genet. Dev. 2008, 18, 54–61. [Google Scholar] [CrossRef] [Green Version]
- Commisso, C.; Davidson, S.M.; Soydaner-Azeloglu, R.G.; Parker, S.J.; Kamphorst, J.J.; Hackett, S.; Grabocka, E.; Nofal, M.; Drebin, J.A.; Thompson, C.B.; et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 2013, 497, 633–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maltese, W.A.; Overmeyer, J.H. Non-apoptotic cell death associated with perturbations of macropinocytosis. Front. Physiol. 2015, 6, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Recouvreux, M.V.; Commisso, C. Macropinocytosis: A Metabolic Adaptation to Nutrient Stress in Cancer. Front. Endocrinol. 2017, 8, 261. [Google Scholar] [CrossRef] [Green Version]
- Bar-Sagi, D.; Feramisco, J.R. Induction of membrane ruffling and fluid-phase pinocytosis in quiescent fibroblasts by ras proteins. Science 1986, 233, 1061. [Google Scholar] [CrossRef]
- Amyere, M.; Payrastre, B.; Krause, U.; Van Der Smissen, P.; Veithen, A.; Courtoy, P.J. Constitutive macropinocytosis in oncogene-transformed fibroblasts depends on sequential permanent activation of phosphoinositide 3-kinase and phospholipase C. Mol. Biol. Cell 2000, 11, 3453–3467. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.M.; Nguyen, T.T.; Ravi, A.; Kubiniok, P.; Finicle, B.T.; Jayashankar, V.; Malacrida, L.; Hou, J.; Robertson, J.; Gao, D.; et al. PTEN Deficiency and AMPK Activation Promote Nutrient Scavenging and Anabolism in Prostate Cancer Cells. Cancer Discov. 2018, 8, 866–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hobbs, G.A.; Baker, N.M.; Miermont, A.M.; Thurman, R.D.; Pierobon, M.; Tran, T.H.; Anderson, A.O.; Waters, A.M.; Diehl, J.N.; Papke, B.; et al. Atypical KRASG12R Mutant Is Impaired in PI3K Signaling and Macropinocytosis in Pancreatic Cancer. Cancer Discov. 2020, 10, 104. [Google Scholar] [CrossRef] [PubMed]
- Love, K.T.; Mahon, K.P.; Levins, C.G.; Whitehead, K.A.; Querbes, W.; Dorkin, J.R.; Qin, J.; Cantley, W.; Qin, L.L.; Racie, T.; et al. Lipid-like materials for low-dose, in vivo gene silencing. Proc. Natl. Acad. Sci. USA 2010, 107, 1864–1869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilleron, J.; Querbes, W.; Zeigerer, A.; Borodovsky, A.; Marsico, G.; Schubert, U.; Manygoats, K.; Seifert, S.; Andree, C.; Stöter, M.; et al. Image-based analysis of lipid nanoparticle-mediated siRNA delivery, intracellular trafficking and endosomal escape. Nat. Biotechnol. 2013, 31, 638–646. [Google Scholar] [CrossRef] [PubMed]
- Sahay, G.; Querbes, W.; Alabi, C.; Eltoukhy, A.; Sarkar, S.; Zurenko, C.; Karagiannis, E.; Love, K.; Chen, D.; Zoncu, R.; et al. Efficiency of siRNA delivery by lipid nanoparticles is limited by endocytic recycling. Nat. Biotechnol. 2013, 31, 653–658. [Google Scholar] [CrossRef] [Green Version]
- Asai, T.; Tsuzuku, T.; Takahashi, S.; Okamoto, A.; Dewa, T.; Nango, M.; Hyodo, K.; Ishihara, H.; Kikuchi, H.; Oku, N. Cell-penetrating peptide-conjugated lipid nanoparticles for siRNA delivery. Biochem. Biophys. Res. Commun. 2014, 444, 599–604. [Google Scholar] [CrossRef]
- Dong, Y.; Love, K.T.; Dorkin, J.R.; Sirirungruang, S.; Zhang, Y.; Chen, D.; Bogorad, R.L.; Yin, H.; Chen, Y.; Vegas, A.J.; et al. Lipopeptide nanoparticles for potent and selective siRNA delivery in rodents and nonhuman primates. Proc. Natl. Acad. Sci. USA 2014, 111, 3955–3960. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.L.; Jiang, G.; Song, Q.X.; Gu, X.; Hu, M.; Wang, X.L.; Song, H.H.; Chen, L.P.; Lin, Y.Y.; Jiang, D.; et al. Lipoprotein-biomimetic nanostructure enables efficient targeting delivery of siRNA to Ras-activated glioblastoma cells via macropinocytosis. Nat. Commun. 2017, 8, 15144. [Google Scholar] [CrossRef] [Green Version]
- Hamasaki, M.; Araki, N.; Hatae, T. Association of early endosomal autoantigen 1 with macropinocytosis in EGF-stimulated A431 cells. Anat. Rec. Part A Discov. Mol. Cell. Evol. Biol. 2004, 277, 298–306. [Google Scholar] [CrossRef]
- Hewlett, L.J.; Prescott, A.R.; Watts, C. The coated pit and macropinocytic pathways serve distinct endosome populations. J. Cell Biol. 1994, 124, 689–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liberali, P.; Kakkonen, E.; Turacchio, G.; Valente, C.; Spaar, A.; Perinetti, G.; Böckmann, R.A.; Corda, D.; Colanzi, A.; Marjomaki, V.; et al. The closure of Pak1-dependent macropinosomes requires the phosphorylation of CtBP1/BARS. EMBO J. 2008, 27, 970–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominska, M.; Dykxhoorn, D.M. Breaking down the barriers: siRNA delivery and endosome escape. J. Cell Sci. 2010, 123, 1183–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juliano, R.L. Intracellular Trafficking and Endosomal Release of Oligonucleotides: What We Know and What We Don’t. Nucleic Acid Ther. 2018, 28, 166–177. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.I. Pharmacological inhibition of endocytic pathways: Is it specific enough to be useful? Methods Mol. Biol. 2008, 440, 15–33. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.P.; Mintern, J.D.; Gleeson, P.A. Macropinocytosis in Different Cell Types: Similarities and Differences. Membranes 2020, 10, 177. [Google Scholar] [CrossRef] [PubMed]
- Dias Carvalho, P.; Machado, A.L.; Martins, F.; Seruca, R.; Velho, S. Targeting the Tumor Microenvironment: An Unexplored Strategy for Mutant KRAS Tumors. Cancers 2019, 11, 2010. [Google Scholar] [CrossRef] [Green Version]
- Monty, M.A.; Islam, M.A.; Nan, X.; Tan, J.; Tuhin, I.J.; Tang, X.; Miao, M.; Wu, D.; Yu, L. Emerging role of RNA interference in immune cells engineering and its therapeutic synergism in immunotherapy. Br. J. Pharmacol. 2021, 178, 1741–1755. [Google Scholar] [CrossRef]
- Hu, B.; Zhong, L.; Weng, Y.; Peng, L.; Huang, Y.; Zhao, Y.; Liang, X.J. Therapeutic siRNA: State of the art. Signal Transduct. Target Ther. 2020, 5, 101. [Google Scholar] [CrossRef]
- Herreros-Villanueva, M.; Hijona, E.; Cosme, A.; Bujanda, L. Mouse models of pancreatic cancer. World J. Gastroenterol. 2012, 18, 1286–1294. [Google Scholar] [CrossRef] [PubMed]
- Tomas-Bort, E.; Kieler, M.; Sharma, S.; Candido, J.B.; Loessner, D. 3D approaches to model the tumor microenvironment of pancreatic cancer. Theranostics 2020, 10, 5074–5089. [Google Scholar] [CrossRef] [PubMed]
- Zeeberg, K.; Cardone, R.A.; Greco, M.R.; Saccomano, M.; Nohr-Nielsen, A.; Alves, F.; Pedersen, S.F.; Reshkin, S.J. Assessment of different 3D culture systems to study tumor phenotype and chemosensitivity in pancreatic ductal adenocarcinoma. Int. J. Oncol. 2016, 49, 243–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.C.; Korc, M. Designer hydrogels: Shedding light on the physical chemistry of the pancreatic cancer microenvironment. Cancer Lett. 2018, 436, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Shichi, Y.; Sasaki, N.; Michishita, M.; Hasegawa, F.; Matsuda, Y.; Arai, T.; Gomi, F.; Aida, J.; Takubo, K.; Toyoda, M.; et al. Enhanced morphological and functional differences of pancreatic cancer with epithelial or mesenchymal characteristics in 3D culture. Sci. Rep. 2019, 9, 10871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beer, M.; Kuppalu, N.; Stefanini, M.; Becker, H.; Schulz, I.; Manoli, S.; Schuette, J.; Schmees, C.; Casazza, A.; Stelzle, M.; et al. A novel microfluidic 3D platform for culturing pancreatic ductal adenocarcinoma cells: Comparison with in vitro cultures and in vivo xenografts. Sci. Rep. 2017, 7, 1325. [Google Scholar] [CrossRef]
- Norberg, K.J.; Liu, X.; Fernandez Moro, C.; Strell, C.; Nania, S.; Blumel, M.; Balboni, A.; Bozoky, B.; Heuchel, R.L.; Lohr, J.M. A novel pancreatic tumour and stellate cell 3D co-culture spheroid model. BMC Cancer 2020, 20, 475. [Google Scholar] [CrossRef]
| In Vitro | In Vivo | ||||
|---|---|---|---|---|---|
| Models | Cell Line | Patient-Derived Organoid | Cell Line Xenograft | Patient-Derived Xenograft | Genetically Modified Mouse Model (GEMM) |
| TME | - | +++ | + | ++ | ++ |
| Immune system | - | ++ | + | + | +++ |
| Pros |
|
|
|
|
|
| Cons |
|
|
|
|
|
| miRNA | Target | PC Cell Line/Model | Combination Therapy | Reference |
|---|---|---|---|---|
| miR-150 | MUC4 and HER2 | Colo-357 and HPAF cells | [74] | |
| miR-155 | SOD2, CAT, and DCK | MiaPaCa and Colo-357 cells | Gemcitabine | [75] |
| miR-199a ASO | RPS18, Acta-2, Collagen1α1, PDGFR-β, and mTOR | hPSCs | [76] | |
| miR-21 and miR-221 ASO | CDK6, IRAK3, NRP1, SMAD7, SOCS6, C5ORF41, KLF12, MAPK10, EFNA1 | L3.6plGres-SP orthotopic | [77] | |
| miR-21 ASO | PDCD4 and PTEN | MIA PaCa-2 s.c. | Gemcitabine | [78] |
| miR-212 | USP9X | PDX | Doxorubicin | [79] |
| miR-345 | SHH, Gli-1, MUC4, and Ki67 | Capan-1 and CD18/HDAF s.c. | Gemcitabine | [80] |
| miR-34a | E2F3, Bcl-2, c-myc, and cyclin D1 | PANC-1 s.c. | [81] | |
| miR-34a and miR-143/145 | SIRT1, CD44, aldehyde dehydrogenase, KRAS2, and RREB1 | MiaPaCa-2 s.c. and orthotopic | [82] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, M.J.; Chang, H.; Nam, G.; Ko, Y.; Kim, S.H.; Roberts, T.M.; Ryu, J.H. RNAi-Based Approaches for Pancreatic Cancer Therapy. Pharmaceutics 2021, 13, 1638. https://doi.org/10.3390/pharmaceutics13101638
Kim MJ, Chang H, Nam G, Ko Y, Kim SH, Roberts TM, Ryu JH. RNAi-Based Approaches for Pancreatic Cancer Therapy. Pharmaceutics. 2021; 13(10):1638. https://doi.org/10.3390/pharmaceutics13101638
Chicago/Turabian StyleKim, Min Ju, Hyeyoun Chang, Gihoon Nam, Youngji Ko, Sun Hwa Kim, Thomas M. Roberts, and Ju Hee Ryu. 2021. "RNAi-Based Approaches for Pancreatic Cancer Therapy" Pharmaceutics 13, no. 10: 1638. https://doi.org/10.3390/pharmaceutics13101638
APA StyleKim, M. J., Chang, H., Nam, G., Ko, Y., Kim, S. H., Roberts, T. M., & Ryu, J. H. (2021). RNAi-Based Approaches for Pancreatic Cancer Therapy. Pharmaceutics, 13(10), 1638. https://doi.org/10.3390/pharmaceutics13101638