
Drug Therapeutic-Use Class Prediction and Repurposing Using Graph Convolutional Networks




Abstract
:1. Introduction
2. Materials and Methods
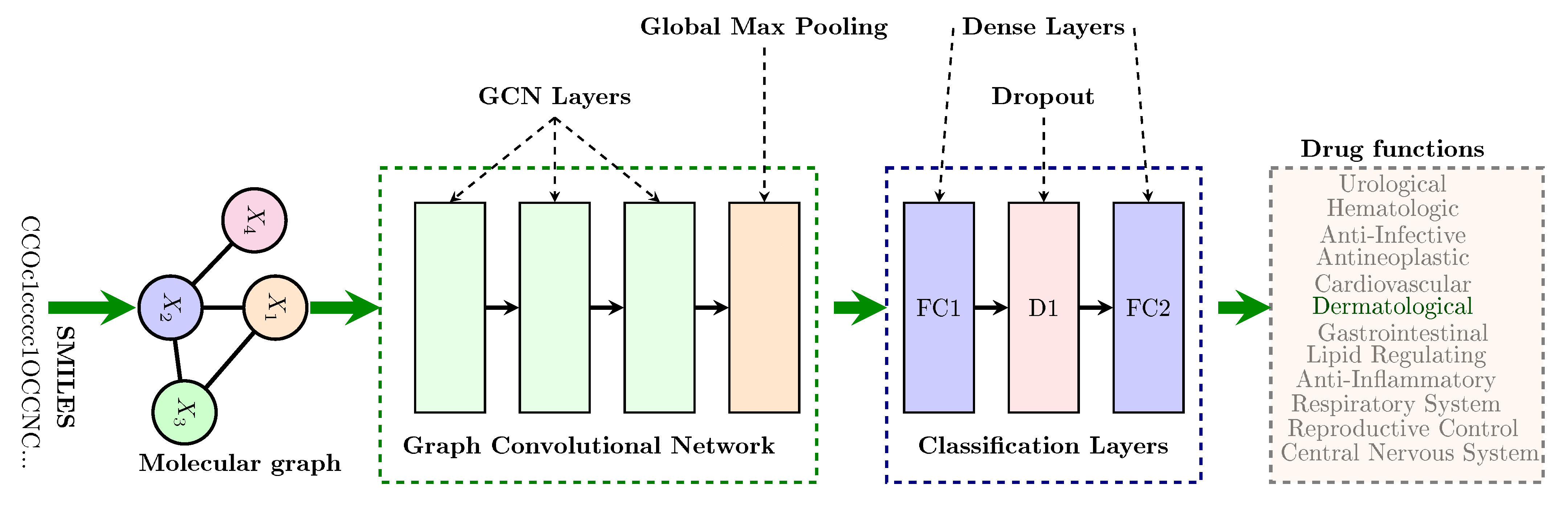
2.1. Overview of Our Approach
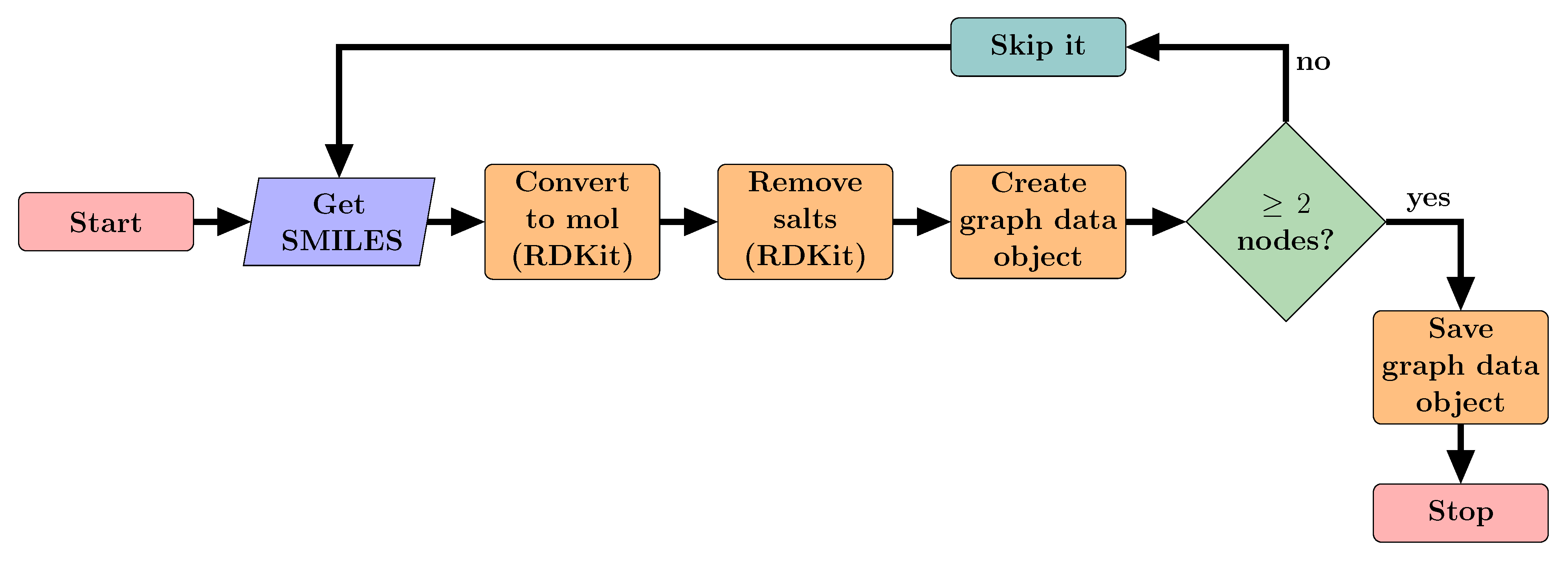
2.2. Data Preparation and Processing
2.2.1. Acquiring Data
2.2.2. Converting SMILES into Graphs
2.3. Graph Convolution Networks
2.4. Training Details
2.5. Performance Evaluation Metrics
2.6. Model and Representation Comparisons
3. Results
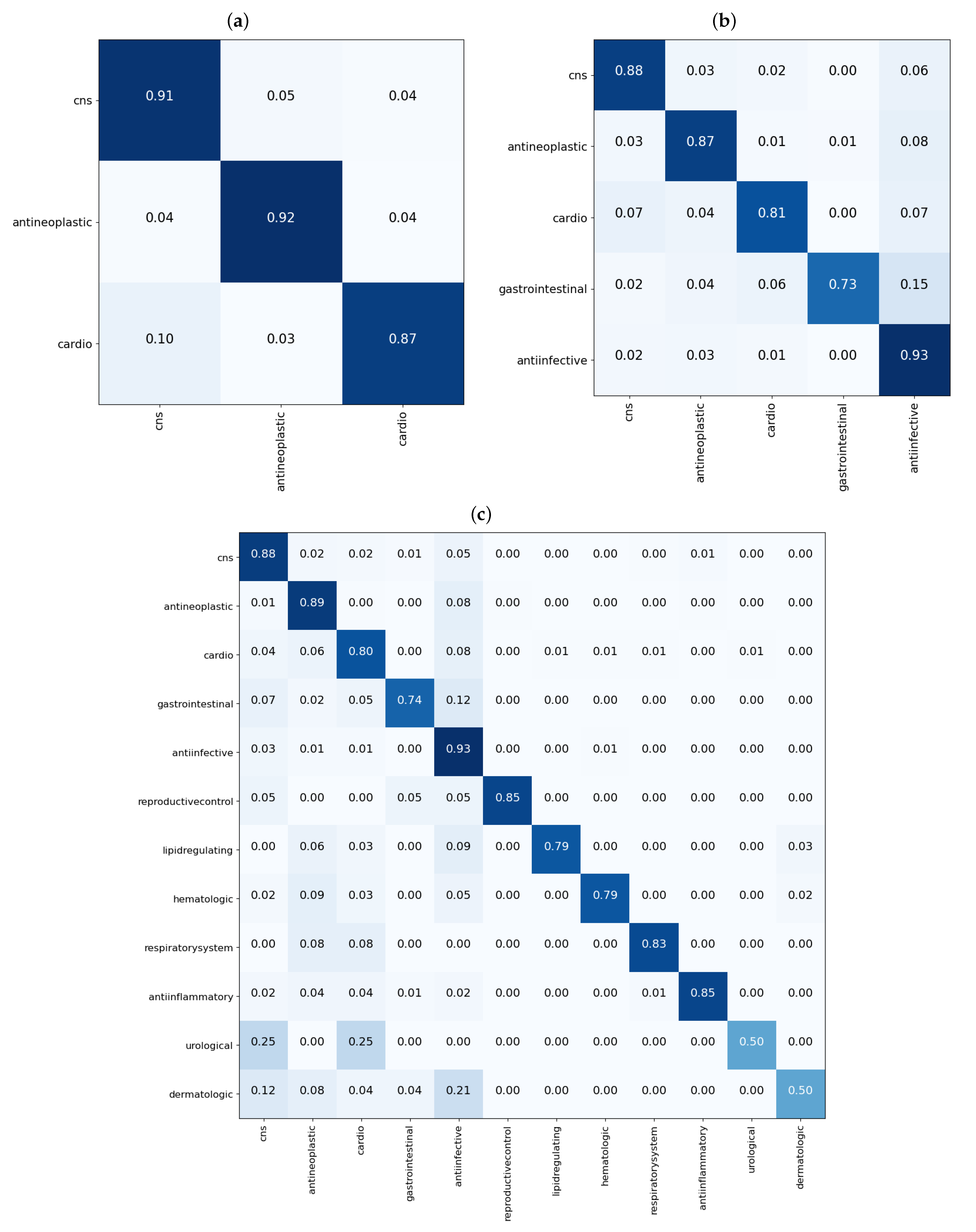
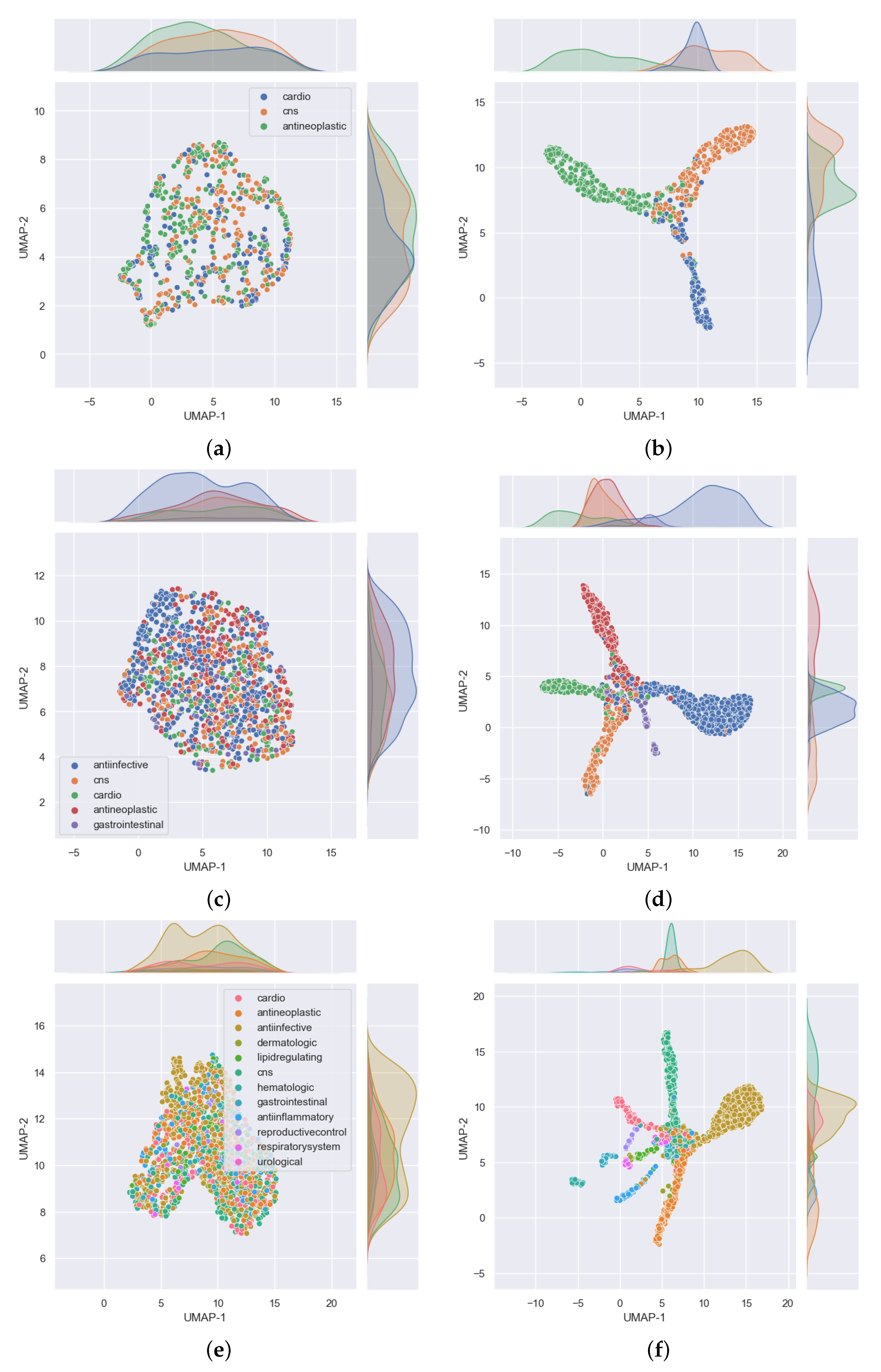
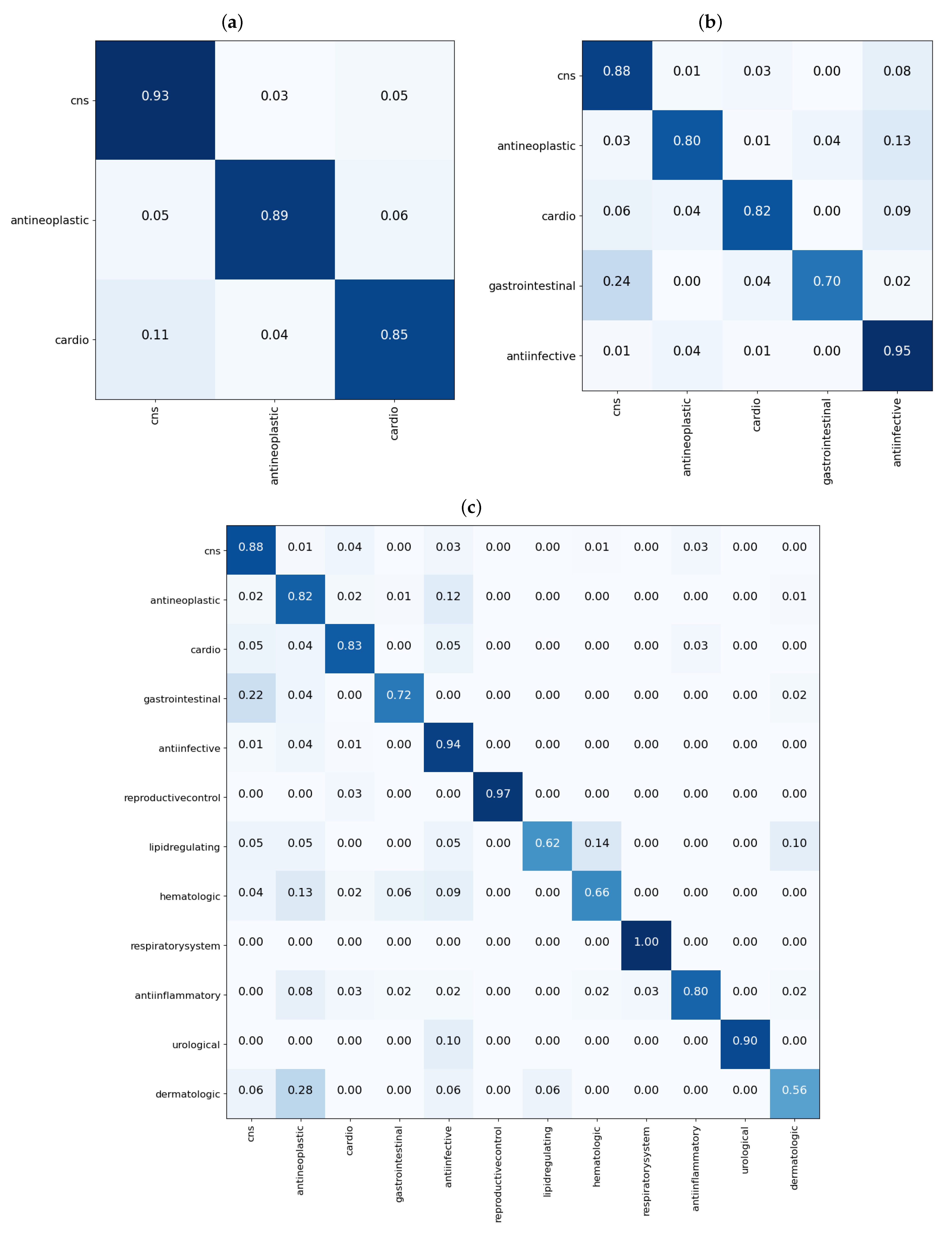
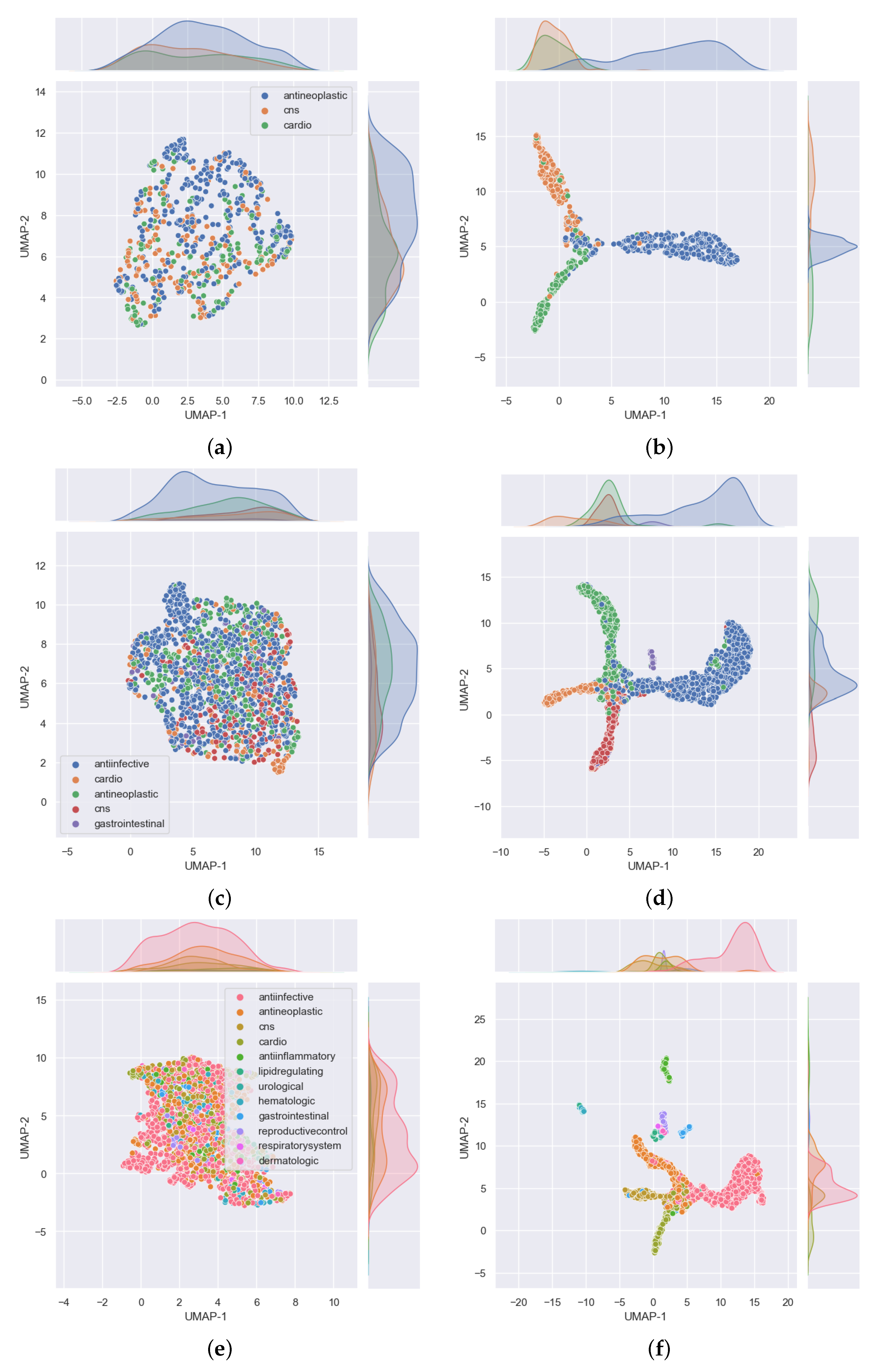
3.1. Single Label Classification Results for Cross Validation
3.2. Single Label Classification Results on Test Data
3.3. Multi-Label Classification Results on Validation and Test Data
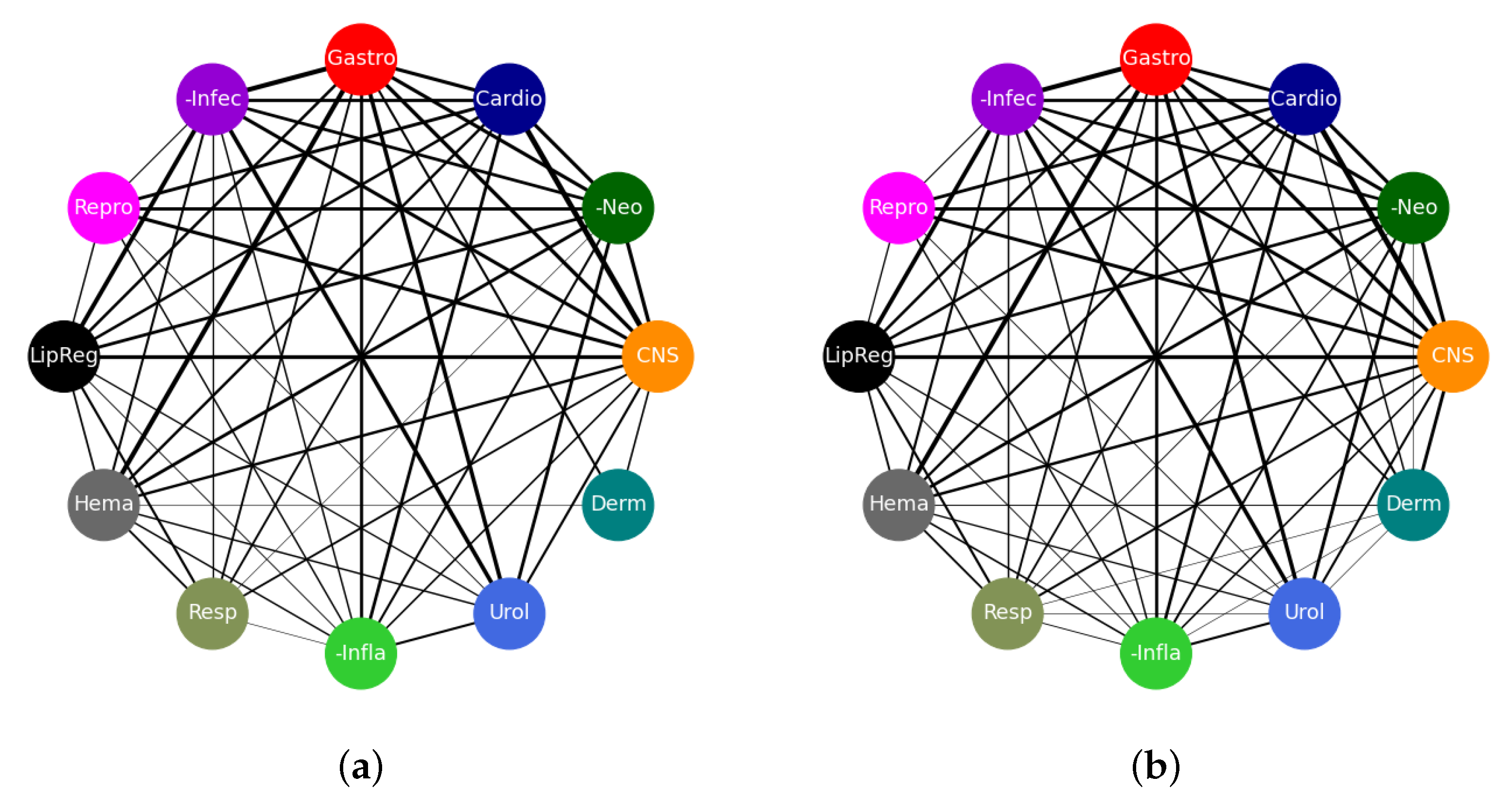
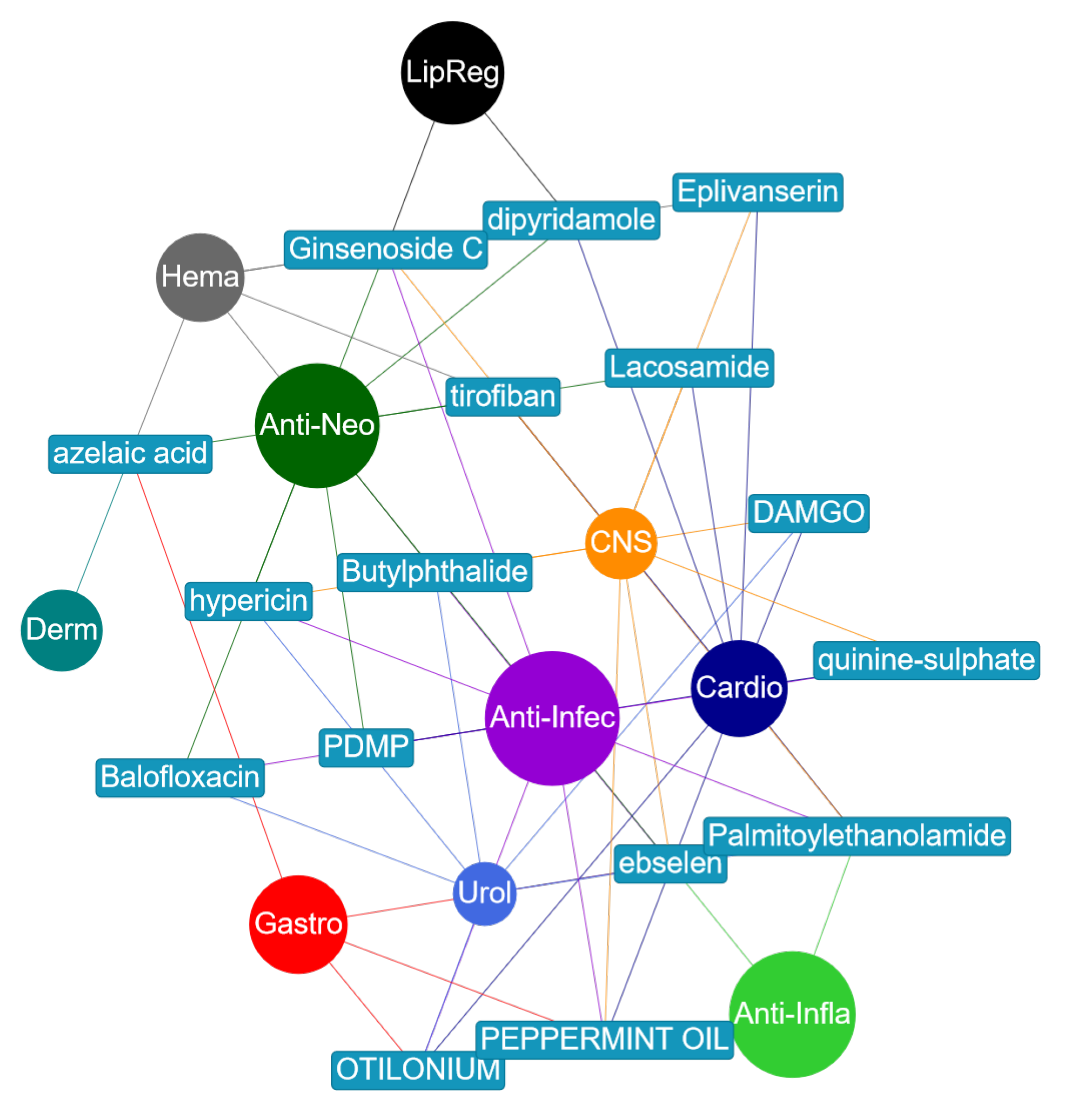
3.4. Drug Repurposing Opportunities
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| MeSH | Medical SubHeading |
| GCN | Graph Convolutional Networks |
| CNN | Convolutional Neural Networks |
| UMAP | Uniform Manifold Approximation and Projection |
| RF | Random Forest |
| ReLU | Rectified Linear Unit |
| FC | Fully Connected |
| SMILES | Simplified Molecular Input Line Entry System |
| CNS | Central Nervous System |
| siRNA | Small interfering RNA |
| ALS | Amyotrophic Lateral Sclerosis |
| PEA | PalmitoylEthanolAmide |
| DIP | Dipyridamole |
| UTI | Urinary Tract Infection |
| HMGCS1 | 3-hydroxy-3-methylglutaryl-CoA synthase 1 |
| SOD1 | Cu,Zn-superoxide dismutase gene |
| API | Application Programming Interface |
References
- Loging, W.; Harland, L.; Williams-Jones, B. High-throughput electronic biology: Mining information for drug discovery. Nat. Rev. Drug Discov. 2007, 6, 220–230. [Google Scholar] [CrossRef]
- Kirchmair, J.; Göller, A.H.; Lang, D.; Kunze, J.; Testa, B.; Wilson, I.D.; Glen, R.C.; Schneider, G. Predicting drug metabolism: Experiment and/or computation? Nat. Rev. Drug Discov. 2015, 14, 387–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickson, M.; Gagnon, J.P. The cost of new drug discovery and development. Discov. Med. 2009, 4, 172–179. [Google Scholar]
- DiMasi, J.A.; Hansen, R.W.; Grabowski, H.G. The price of innovation: New estimates of drug development costs. J. Health Econ. 2003, 22, 151–185. [Google Scholar] [CrossRef] [Green Version]
- Iskar, M.; Zeller, G.; Blattmann, P.; Campillos, M.; Kuhn, M.; Kaminska, K.H.; Runz, H.; Gavin, A.C.; Pepperkok, R.; Van Noort, V.; et al. Characterization of drug-induced transcriptional modules: Towards drug repositioning and functional understanding. Mol. Syst. Biol. 2013, 9, 662. [Google Scholar] [CrossRef]
- Kutalik, Z.; Beckmann, J.S.; Bergmann, S. A modular approach for integrative analysis of large-scale gene-expression and drug-response data. Nat. Biotechnol. 2008, 26, 531–539. [Google Scholar] [CrossRef]
- Herrera-Ruiz, D.; Faria, T.N.; Bhardwaj, R.K.; Timoszyk, J.; Gudmundsson, O.S.; Moench, P.; Wall, D.A.; Smith, R.L.; Knipp, G.T. A novel hPepT1 stably transfected cell line: Establishing a correlation between expression and function. Mol. Pharm. 2004, 1, 136–144. [Google Scholar] [CrossRef]
- Aliper, A.; Plis, S.; Artemov, A.; Ulloa, A.; Mamoshina, P.; Zhavoronkov, A. Deep learning applications for predicting pharmacological properties of drugs and drug repurposing using transcriptomic data. Mol. Pharm. 2016, 13, 2524–2530. [Google Scholar] [CrossRef] [Green Version]
- Meyer, J.G.; Liu, S.; Miller, I.J.; Coon, J.J.; Gitter, A. Learning drug functions from chemical structures with convolutional neural networks and random forests. J. Chem. Inf. Model. 2019, 59, 4438–4449. [Google Scholar] [CrossRef] [Green Version]
- Landrum, G. RDKit: Open-Source Cheminformatics. 2006. Available online: http://www.rdkit.org (accessed on 30 January 2021).
- Rogers, D.; Hahn, M. Extended-connectivity fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Zitnik, M.; Agrawal, M.; Leskovec, J. Modeling polypharmacy side effects with graph convolutional networks. Bioinformatics 2018, 34, i457–i466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gysi, D.M.; Do Valle, Í.; Zitnik, M.; Ameli, A.; Gan, X.; Varol, O.; Sanchez, H.; Baron, R.M.; Ghiassian, D.; Loscalzo, J.; et al. Network medicine framework for identifying drug repurposing opportunities for COVID-19. Proc. Natl. Acad. Sci. USA 2020, 118, e2025581118. [Google Scholar] [CrossRef]
- Morris, C.; Ritzert, M.; Fey, M.; Hamilton, W.L.; Lenssen, J.E.; Rattan, G.; Grohe, M. Weisfeiler and leman go neural: Higher-order graph neural networks. In Proceedings of the AAAI Conference on Artificial Intelligence, Honolulu, HI, USA, 27 January–1 February 2019; Volume 33, pp. 4602–4609. [Google Scholar]
- Goh, G.B.; Siegel, C.; Vishnu, A.; Hodas, N.; Baker, N. How much chemistry does a deep neural network need to know to make accurate predictions? In Proceedings of the 2018 IEEE Winter Conference on Applications of Computer Vision (WACV), Lake Tahoe, NV, USA, 12–15 March 2018; pp. 1340–1349. [Google Scholar]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem substance and compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef]
- Fey, M.; Lenssen, J.E. Fast graph representation learning with PyTorch Geometric. arXiv 2019, arXiv:1903.02428. [Google Scholar]
- Ramsundar, B.; Eastman, P.; Walters, P.; Pande, V.; Leswing, K.; Wu, Z. Deep Learning for the Life Sciences; O’Reilly Media: Newton, MA, USA, 2019. [Google Scholar]
- Nguyen, T.; Le, H.; Venkatesh, S. GraphDTA: Prediction of drug–target binding affinity using graph convolutional networks. BioRxiv 2019, 684662. [Google Scholar] [CrossRef]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Paszke, A.; Gross, S.; Chintala, S.; Chanan, G.; Yang, E.; DeVito, Z.; Lin, Z.; Desmaison, A.; Antiga, L.; Lerer, A. Automatic differentiation in pytorch. In Proceedings of the 31st Conference on Neural Information Processing Systems (NIPS 2017), Long Beach, CA, USA, 4–9 December 2017. [Google Scholar]
- Akiba, T.; Sano, S.; Yanase, T.; Ohta, T.; Koyama, M. Optuna: A next-generation hyperparameter optimization framework. In Proceedings of the 25th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, Anchorage, AK, USA, 4–8 August 2019; pp. 2623–2631. [Google Scholar]
- Szymański, P.; Kajdanowicz, T. A scikit-based Python environment for performing multi-label classification. arXiv 2017, arXiv:1702.01460. [Google Scholar]
- McInnes, L.; Healy, J.; Melville, J. Umap: Uniform manifold approximation and projection for dimension reduction. arXiv 2018, arXiv:1802.03426. [Google Scholar]
- Waskom, M.L. Seaborn: Statistical data visualization. J. Open Source Softw. 2021, 6, 3021. [Google Scholar] [CrossRef]
- Marai, G.E.; Pinaud, B.; Bühler, K.; Lex, A.; Morris, J.H. Ten simple rules to create biological network figures for communication. PLoS Comput. Biol. 2019, 15, e1007244. [Google Scholar] [CrossRef]
- Hagberg, A.; Swart, P.; S Chult, D. Exploring Network Structure, Dynamics, and Function Using NetworkX; Technical Report; Los Alamos National Lab.(LANL): Los Alamos, NM, USA, 2008. [Google Scholar]
- Perrone, G.; Unpingco, J.; Lu, H.M. Network visualizations with Pyvis and VisJS. arXiv 2020, arXiv:2006.04951. [Google Scholar]
- Song, Z.; Kong, K.; Wu, H.; Maricic, N.; Ramalingam, B.; Priestap, H.; Schneper, L.; Quirke, J.; Høiby, N.; Mathee, K. Panax ginseng has anti-infective activity against opportunistic pathogen Pseudomonas aeruginosa by inhibiting quorum sensing, a bacterial communication process critical for establishing infection. Phytomedicine 2010, 17, 1040–1046. [Google Scholar] [CrossRef] [Green Version]
- Hastings, J.; Owen, G.; Dekker, A.; Ennis, M.; Kale, N.; Muthukrishnan, V.; Turner, S.; Swainston, N.; Mendes, P.; Steinbeck, C. ChEBI in 2016: Improved services and an expanding collection of metabolites. Nucleic Acids Res. 2016, 44, D1214–D1219. [Google Scholar] [CrossRef]
- Kim, Y.H.; Park, K.H.; Rhoxy, H.M. Transcriptional activation of the Cu, Zn-superoxide dismutase gene through the AP2 site by ginsenoside Rb2 extracted from a medicinal plant, Panax ginseng. J. Biol. Chem. 1996, 271, 24539–24543. [Google Scholar] [CrossRef] [Green Version]
- Radad, K.; Moldzio, R.; Rausch, W.D. Ginsenosides and their CNS targets. CNS Neurosci. Ther. 2011, 17, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Horiba, M.; Ishikawa, K.; Katoh, S.; Naide, Y.; Yanaoka, M.; Andoh, S. Laboratory and clinical study of balofloxacin (Q-35), a new fluoroquinolone, in urinary tract infection. Drugs 1995, 49, 376–378. [Google Scholar] [CrossRef]
- Ge, S.M.; Zhan, D.L.; Zhang, S.H.; Song, L.Q.; Han, W.W. Reverse screening approach to identify potential anti-cancer targets of dipyridamole. Am. J. Transl. Res. 2016, 8, 5187. [Google Scholar] [PubMed]
- Zhou, S.; Xu, H.; Tang, Q.; Xia, H.; Bi, F. Dipyridamole enhances the cytotoxicities of trametinib against colon cancer cells through combined targeting of HMGCS1 and MEK pathway. Mol. Cancer Ther. 2020, 19, 135–146. [Google Scholar] [CrossRef] [Green Version]
- Esquejo, R.M.; Roqueta-Rivera, M.; Shao, W.; Phelan, P.E.; Seneviratne, U.; Am Ende, C.W.; Hershberger, P.M.; Machamer, C.E.; Espenshade, P.J.; Osborne, T.F. Dipyridamole Inhibits Lipogenic Gene Expression by Retaining SCAP-SREBP in the Endoplasmic Reticulum. Cell Chem. Biol. 2021, 28, 169–179. [Google Scholar] [CrossRef]
- Roelants, M.; Van Cleynenbreugel, B.; Lerut, E.; Van Poppel, H.; de Witte, P.A. Human serum albumin as key mediator of the differential accumulation of hypericin in normal urothelial cell spheroids versus urothelial cell carcinoma spheroids. Photochem. Photobiol. Sci. 2011, 10, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Sim, H.G.; Lau, W.K.; Olivo, M.; Tan, P.H.; Cheng, C.W. Is photodynamic diagnosis using hypericin better than white-light cystoscopy for detecting superficial bladder carcinoma? BJU Int. 2005, 95, 1215–1218. [Google Scholar] [CrossRef] [PubMed]
- D’Hallewin, M.A.; Kamuhabwa, A.; Roskams, T.; De Witte, P.; Baert, L. Hypericin-based fluorescence diagnosis of bladder carcinoma. BJU Int. 2002, 89, 760–763. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, A.; Donzelli, S.; Girgenti, V.; Sacconi, A.; Vasco, C.; Salmaggi, A.; Blandino, G.; Maschio, M.; Ciusani, E. In vitro antineoplastic effects of brivaracetam and lacosamide on human glioma cells. J. Exp. Clin. Cancer Res. 2017, 36, 1–13. [Google Scholar] [CrossRef]
- Rudà, R.; Pellerino, A.; Franchino, F.; Bertolotti, C.; Bruno, F.; Mo, F.; Migliore, E.; Ciccone, G.; Soffietti, R. Lacosamide in patients with gliomas and uncontrolled seizures: Results from an observational study. J. Neuro Oncol. 2018, 136, 105–114. [Google Scholar] [CrossRef]
- Rudà, R.; Houillier, C.; Maschio, M.; Reijneveld, J.C.; Hellot, S.; De Backer, M.; Chan, J.; Joeres, L.; Leunikava, I.; Glas, M.; et al. Effectiveness and tolerability of lacosamide as add-on therapy in patients with brain tumor–related epilepsy: Results from a prospective, noninterventional study in European clinical practice (VIBES). Epilepsia 2020, 61, 647–656. [Google Scholar] [CrossRef]
- Zhou, L.; She, P.; Tan, F.; Li, S.; Zeng, X.; Chen, L.; Luo, Z.; Wu, Y. Repurposing Antispasmodic Agent Otilonium Bromide for Treatment of Staphylococcus aureus Infections. Front. Microbiol. 2020, 11, 1720. [Google Scholar] [CrossRef]
- Guillamat-Prats, R.; Rinne, P.; Rami, M.; Ring, L.; Van Der Vorst, E.; Steffens, S. P692Palmitoylethanolamide promotes an anti-inflammatory macrophage phenotype and attenuates atherosclerotic plaque formation in mice. Eur. Heart J. 2017, 38, ehx501.P692. [Google Scholar] [CrossRef] [Green Version]
- Quagliariello, V.; Paccone, A.; Buccolo, S.; Iovine, M.; Botti, G.; Maurea, N. The Combination of Palmitoylethanolamide and Polydatin Reduces Inflammation in Cardiac and Vascular Endothelial Cells Exposed to Doxorubicin through Peroxisome Proliferator-Activated Receptor-A. 2021. Available online: https://esc365.escardio.org/presentation/233593?query=Quagliariello (accessed on 20 September 2021).
- Singh, R.; Shushni, M.A.; Belkheir, A. Antibacterial and antioxidant activities of Mentha piperita L. Arab. J. Chem. 2015, 8, 322–328. [Google Scholar] [CrossRef] [Green Version]
- Mahdavikian, S.; Rezaei, M.; Modarresi, M.; Khatony, A. Comparing the effect of aromatherapy with peppermint and lavender on the sleep quality of cardiac patients: A randomized controlled trial. Sleep Sci. Pract. 2020, 4, 1–8. [Google Scholar] [CrossRef]
- Parikka, M.; Nissinen, L.; Kainulainen, T.; Bruckner-Tuderman, L.; Salo, T.; Heino, J.; Tasanen, K. Collagen XVII promotes integrin-mediated squamous cell carcinoma transmigration—A novel role for αIIb integrin and tirofiban. Exp. Cell Res. 2006, 312, 1431–1438. [Google Scholar] [CrossRef]
- Bruno, A.; Dovizio, M.; Tacconelli, S.; Contursi, A.; Ballerini, P.; Patrignani, P. Antithrombotic agents and cancer. Cancers 2018, 10, 253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Sun, C.; Wang, L.; Lian, Y.; Xie, N.; Huang, S.; Zhao, W.; Ren, M.; Wu, D.; Ding, J.; et al. Low-dose tirofiban treatment improves neurological deterioration outcome after intravenous thrombolysis. Stroke 2019, 50, 3481–3487. [Google Scholar] [CrossRef] [PubMed]
- Kokhlikyan, N.; Miglani, V.; Martin, M.; Wang, E.; Alsallakh, B.; Reynolds, J.; Melnikov, A.; Kliushkina, N.; Araya, C.; Yan, S.; et al. Captum: A unified and generic model interpretability library for pytorch. arXiv 2020, arXiv:2009.07896. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MeSH Class | Label | Training Samples | Test Samples * | Task Subgroups |
|---|---|---|---|---|
| Central Nervous System | 0 | 1139 | 176 (0.13) | 3, 5, 12 |
| Antineoplastic | 1 | 1177 | 347 (0.23) | 3, 5, 12 |
| Cardiovascular | 2 | 788 | 152 (0.16) | 3, 5, 12 |
| Gastrointestinal | 3 | 258 | 46 (0.15) | 5, 12 |
| Anti-infective | 4 | 2398 | 776 (0.24) | 5, 12 |
| Reproductive Control | 5 | 148 | 33 (0.18) | 12 |
| Lipid Regulating | 6 | 164 | 21 (0.11) | 12 |
| Hematologic | 7 | 267 | 47 (0.15) | 12 |
| Respiratory System † | 8 | 101 | 8 (0.07) | 12 |
| Anti-inflammatory | 9 | 373 | 64 (0.15) | 12 |
| Urological | 10 | 26 | 10 (0.28) | 12 |
| Dermatological | 11 | 115 | 18 (0.14) | 12 |
| Setting for Model for Each of the Task Subgroups | ||||
|---|---|---|---|---|
| Hyperparameter | 3 | 5 | 12 | m |
| Learning rate | 0.0005 | 0.0005 | 0.0005 | 0.0005 |
| Batch size | 256 | 256 | 512 | 512 |
| Optimizer | Adam | Adam | Adam | Adam |
| GCN layers | 3 | 3 | 3 | 3 |
| Dense layers | 2 | 2 | 2 | 2 |
| Dropout | 0.4 | 0.4 | 0.25 | 0.25 |
| Task Subgroup | Metric | IMG + CNN Val | MFP + RF Val | GCN Val | GCN Test |
|---|---|---|---|---|---|
| 3 | Accuracy | ||||
| BAC | |||||
| MCC | |||||
| AUROC | |||||
| AP | |||||
| 5 | Accuracy | ||||
| BAC | |||||
| MCC | |||||
| AUROC | |||||
| AP | |||||
| 12 | Accuracy | ||||
| BAC | |||||
| MCC | |||||
| AUROC | |||||
| AP |
| Task Subgroup | Metric | IMG + CNN Val | GCN Val | GCN Test |
|---|---|---|---|---|
| m | Accuracy * | |||
| ( multi-label) | ||||
| AUROC | ||||
| AP |
| Compound | True MeSH Class | Predicted MeSH Class | Evidence of Prediction |
|---|---|---|---|
| Ginsenoside Rb2 | antineoplastic | anti-infective | [30,31,32,33] |
| lipid regulating | CNS | ||
| Balofloxacin | anti-infective | urological | [34] |
| antineoplastic | antineoplastic | ||
| Dipyridamole | cardiovascular | antineoplastic | [35,36,37] |
| hematologic | lipid regulating | ||
| Hypericin | anti-infective | anti-infective | [38,39,40] |
| antineoplastic | antineoplastic | ||
| CNS | urological | ||
| Lacosamide | cardiovascular | antineoplastic | [41,42,43] |
| CNS | CNS | ||
| Otilonium bromide | cardiovascular | anti-infective | [44] |
| gastrointestinal | urological | ||
| Palmitoylethanolamide | anti-infective | anti-infective | [45,46] |
| anti-inflammatory | cardiovascular | ||
| CNS | urological | ||
| Peppermint oil | CNS | anti-infective | [47,48] |
| gastrointestinal | cardiovascular | ||
| Tirofiban | cardiovascular | CNS | [49,50,51] |
| hematologic | antineoplastic |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chipofya, M.; Tayara, H.; Chong, K.T. Drug Therapeutic-Use Class Prediction and Repurposing Using Graph Convolutional Networks. Pharmaceutics 2021, 13, 1906. https://doi.org/10.3390/pharmaceutics13111906
Chipofya M, Tayara H, Chong KT. Drug Therapeutic-Use Class Prediction and Repurposing Using Graph Convolutional Networks. Pharmaceutics. 2021; 13(11):1906. https://doi.org/10.3390/pharmaceutics13111906
Chicago/Turabian StyleChipofya, Mapopa, Hilal Tayara, and Kil To Chong. 2021. "Drug Therapeutic-Use Class Prediction and Repurposing Using Graph Convolutional Networks" Pharmaceutics 13, no. 11: 1906. https://doi.org/10.3390/pharmaceutics13111906
APA StyleChipofya, M., Tayara, H., & Chong, K. T. (2021). Drug Therapeutic-Use Class Prediction and Repurposing Using Graph Convolutional Networks. Pharmaceutics, 13(11), 1906. https://doi.org/10.3390/pharmaceutics13111906