
Targeting HER2 Expressing Tumors with a Potent Drug Conjugate Based on an Albumin Binding Domain-Derived Affinity Protein

 , , , ,
, , , ,  ,
,  , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. General
2.2. Production and Purification of ADAPT Fusion Proteins
2.3. Conjugation with mcDM1
2.4. Biochemical Characterization
2.5. Biosensor Analysis on a Biacore Instrument
2.6. Cell Culture
2.7. In Vitro Cytotoxicity Analysis
2.8. Radiolabeling
2.9. In Vitro Characterization of the 99mTc-Labeled Constructs
2.10. Biodistribution in Tumor Bearing Mice
3. Results
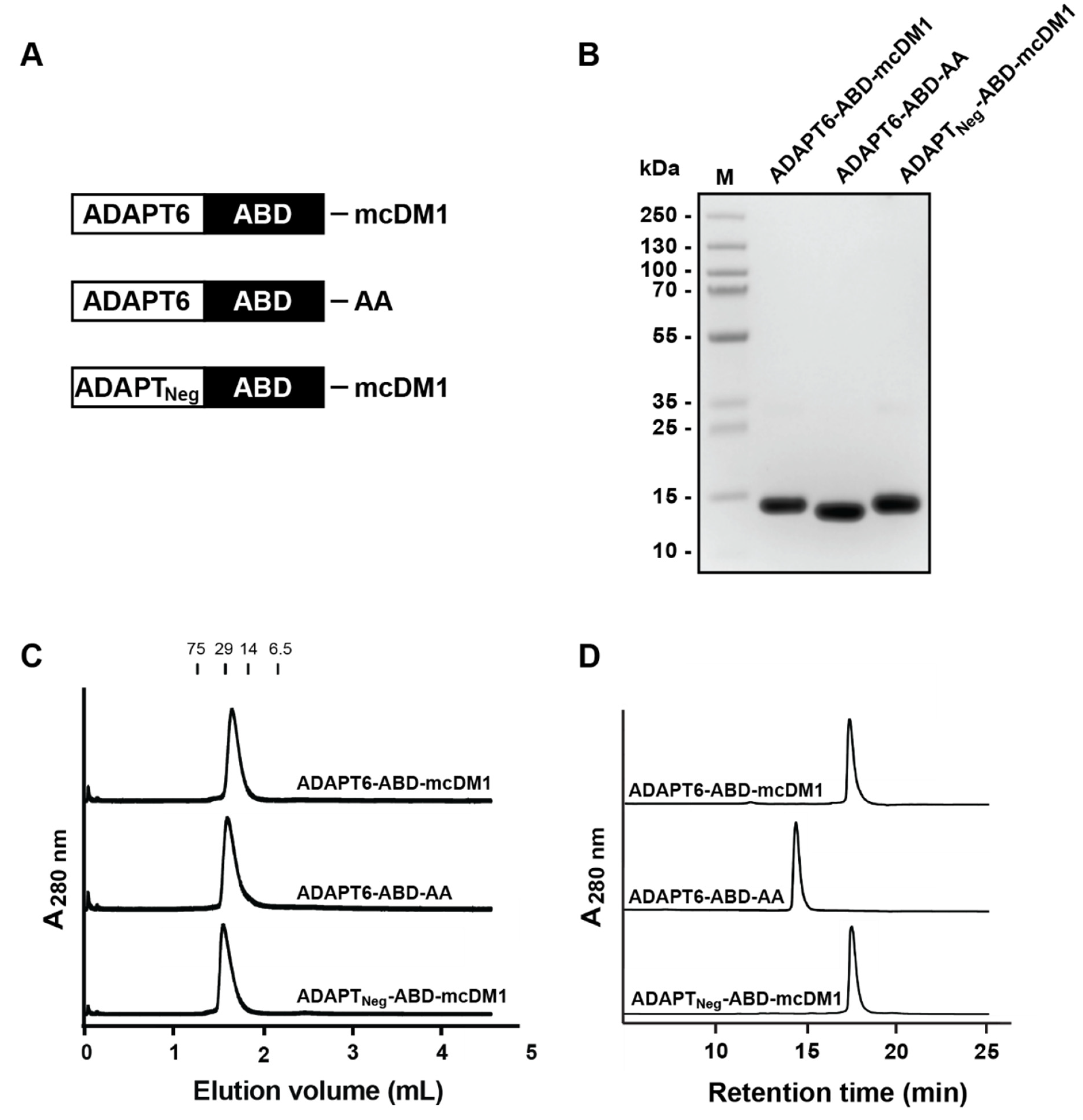
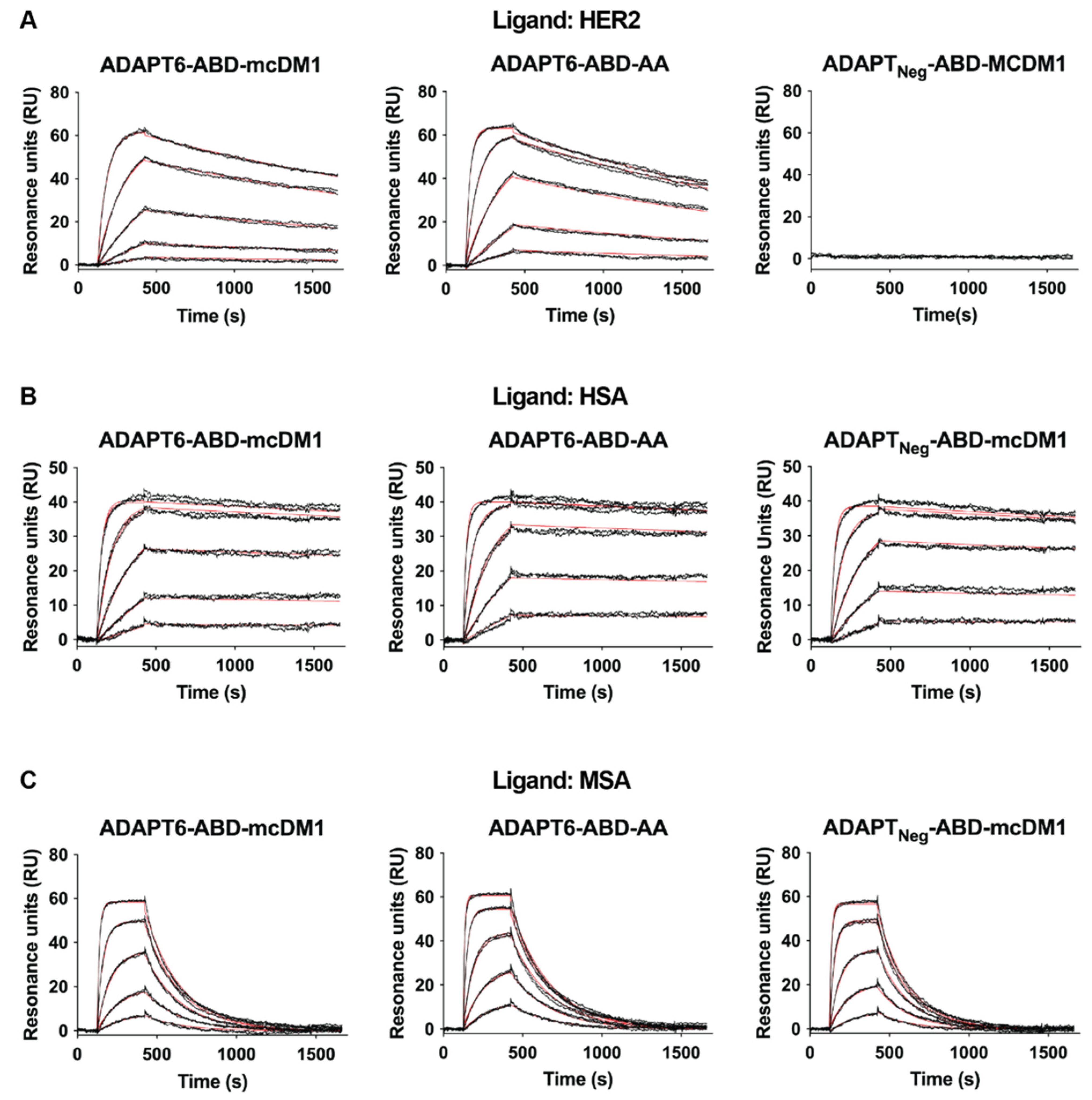
3.1. Production, Purification, Conjugation and Biochemical Characterization
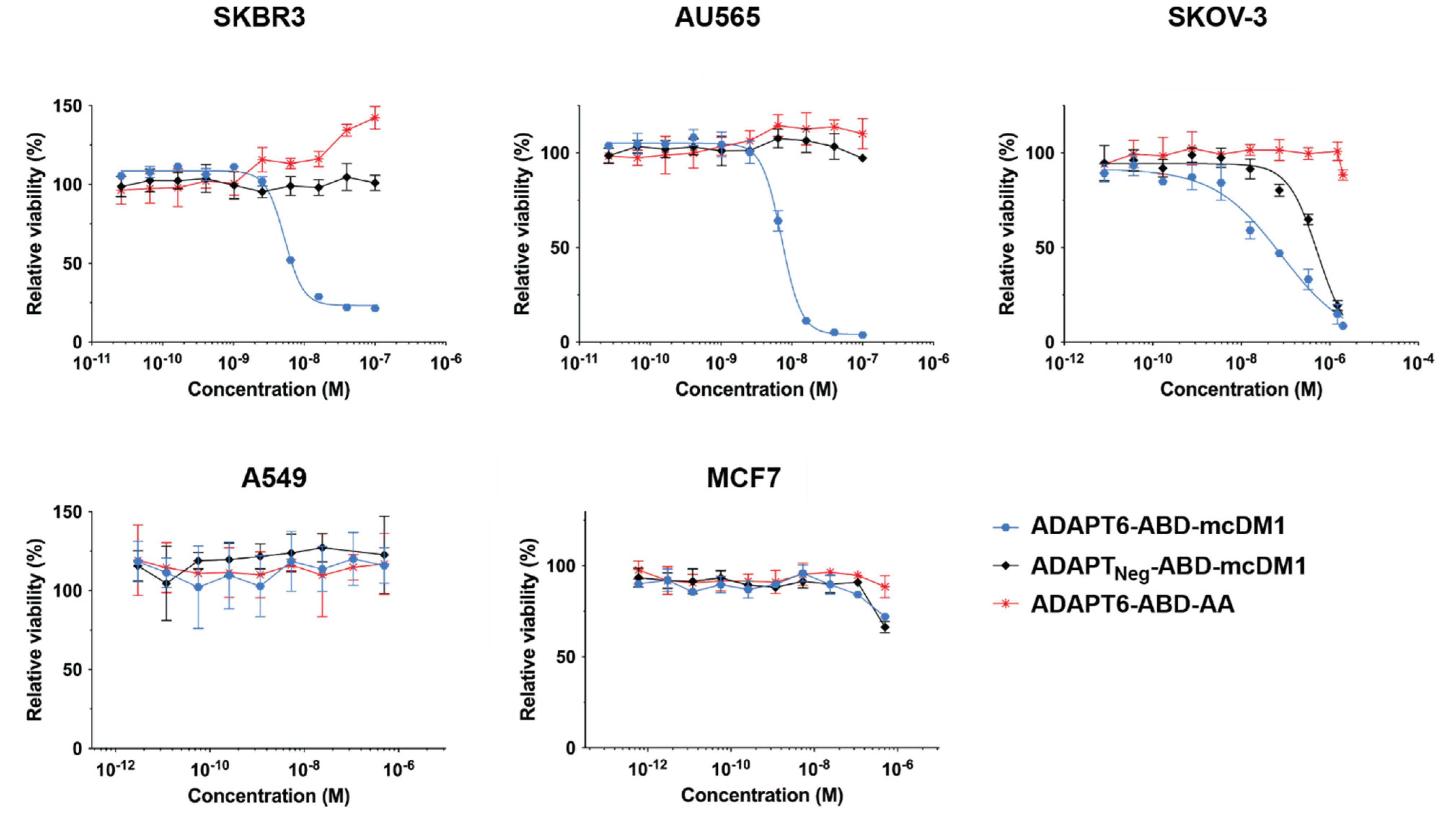
3.2. In Vitro Cytotoxicity Analysis
3.3. Radiolabeling
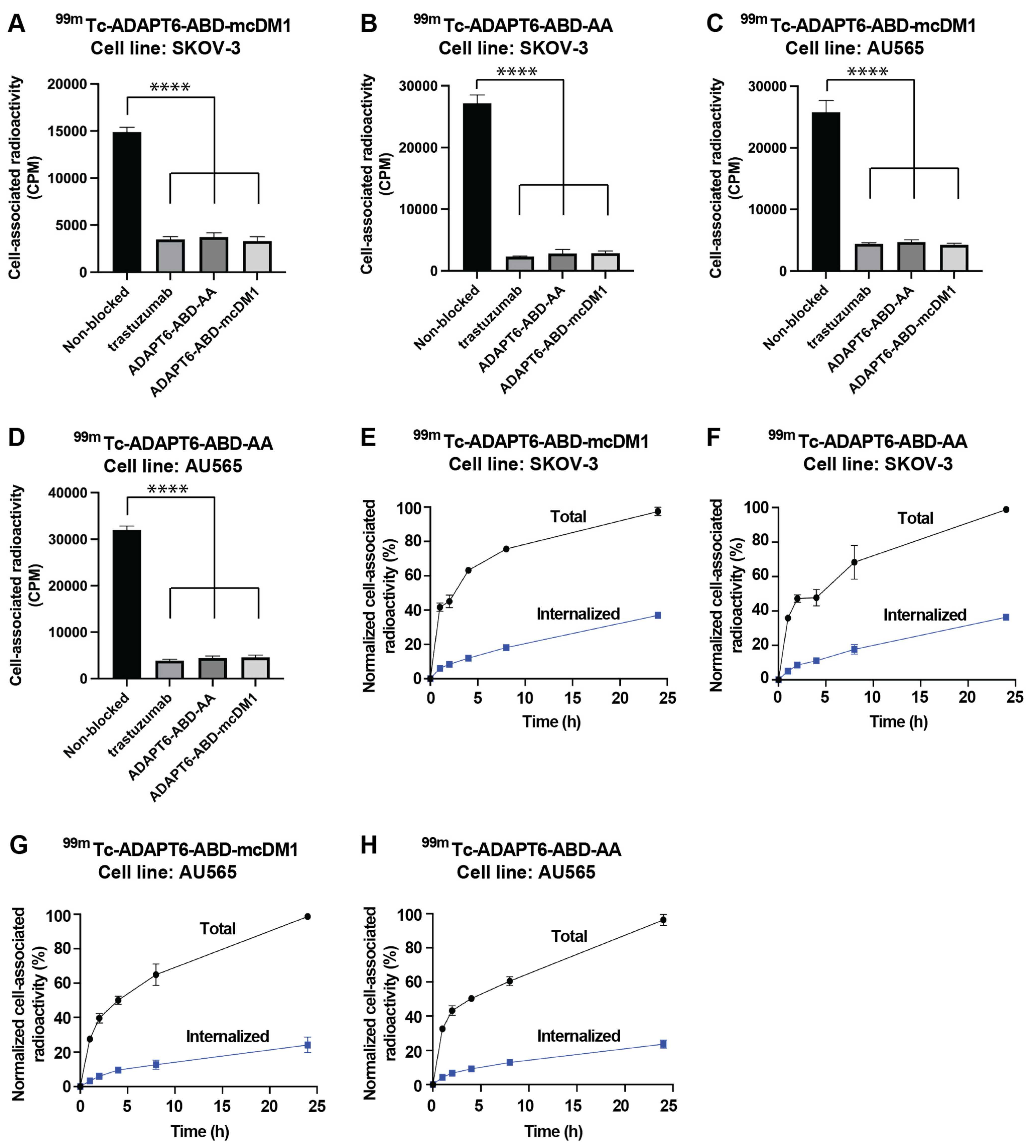
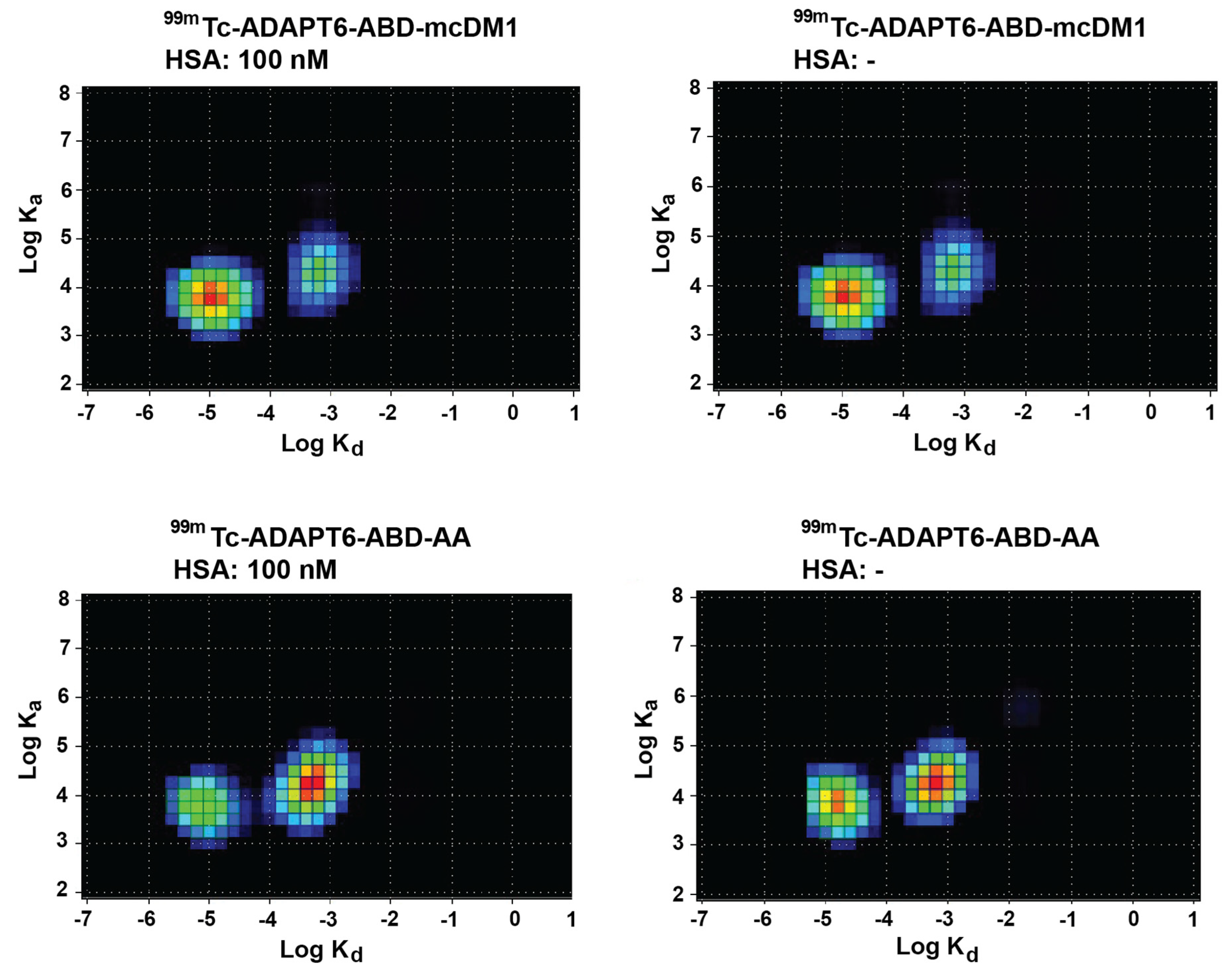
3.4. In Vitro Characterization of the Radiolabeled Constructs
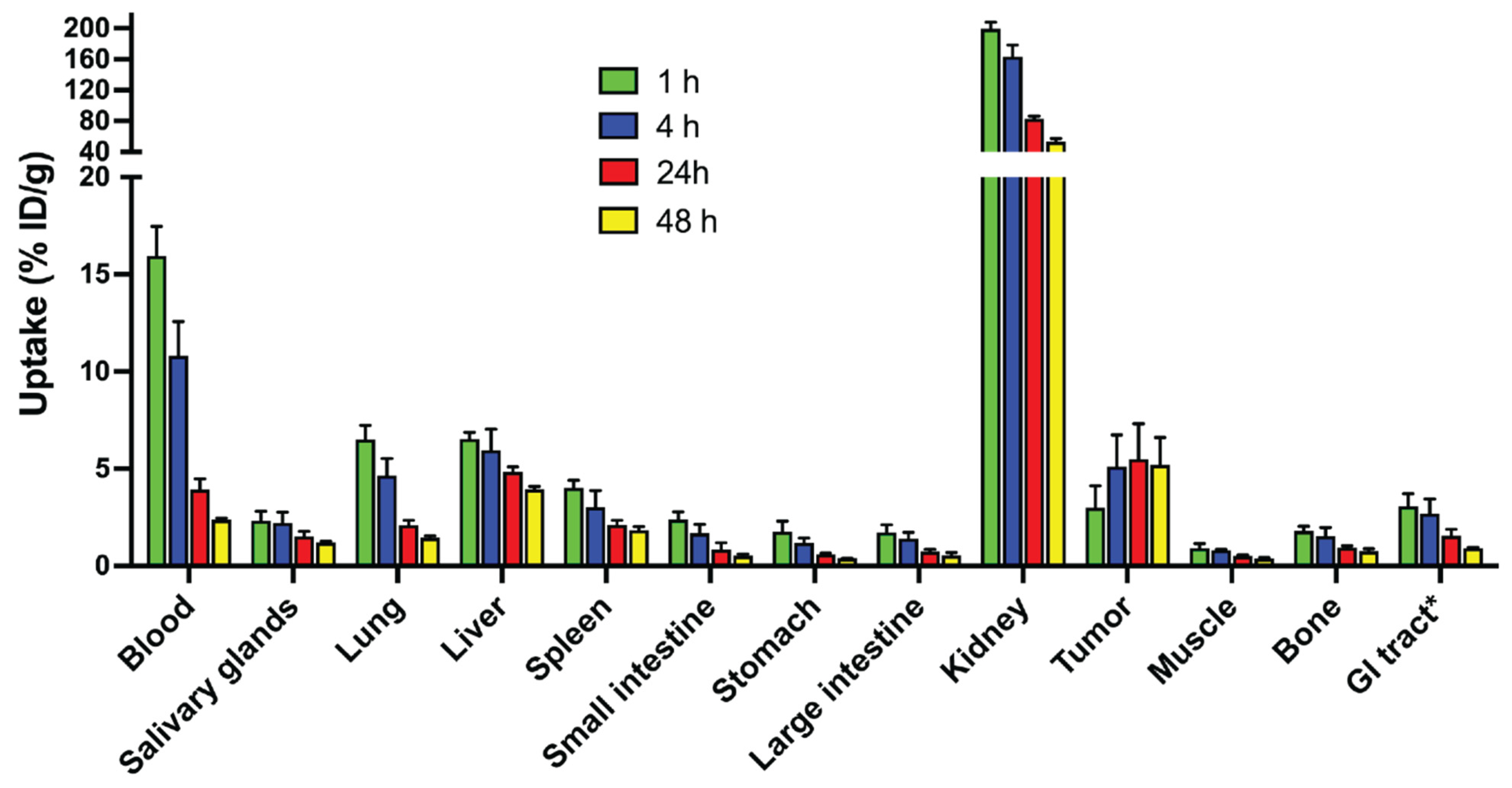
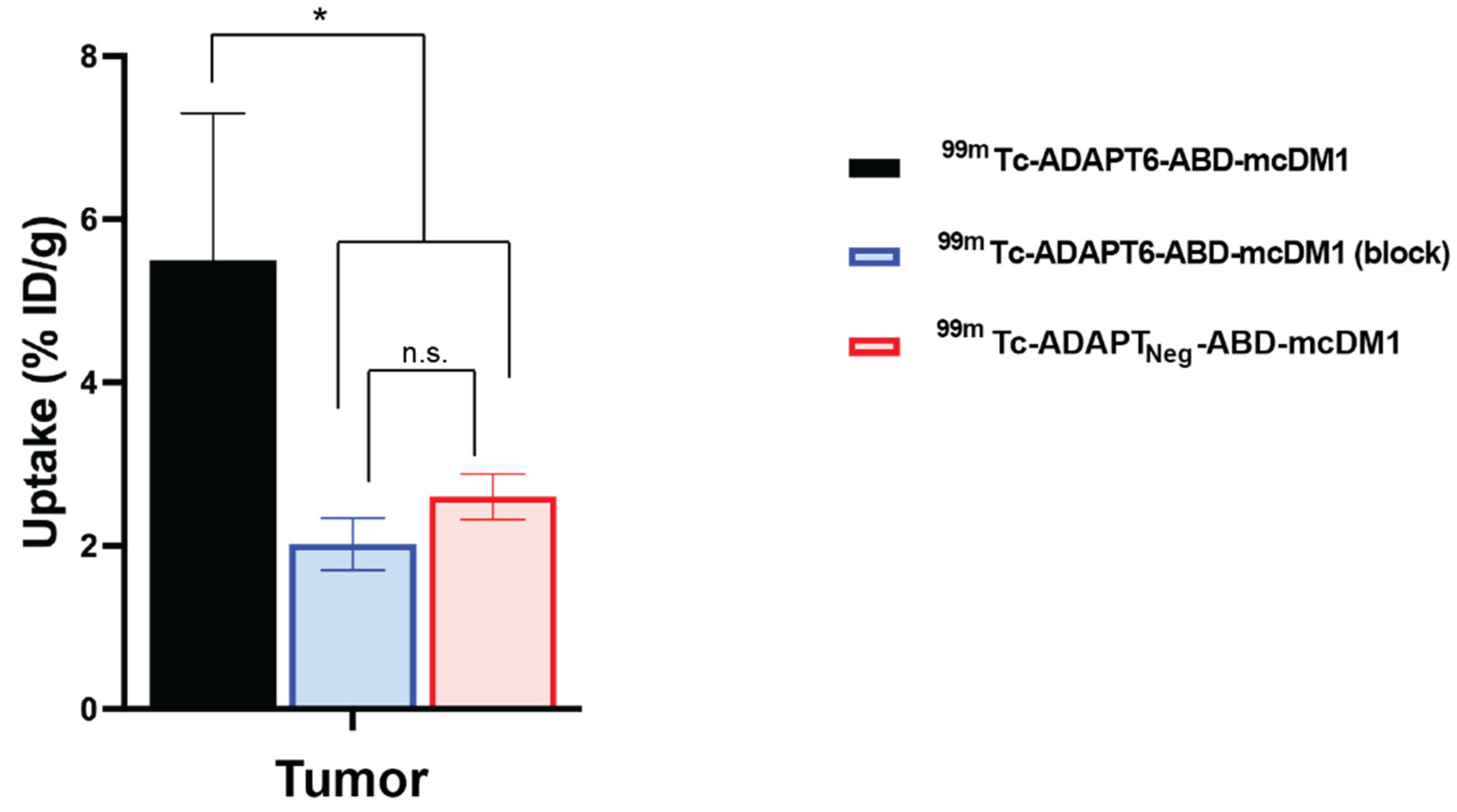
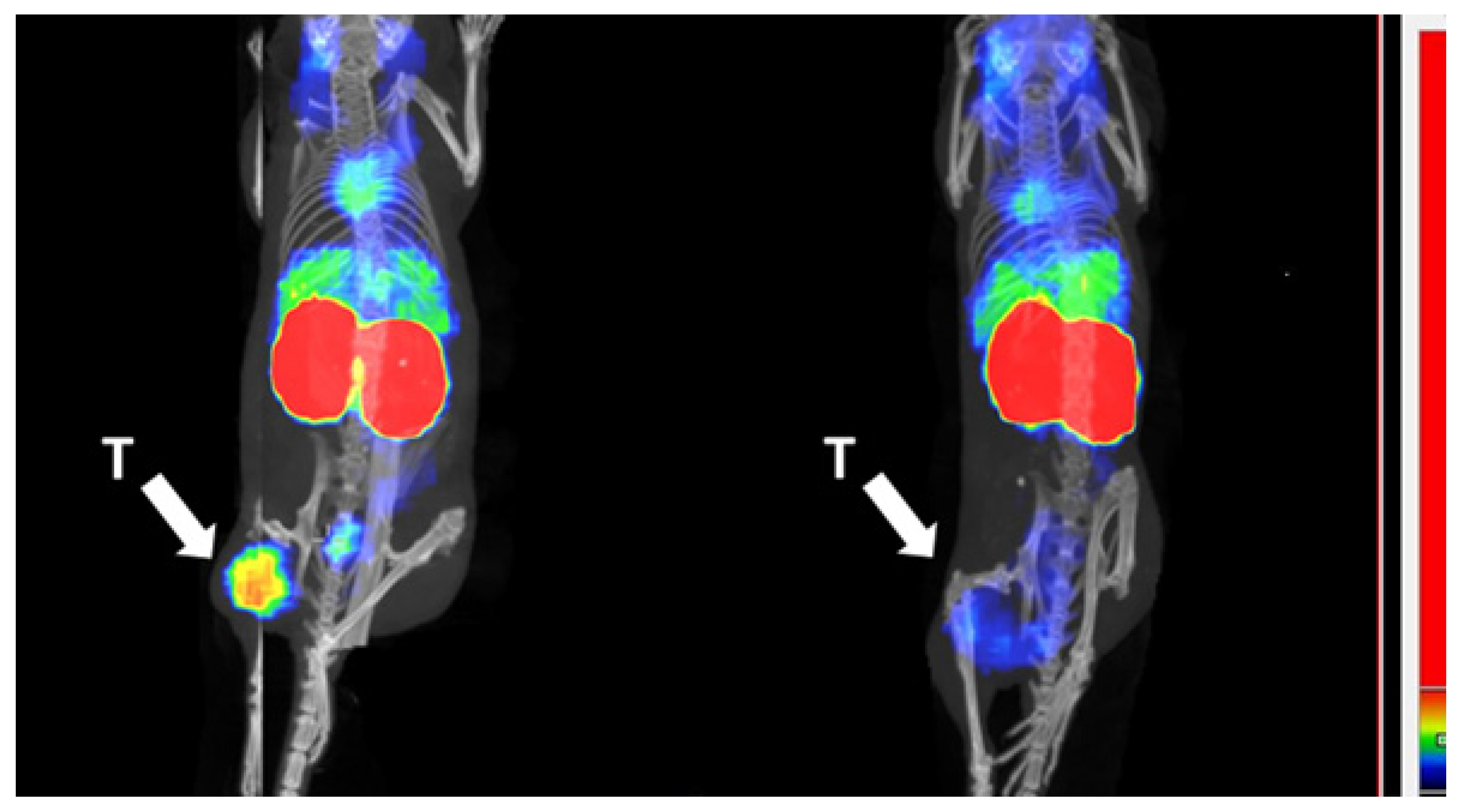
3.5. Biodistribution in Tumor Bearing Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Parakh, S.; Parslow, A.C.; Gan, H.K.; Scott, A.M. Antibody-Mediated Delivery of Therapeutics for Cancer Therapy. Expert Opin. Drug Deliv. 2016, 13, 401–419. [Google Scholar] [CrossRef]
- Tai, W.; Mahato, R.; Cheng, K. The Role of HER2 in Cancer Therapy and Targeted Drug Delivery. J. Control. Release 2010, 146, 264–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, M.; Schwaederle, M.; Arguello, D.; Millis, S.Z.; Gatalica, Z.; Kurzrock, R. HER2 Expression Status in Diverse Cancers: Review of Results from 37,992 Patients. Cancer Metastasis Rev. 2015, 34, 157–164. [Google Scholar] [CrossRef] [Green Version]
- Costa, R.L.B.; Czerniecki, B.J. Clinical Development of Immunotherapies for HER2 + Breast Cancer: A Review of HER2-Directed Monoclonal Antibodies and Beyond. NPJ Breast Cancer 2020, 6, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khongorzul, P.; Ling, C.J.; Khan, F.U.; Ihsan, A.U.; Zhang, J. Antibody–Drug Conjugates: A Comprehensive Review. Mol. Cancer Res. 2020, 18, 3–19. [Google Scholar] [CrossRef] [Green Version]
- Phillips, G.D.L.; Li, G.; Dugger, D.L.; Crocker, L.M.; Parsons, K.L.; Mai, E.; Blättler, W.A.; Lambert, J.M.; Chari, R.V.J.; Lutz, R.J.; et al. Targeting HER2-Positive Breast Cancer with Trastuzumab-DM1, an Antibody–Cytotoxic Drug Conjugate. Cancer Res. 2008, 68, 9280–9290. [Google Scholar] [CrossRef] [Green Version]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and Challenges for the next Generation of Antibody–Drug Conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef] [PubMed]
- Walsh, S.J.; Bargh, J.D.; Dannheim, F.M.; Hanby, A.R.; Seki, H.; Counsell, A.J.; Ou, X.; Fowler, E.; Ashman, N.; Takada, Y.; et al. Site-Selective Modification Strategies in Antibody–Drug Conjugates. Chem. Soc. Rev. 2021, 50, 1305–1353. [Google Scholar] [CrossRef]
- Bennett, G.; Brown, A.; Mudd, G.; Huxley, P.; Rietschoten, K.V.; Pavan, S.; Chen, L.; Watcham, S.; Lahdenranta, J.; Keen, N. MMAE Delivery Using the Bicycle Toxin Conjugate BT5528. Mol. Cancer 2020, 19, 1385–1394. [Google Scholar] [CrossRef]
- Altai, M.; Liu, H.; Ding, H.; Mitran, B.; Edqvist, P.-H.; Tolmachev, V.; Orlova, A.; Gräslund, T. Affibody-Derived Drug Conjugates: Potent Cytotoxic Molecules for Treatment of HER2 over-Expressing Tumors. J. Control Release 2018, 288, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Ding, H.; Vorobyeva, A.; Oroujeni, M.; Orlova, A.; Tolmachev, V.; Gräslund, T. Drug Conjugates Based on a Monovalent Affibody Targeting Vector Can Efficiently Eradicate HER2 Positive Human Tumors in an Experimental Mouse Model. Cancers 2021, 13, 85. [Google Scholar] [CrossRef] [PubMed]
- Sörensen, J.; Sandberg, D.; Sandström, M.; Wennborg, A.; Feldwisch, J.; Tolmachev, V.; Åström, G.; Lubberink, M.; Garske-Román, U.; Carlsson, J.; et al. First-in-Human Molecular Imaging of HER2 Expression in Breast Cancer Metastases Using the 111In-ABY-025 Affibody Molecule. J. Nucl. Med. 2014, 55, 730–735. [Google Scholar] [CrossRef] [Green Version]
- Bragina, O.; Chernov, V.; Schulga, A.; Konovalova, E.; Garbukov, E.; Vorobyeva, A.; Orlova, A.; Tashireva, L.; Sorensen, J.; Zelchan, R.; et al. Phase I Trial of 99mTc-(HE)3-G3, a DARPin-Based Probe for Imaging of HER2 Expression in Breast Cancer. J. Nucl. Med. 2021, 62. [Google Scholar] [CrossRef]
- Bragina, O.; von Witting, E.; Garousi, J.; Zelchan, R.; Sandström, M.; Orlova, A.; Medvedeva, A.; Doroshenko, A.; Vorobyeva, A.; Lindbo, S.; et al. Phase I Study of 99mTc-ADAPT6, a Scaffold Protein–Based Probe for Visualization of HER2 Expression in Breast Cancer. J. Nucl. Med. 2021, 62, 493–499. [Google Scholar] [CrossRef]
- Garousi, J.; Orlova, A.; Frejd, F.Y.; Tolmachev, V. Imaging Using Radiolabelled Targeted Proteins: Radioimmunodetection and Beyond. EJNMMI Radiopharm. Chem. 2020, 5, 16. [Google Scholar] [CrossRef]
- Nilvebrant, J.; Hober, S. The Albumin-Binding Domain As A Scaffold For Protein Engineering. Comput. Struct. Biotechnol. J. 2013, 6, e201303009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilvebrant, J.; Åstrand, M.; Georgieva-Kotseva, M.; Björnmalm, M.; Löfblom, J.; Hober, S. Engineering of Bispecific Affinity Proteins with High Affinity for ERBB2 and Adaptable Binding to Albumin. PLoS ONE 2014, 9, e103094. [Google Scholar] [CrossRef] [Green Version]
- Nilvebrant, J.; Alm, T.; Hober, S.; Löfblom, J. Engineering Bispecificity into a Single Albumin-Binding Domain. PLoS ONE 2011, 6, e25791. [Google Scholar] [CrossRef]
- Garousi, J.; Lindbo, S.; Nilvebrant, J.; Åstrand, M.; Buijs, J.; Sandström, M.; Honarvar, H.; Orlova, A.; Tolmachev, V.; Hober, S. ADAPT, a Novel Scaffold Protein-Based Probe for Radionuclide Imaging of Molecular Targets That Are Expressed in Disseminated Cancers. Cancer Res. 2015, 75, 4364–4371. [Google Scholar] [CrossRef] [Green Version]
- Garousi, J.; Lindbo, S.; Mitran, B.; Buijs, J.; Vorobyeva, A.; Orlova, A.; Tolmachev, V.; Hober, S. Comparative Evaluation of Tumor Targeting Using the Anti-HER2 ADAPT Scaffold Protein Labeled at the C-Terminus with Indium-111 or Technetium-99m. Sci. Rep. 2017, 7, 14780. [Google Scholar] [CrossRef] [Green Version]
- Lindbo, S.; Garousi, J.; Åstrand, M.; Honarvar, H.; Orlova, A.; Hober, S.; Tolmachev, V. Influence of Histidine-Containing Tags on the Biodistribution of ADAPT Scaffold Proteins. Bioconj. Chem. 2016, 27, 716–726. [Google Scholar] [CrossRef] [PubMed]
- Lindbo, S.; Garousi, J.; Mitran, B.; Altai, M.; Buijs, J.; Orlova, A.; Hober, S.; Tolmachev, V. Radionuclide Tumor Targeting Using ADAPT Scaffold Proteins: Aspects of Label Positioning and Residualizing Properties of the Label. J. Nucl. Med. 2018, 59, 93–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindbo, S.; Garousi, J.; Mitran, B.; Vorobyeva, A.; Oroujeni, M.; Orlova, A.; Hober, S.; Tolmachev, V. Optimized Molecular Design of ADAPT-Based HER2-Imaging Probes Labeled with 111In and 68Ga. Mol. Pharm. 2018, 15, 2674–2683. [Google Scholar] [CrossRef]
- von Witting, E.; Garousi, J.; Lindbo, S.; Vorobyeva, A.; Altai, M.; Oroujeni, M.; Mitran, B.; Orlova, A.; Hober, S.; Tolmachev, V. Selection of the Optimal Macrocyclic Chelators for Labeling with 111In and 68Ga Improves Contrast of HER2 Imaging Using Engineered Scaffold Protein ADAPT6. Eur. J. Pharm. Biopharm. 2019, 140, 109–120. [Google Scholar] [CrossRef]
- Tolmachev, V.; Orlova, A.; Pehrson, R.; Galli, J.; Baastrup, B.; Andersson, K.; Sandström, M.; Rosik, D.; Carlsson, J.; Lundqvist, H.; et al. Radionuclide Therapy of HER2-Positive Microxenografts Using a 177Lu-Labeled HER2-Specific Affibody Molecule. Cancer Res. 2007, 67, 2773–2782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonsson, A.; Dogan, J.; Herne, N.; Abrahmsén, L.; Nygren, P.-Å. Engineering of a Femtomolar Affinity Binding Protein to Human Serum Albumin. Protein Eng. Des. Sel. 2008, 21, 515–527. [Google Scholar] [CrossRef]
- Andersen, J.T.; Pehrson, R.; Tolmachev, V.; Daba, M.B.; Abrahmsén, L.; Ekblad, C. Extending Half-Life by Indirect Targeting of the Neonatal Fc Receptor (FcRn) Using a Minimal Albumin Binding Domain. J. Biol. Chem. 2011, 286, 5234–5241. [Google Scholar] [CrossRef] [Green Version]
- Garousi, J.; von Witting, E.; Borin, J.; Vorobyeva, A.; Altai, M.; Vorontsova, O.; Konijnenberg, M.W.; Oroujeni, M.; Orlova, A.; Tolmachev, V.; et al. Radionuclide Therapy Using ABD-Fused ADAPT Scaffold Protein: Proof of Principle. Biomaterials 2021, 266, 120381. [Google Scholar] [CrossRef]
- Babai, S.; Auclert, L.; Le-Louët, H. Safety Data and Withdrawal of Hepatotoxic Drugs. Thérapie 2018. [Google Scholar] [CrossRef]
- Ding, H.; Altai, M.; Rinne, S.S.; Vorobyeva, A.; Tolmachev, V.; Gräslund, T.; Orlova, A. Incorporation of a Hydrophilic Spacer Reduces Hepatic Uptake of HER2-Targeting Affibody–DM1 Drug Conjugates. Cancers 2019, 11, 1168. [Google Scholar] [CrossRef] [Green Version]
- Garousi, J.; Lindbo, S.; Borin, J.; von Witting, E.; Vorobyeva, A.; Oroujeni, M.; Mitran, B.; Orlova, A.; Buijs, J.; Tolmachev, V.; et al. Comparative Evaluation of Dimeric and Monomeric Forms of ADAPT Scaffold Protein for Targeting of HER2-Expressing Tumours. Eur. J. Pharm. Biopharm. 2019, 134, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Deyev, S.; Vorobyeva, A.; Schulga, A.; Proshkina, G.; Güler, R.; Löfblom, J.; Mitran, B.; Garousi, J.; Altai, M.; Buijs, J.; et al. Comparative Evaluation of Two DARPin Variants: Effect of Affinity, Size, and Label on Tumor Targeting Properties. Mol. Pharm. 2019, 16, 995–1008. [Google Scholar] [CrossRef] [PubMed]
- Waibel, R.; Alberto, R.; Willuda, J.; Finnern, R.; Schibli, R.; Stichelberger, A.; Egli, A.; Abram, U.; Mach, J.-P.; Plückthun, A.; et al. Stable One-Step Technetium-99m Labeling of His-Tagged Recombinant Proteins with a Novel Tc(I)–Carbonyl Complex. Nat. Biotechnol. 1999, 17, 897–901. [Google Scholar] [CrossRef]
- Tolmachev, V.; Orlova, A.; Andersson, K. Methods for Radiolabelling of Monoclonal Antibodies. In Human Monoclonal Antibodies: Methods and Protocols; Steinitz, M., Ed.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2014; pp. 309–330. ISBN 978-1-62703-586-6. [Google Scholar]
- Oroujeni, M.; Rinne, S.S.; Vorobyeva, A.; Loftenius, A.; Feldwisch, J.; Jonasson, P.; Chernov, V.; Orlova, A.; Frejd, F.Y.; Tolmachev, V. Preclinical Evaluation of 99mTc-ZHER2:41071, a Second-Generation Affibody-Based HER2-Visualizing Imaging Probe with a Low Renal Uptake. Int. J. Mol. Sci. 2021, 22, 2770. [Google Scholar] [CrossRef] [PubMed]
- Barok, M.; Joensuu, H.; Isola, J. Trastuzumab Emtansine: Mechanisms of Action and Drug Resistance. Breast Cancer Res 2014, 16, 209. [Google Scholar] [CrossRef] [Green Version]
- Kovtun, Y.V.; Audette, C.A.; Mayo, M.F.; Jones, G.E.; Doherty, H.; Maloney, E.K.; Erickson, H.K.; Sun, X.; Wilhelm, S.; Ab, O.; et al. Antibody-Maytansinoid Conjugates Designed to Bypass Multidrug Resistance. Cancer Res 2010, 70, 2528–2537. [Google Scholar] [CrossRef] [Green Version]
- von Schwarzenberg, K.; Lajtos, T.; Simon, L.; Müller, R.; Vereb, G.; Vollmar, A.M. V-ATPase Inhibition Overcomes Trastuzumab Resistance in Breast Cancer. Mol. Oncol. 2014, 8, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Altai, M.; Yin, W.; Lindbo, S.; Liu, H.; Garousi, J.; Xu, T.; Orlova, A.; Tolmachev, V.; Hober, S.; et al. HER2-Specific Pseudomonas Exotoxin A PE25 Based Fusions: Influence of Targeting Domain on Target Binding, Toxicity, and In Vivo Biodistribution. Pharmaceutics 2020, 12, 391. [Google Scholar] [CrossRef]
- Barta, P.; Malmberg, J.; Melicharova, L.; Strandgård, J.; Orlova, A.; Tolmachev, V.; Laznicek, M.; Andersson, K. Protein Interactions with HER-Family Receptors Can Have Different Characteristics Depending on the Hosting Cell Line. Int. J. Oncol. 2012, 40, 1677–1682. [Google Scholar] [CrossRef]
- Liu, Y.; Vorobyeva, A.; Xu, T.; Orlova, A.; Loftenius, A.; Bengtsson, T.; Jonasson, P.; Tolmachev, V.; Frejd, F.Y. Comparative Preclinical Evaluation of HER2-Targeting ABD-Fused Affibody® Molecules 177Lu-ABY-271 and 177Lu-ABY-027: Impact of DOTA Position on ABD Domain. Pharmaceutics 2021, 13, 839. [Google Scholar] [CrossRef]
- Björkelund, H.; Gedda, L.; Barta, P.; Malmqvist, M.; Andersson, K. Gefitinib Induces Epidermal Growth Factor Receptor Dimers Which Alters the Interaction Characteristics with 125I-EGF. PLoS ONE 2011, 6, e24739. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | Ligand | ka (M−1·s−1) | kd (s−1) | KD (M) |
|---|---|---|---|---|
| ADAPT6-ABD-mcDM1 | HER2 | 5.52 × 104 | 3.12 × 10−4 | 5.65 × 10−9 |
| HSA | 1.08 × 105 | 5.89 × 10−5 | 5.43 × 10−10 | |
| MSA | 2.11 × 105 | 4.42 × 10−3 | 2.09 × 10−8 | |
| ADAPT6-ABD-AA | HER2 | 1.16 × 105 | 4.00 × 10−4 | 3.44 × 10−9 |
| HSA | 5.47 × 105 | 5.22 × 10−5 | 9.54 × 10−11 | |
| MSA | 1.03 × 106 | 4.02 × 10−3 | 3.90 × 10−9 | |
| ADAPTNeg-ABD-mcDM1 | HER2 | ND a | ND | ND |
| HSA | 4.11 × 105 | 6.80 × 10−5 | 1.65 × 10−10 | |
| MSA | 7.90 × 105 | 5.18 × 10−3 | 6.55 × 10−9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garousi, J.; Ding, H.; von Witting, E.; Xu, T.; Vorobyeva, A.; Oroujeni, M.; Orlova, A.; Hober, S.; Gräslund, T.; Tolmachev, V. Targeting HER2 Expressing Tumors with a Potent Drug Conjugate Based on an Albumin Binding Domain-Derived Affinity Protein. Pharmaceutics 2021, 13, 1847. https://doi.org/10.3390/pharmaceutics13111847
Garousi J, Ding H, von Witting E, Xu T, Vorobyeva A, Oroujeni M, Orlova A, Hober S, Gräslund T, Tolmachev V. Targeting HER2 Expressing Tumors with a Potent Drug Conjugate Based on an Albumin Binding Domain-Derived Affinity Protein. Pharmaceutics. 2021; 13(11):1847. https://doi.org/10.3390/pharmaceutics13111847
Chicago/Turabian StyleGarousi, Javad, Haozhong Ding, Emma von Witting, Tianqi Xu, Anzhelika Vorobyeva, Maryam Oroujeni, Anna Orlova, Sophia Hober, Torbjörn Gräslund, and Vladimir Tolmachev. 2021. "Targeting HER2 Expressing Tumors with a Potent Drug Conjugate Based on an Albumin Binding Domain-Derived Affinity Protein" Pharmaceutics 13, no. 11: 1847. https://doi.org/10.3390/pharmaceutics13111847
APA StyleGarousi, J., Ding, H., von Witting, E., Xu, T., Vorobyeva, A., Oroujeni, M., Orlova, A., Hober, S., Gräslund, T., & Tolmachev, V. (2021). Targeting HER2 Expressing Tumors with a Potent Drug Conjugate Based on an Albumin Binding Domain-Derived Affinity Protein. Pharmaceutics, 13(11), 1847. https://doi.org/10.3390/pharmaceutics13111847