1. Introduction
In recent years, antibody–drug conjugates (ADCs) have become promising antitumor agents in the context of personalized medicine [
1,
2]. These therapeutics are developed through the conjugation of antibodies to highly potent cytotoxic drugs. To date, nine ADCs have been approved for clinical use, and almost eighty different ADC candidates are being tested in phase I to III clinical trials [
3]. Many ADCs target growth factor receptors expressed at the surface of cancer cells [
4].
HER-3 belongs to the epidermal growth factor receptor family of tyrosine kinases (ErbB). The family consists of four members: HER-1 (ErbB-1), HER-2 (ErbB-2), HER-3 (ErbB-3) and HER-4 (ErbB-4). Many studies have demonstrated a critical role for ErbB receptors in cell survival, proliferation and differentiation, as well as malignant transformation [
5,
6], and therapeutic agents against HER-1 and HER-2 are considered among the most successful examples of targeted therapy. Indeed, both small molecules inhibiting the tyrosine kinase activity and therapeutic monoclonal antibodies targeting EGFR have been approved for clinical use in NSCLC (Non Small Cell Lung Cancer) and colorectal cancer, whereas agents targeting HER-2 are approved for HER-2-overexpressing breast and gastric cancer. Among these, two different anti-HER-2 antibody–drug conjugates (ADCs) are now approved, Kadcyla and Enhertu, with additional HER-2-targeted ADCs currently in phase III clinical trials. Moreover, recent studies have highlighted a key role for HER-3 in tumor progression and drug resistance [
7]. Elevated expression of HER-3 has been observed in a wide variety of human cancers and has been associated with a worse survival in cancer patients with solid tumors. Studies on the underlying mechanisms implicate HER-3 expression as a major cause of treatment failure in cancer therapy. Activation of HER-3 signaling has also been shown to promote cancer metastasis [
8]. These data strongly support the notion that therapeutic inactivation of HER-3 and/or its downstream signaling is required to overcome treatment resistance and improve the outcomes of cancer patients. Unlike the other family members, the functions of HER-3 are not mediated by enzymatic catalytic activity, indicating that the ATP analog class of TKIs would not be suitable for this target. The only possibility of blocking HER-3 is by targeting its extracellular domain (ECD) by specific antibodies [
9].
Previously, we developed a HER-3 targeting ADC composed of EV20, a humanized anti-HER-3 antibody [
10,
11,
12,
13], conjugated with the tubulin inhibitor monomethyl auristatin F (MMAF) via a non-cleavable linker. This ADC, named EV20/MMAF, showed potent therapeutic activity against melanoma and breast cancer, in particular on a variant with acquired resistance to anti-HER2 therapies [
14,
15]. In the present paper, we have generated a novel EV20-based ADC, named EV20/NMS-P945, consisting of EV20 conjugated with a duocarmycin-like thienoindole derivative NMS-P528, using a peptide cleavable linker. There is a growing body of evidence suggesting that DNA-damaging agents, such as duocarmycins, have potential advantages over the microtubule-targeting agent MMAF as a loading agent for ADC development; first, DNA-damaging agents show higher cell killing activity, enabling ADCs to efficiently target tumor cells expressing low amounts of the antigen; secondly, these cytotoxic agents are active throughout the various cell-cycle phases, demonstrating high potency against both dividing and nondividing cells [
16]. Moreover, in contrast to MMAF, which is hydrophilic and cannot permeate cell membranes, the NMS-P945 payload possesses bystander activity, which enables an efficient targeting of heterogeneous solid tumors [
17]. Duocarmycins have been explored for ADC production in the past due to their high cytotoxic activity; however, their potential as payloads for ADC production has been hampered by unfavorable physicochemical properties, such as low solubility, causing poor conjugation levels and aggregation of the final products. More recently, ADC SYD985, a duocarmycin derivative conjugated to trastuzumab reached phase III clinical trials in HER2-positive breast cancers [
18,
19], confirming the validity of this class of toxins as ADC payloads. NMS-P528 is a new duocarmycin-like molecule developed at Nerviano Medical Sciences (NMS), which is suitable for ADC production due to its favorable physicochemical properties [
17]. NMS-P945, a drug linker containing NMS-P528, proved to be suitable for antibody conjugation, obtaining monomeric ADCs with an average Drug-to–Antibody Ratio (DAR) > 3.5 [
17]. In the present study, we explored the anti-cancer effects of EV20/NMS-P945 against human cancers in vitro and in vivo. EV20/NMS-P945 inhibited cell proliferation in multiple tumor cell lines in a target- and dose-dependent manner. In vivo EV20/NMS-P945 treatment effectively inhibited the growth of cancer cell-derived xenograft tumors in athymic nude mice. This novel ADC revealed a favorable pharmacokinetics, with terminal half-life (t
1/2) and distribution volumes comparable to those of the naked antibody. Finally, administration to up to 8 mg/kg was well tolerated in cynomolgus monkeys. These data suggest that EV20/NMS-P945 may be a promising therapeutic agent for the treatment of HER-3 expressing tumors.
3. Discussion
Over the past two decades, there has been a significant increase in interest in developing new cancer therapeutic agents specifically targeting and hampering the HER-3 receptor [
6]. There are essentially two reasons behind this therapeutic strategy: first, HER-3 is overexpressed in many cancers [
5]; second, the receptor acts as the main hub for escape mechanisms during the emergence of resistance to anticancer drugs [
22,
23,
24]. In addition, somatic HER-3 oncogenic mutations have been identified in gastric and colorectal cancer patients [
25]. Initial strategies adopted to block HER-3 activity included the use of small-molecule inhibitors (such as lapatinib, erlotinib, gefitinib, afatinib and neratinib), which are able to abrogate its dimerization partners’ kinase activity, or direct targeting of its extracellular domain through the blocking of monoclonal antibodies [
26]. In particular, a panel of naked antibodies has been developed and entered into clinical trials both as a monotherapy and/or in combination with approved anticancer drugs known to induce HER-3 receptor upregulation [
7]. The results of these studies showed no therapeutic improvement. More recently, the use of antibody–drug conjugates (ADCs) has been proposed as a different therapeutic approach to target HER-3-positive tumors efficiently. ADCs may be developed using monoclonal antibodies with high internalization capacity, as they mediate the efficient delivery of the conjugated, potent toxins inside target tumor cells [
2].
To date, two HER-3-targeting ADCs have been developed. One of these is based on MMAE conjugated to the anti-HER-3 antibody 9F7–F11. This ADC, which is at the preclinical stage, was able to improve chemoradiation in PDAC [
27]. A second ADC, named U3-1402, is composed of a humanized anti-HER-3 IgG1 antibody (patritumab) coupled to a novel membrane-permeable topoisomerase-I inhibitor DXd (DX-8951 derivative) through an enzymatically cleavable peptide-linker. U3-1402 is currently in Phase I/II clinical trials in metastatic breast, colorectal and non-small cell lung cancer [
28,
29,
30].
Recently, we have developed HER-3-targeting ADCs by conjugating monomethyl-Auristatin F (MMAF) to humanized EV20 antibody using both a non-cleavable and (val-cit)-cleavable linker. These ADCs, called EV20/MMAF and EV20-sss-vc/MMAF respectively, displayed potent antitumor activity in melanoma and breast carcinoma resistant to anti-HER-2 therapies [
13,
14] and liver cancer [
28]. Here, we aimed to increase the potential of EV20 as a scaffold for the specific delivery of cytotoxic agents by generating EV20/NMS-P945, which is composed of EV20 coupled to the novel thienoindole derivative NMS-P528 through a peptidic cleavable drug linker and a self-immolating spacer. A similar payload and conjugation chemistry has recently been successfully applied for the generation of an ADC targeting the ALK receptor in neuroblastoma [
31] and HER-2 [
17]. The results presented here demonstrate that EV20/NMS-P945 is obtained as a monomeric ADC with an average DAR of 3.6. The ADC has proven to be stable in storage buffer up to 2 months at 4 °C and in monkey and human plasma at 37 °C up to 30 days. Compared to the naked antibody, EV20/NMS-P945 ADC maintained the same in vitro and in vivo binding activity, internalization properties and ability to hamper receptor signaling.
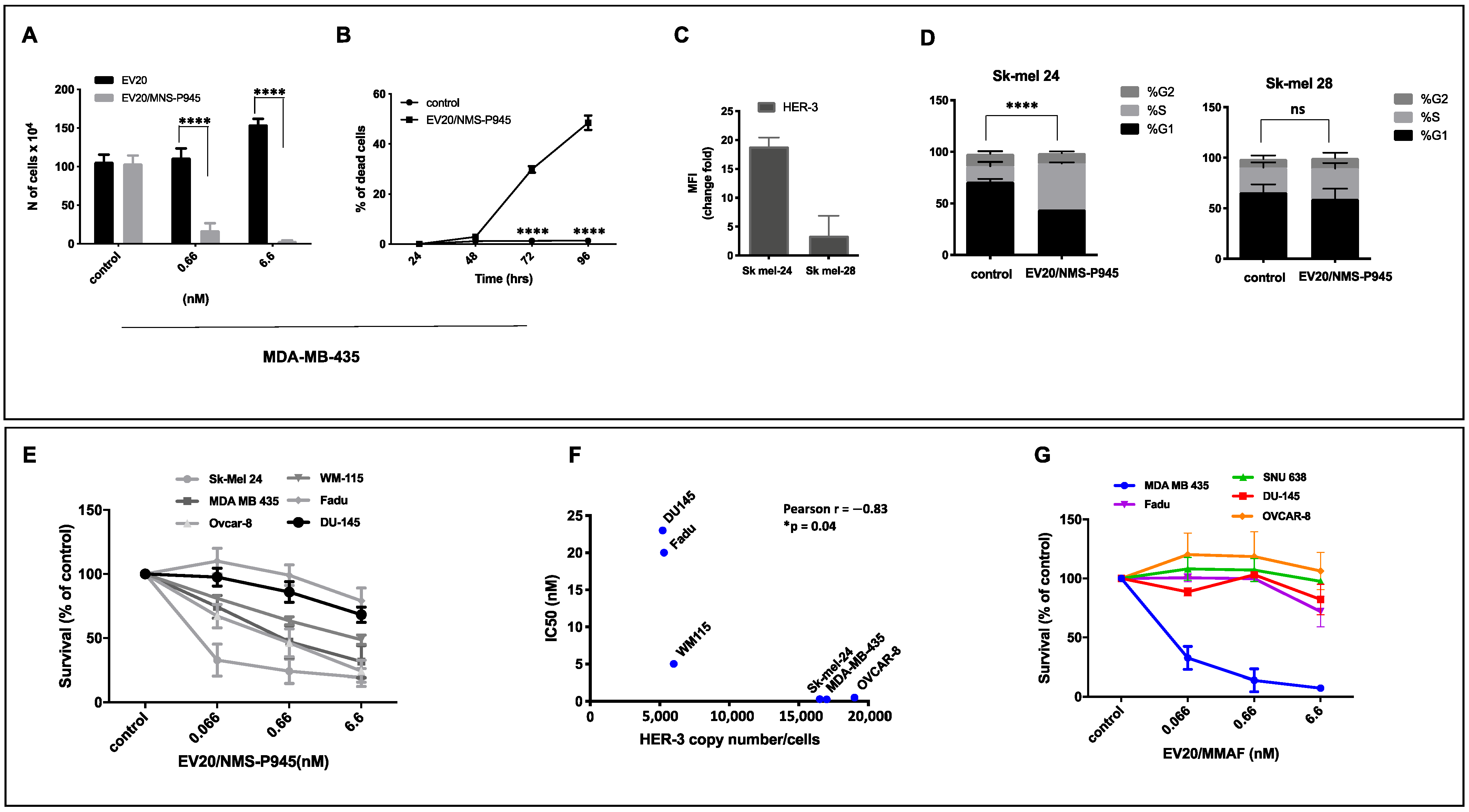
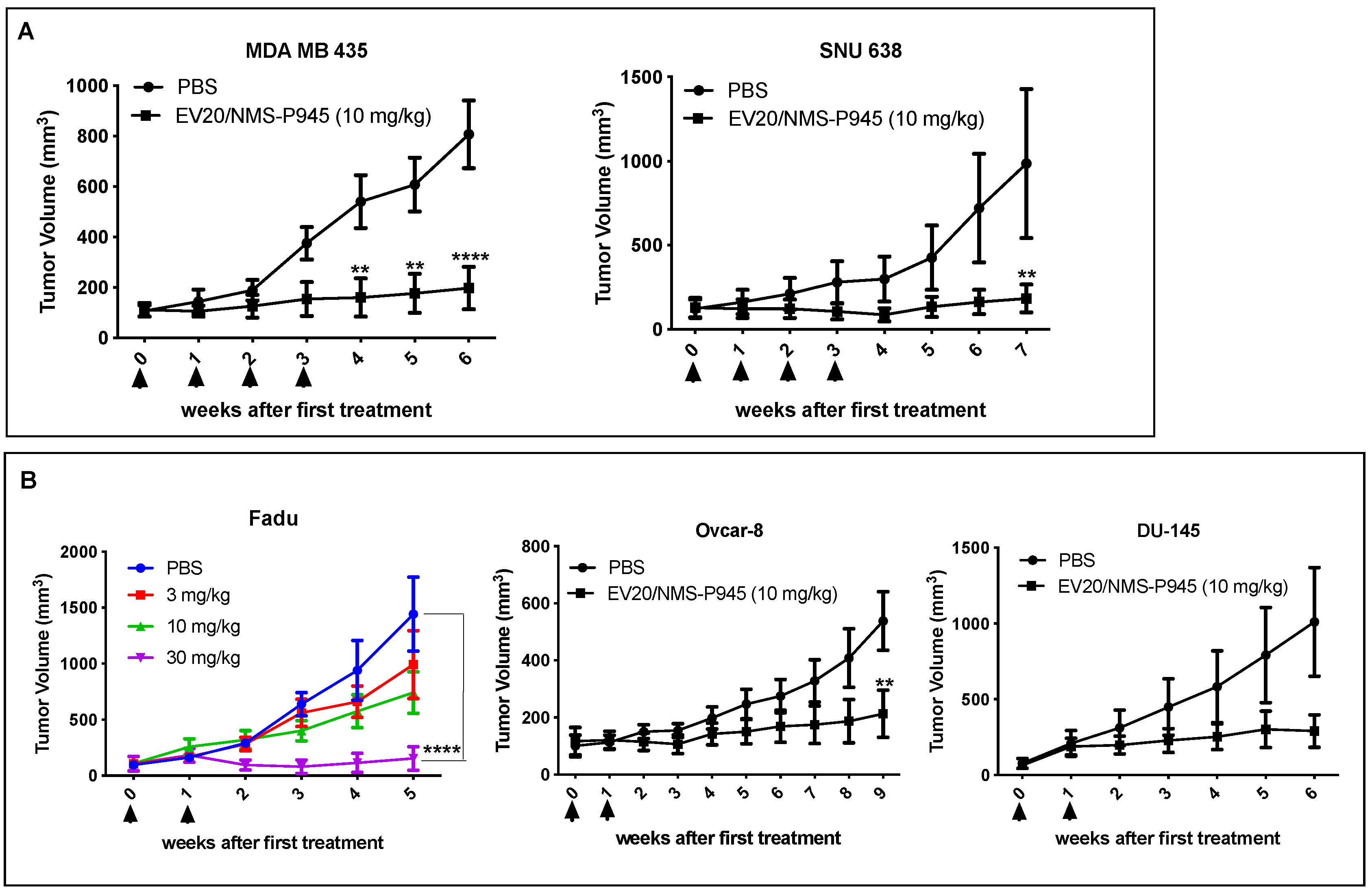
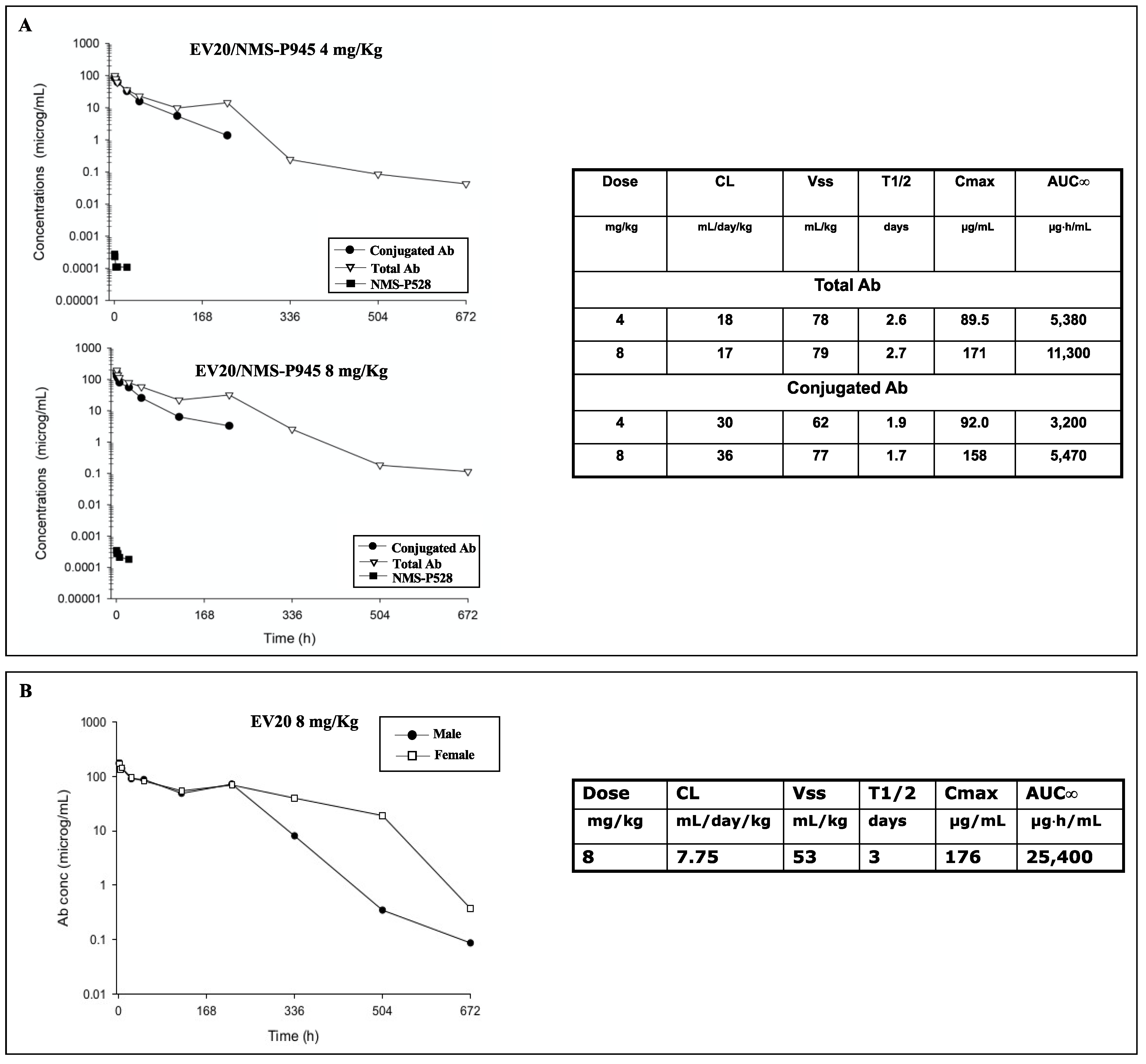
As for ADC potency, EV20/NMS-P945 displayed a potent and target-dependent antitumor activity in vitro, which was positively correlated to surface HER-3 receptor density, in a wide range of solid human cancers. Importantly, this novel ADC showed in vitro superior cytotoxic activity compared to EV20/MMAF in some HER-3 positive solid tumors like gastric, ovarian, head and neck and prostatic cancer. A possible explanation could lie in the fact that the thienoindole derivative NMS-P528 is highly active in heterogenous tumors because of the bystander effect exerted by the toxin in cancer cells with low cell doubling time due to its mechanism of action, thus representing a good therapeutic option for solid tumors. TCR analysis using MP-RM-1, murine analogue of EV20, showed a restricted HER-3 expression pattern among normal human tissues, which was superimposable with non-human primate tissues, demonstrating cross-reactivity of EV20 with monkey HER-3. An exploratory safety study in cynomolgus monkeys demonstrated that the ADC is well tolerated at the dosage of 4 and 8 mg/kg and possesses a favorable pharmacokinetic profile, similar to that of commonly measured ADC analytes [
21], such as Adcetris and Kadcyla. Of note, EV20/NMS-P945 demonstrated high stability in vivo in monkey plasma and free toxin was detected at very low levels in plasma and was present no longer than 24 h after ADC dosing. Duocarmycin-based ADCs represent a good therapeutic strategy, as demonstrated by (vic-)trastuzumab duocarmazine (SYD985), constituted by anti-HER2 antibody conjugated with a cleavable linker-duocarmycin analog, vc-secoDUBA, which recently entered phase III evaluation in metastatic breast cancer [
3].
In summary, the data presented here position EV20/NMS-P945 as a promising therapeutic agent for the treatment of HER-3-expressing cancers.
4. Materials and Methods
4.1. Reagents
Antibodies used in the present study were as follows: phosphorylated ErbB-3 (Clone 21D3, Tyr1289, #4791), ErbB-3 (Clone D22C5, #12708), phosphorylated Akt (Ser473, Clone D9E, #4060), Akt (#9272), all from Cell Signaling Technology, Inc. Neuregulin-1β (NRG-1β, #5218SC) was purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA). EV20 antibody was produced as previously described [
13]. MP-RM-1 antibody was produced as previously described [
20]. Recombinant human ECD HER-3 was from ACROBiosystems (Bethesda, MD, USA). (3-(4,5-Dimethylthiazole-2-yl)-2,5 diphenyltetrazolium bromide (MTT) was purchased from Sigma-Aldrich Corporation (St. Louis, MO, USA).
4.2. Cell Lines
Prostate cancer (DU145 (HTB-81)), head and neck squamous cell carcinoma (Fadu (HTB-43)), pancreatic cancer (BxPC3 (CRL-1687)), melanoma (A375M (CRL-3222), MDA-MB-435 (HTB-131), Sk-mel 24 (HTB-71), Sk-mel 28 (HTB-72), WM115 (CRL-1675)), cells were purchased from American Type Culture Collection (Rockville, MD, USA). Ovarian cancer (OVCAR-8) were from the National Cancer Institute/National Institutes of Health (NCI/NIH; Frederick, MD, USA). Gastric cancer cells (SNU638) were purchased from the Korean Cell Line Bank (KCLB). All cell lines were cultured less than 3 months after resuscitation. The cells were cultured according to manufacturers’ instructions, using EMEM (Eagle’s Minimum Essential Medium) from Thermo Fisher Scientific, Inc., (Waltham, MA, USA) for DU145, Fadu, Sk-mel 28, Sk-mel 24 and WM115 cells, DMEM (Dulbecco’s Modified Eagle’s Medium) from Thermo Fisher Scientific, Inc. for A375M, and RPMI-1640 medium (Thermo Fisher Scientific, Inc.) for MDA-MB-435, OVCAR-8 and SNU638 cells, supplemented with 10% heat-inactivated fetal bovine serum (FBS, Invitrogen, Carlsbad, CA, USA), l-glutamine, 100 units/mL penicillin and 100 μg/mL streptomycin (Sigma-Aldrich Corporation, St. Louis, MO, USA), and incubated at 37 °C in humidified air with 5% CO2.
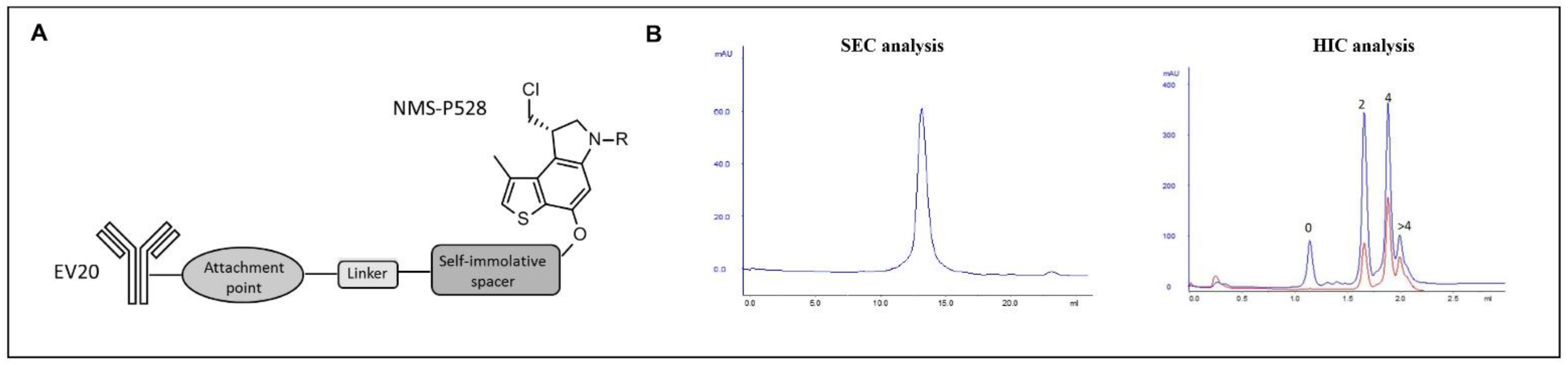
4.3. Generation of EV20/NMS-P945
NMS-P528 was conjugated to the EV20 antibody through a partial interchain disulfide reduction, followed by derivatization with the drug with the aim of reaching an average DAR > 3.5. The resulting ADC was characterized by size exclusion chromatography (SEC) to check for aggregated material, by hydrophobic interaction chromatography (HIC) to evaluate the distribution of the different isoforms, and by reducing and nonreducing SDS-PAGE to assess purity. Liquid chromatography–mass spectrometry analysis of the reduced product allowed for DAR determination.
4.4. EV20/NMS-P945 Stability Evaluation
For stability evaluation in storage buffer, EV20/NMS-P945 was incubated in PBS at two different concentrations: 2 mg/mL and 4 mg/mL at 4 °C for 1 week, 2 weeks, 1 month and 2 months. Aggregation state, protein concentration and drug–antibody ratio (DAR) were analyzed using SEC and HIC analysis. For stability evaluation in plasma, EV20/NMS-P945 at the concentration of 225 μg/mL was incubated in monkey and human plasma at 37 °C for 0, 3 h, 24 h, 3 days, 5 days, 8 days, 15 days, 22 days and 30 days and aliquots were taken and analyzed by sandwich ELISA assay. The amount of total antibody and ADC were assayed by sandwich ELISA using total anti-human IgG and an anti-NMS-P945 antibody as coating agents, respectively. A 96-well plate was coated with anti-human IgG (Bethyl Lab. Cat n A80-319A) or anti-NMS-P945 antibody (custom made). After overnight coating at 4 °C, the plate was blocked with 100 μL of 2% BSA in PBS containing 0.05% Tween 20 (PBS-T) with agitation at room temperature for 1 h. Subsequently, the solution was removed and each ADC sample was added to each well, and the plate was incubated at room temperature for 2 h. After each well was washed three times with 100 μL of PBS-T, 100 μL of goat anti-Human IgG-h + l HRP Conjugated Antibody (Bethyl Lab. Cat n A80-319P) was added. After being incubated at room temperature for 1 h, the plate was washed three times with 100 μL of PBS-T and 100 μL of 3,3′,5,5′-tetramethylbenzidine (TMB) substrate was added. After color was developed for 10−30 min, acid solution was added to each well and then the absorbance at 450 nm was recorded using a plate reader.
4.5. Immunohistochemistry
Normal frozen tissues from cynomolgus monkeys were purchased as microarrays from US, Biomax, Inc. For the immunohistochemical analysis, frozen tissue sections underwent immunoperoxidase staining by incubation overnight at 4 °C (refrigerator) with the anti-HER-3 MP-RM-1 monoclonal antibody (mAb). After removal of the primary mAb and washing twice with cold PBS, sections were stained utilizing the Vectastain ABC kit (Vector Laboratories, Inc., Burlingame, CA, USA) according to the manufacturer’s instructions, using chromogens aminoethylcarbazole and 3,3′-diaminobenzidine (DAB), as appropriate. Nuclear staining was done using filtered Mayer’s hematoxylin (Whatman paper 541) for 5 min, followed by washing of the slides with filtered distilled water (Whatman paper 541). Finally, slides were mounted in aqueous mounting medium with no sealing of the coverslips.
The experiments included appropriate negative (isotype-matched primary irrelevant mAb) and positive (archival tissue section displaying HER-3 expression) controls. Immunohistochemical findings were recorded blindly by two investigators. Normal tissues were scored as “negative” when no cellular or interstitial component was stained on a wide range of primary antibody concentrations (10–50 µg/mL). Normal tissues were scored as “positive” when unequivocal HER-3 staining was observed.
4.6. ELISA
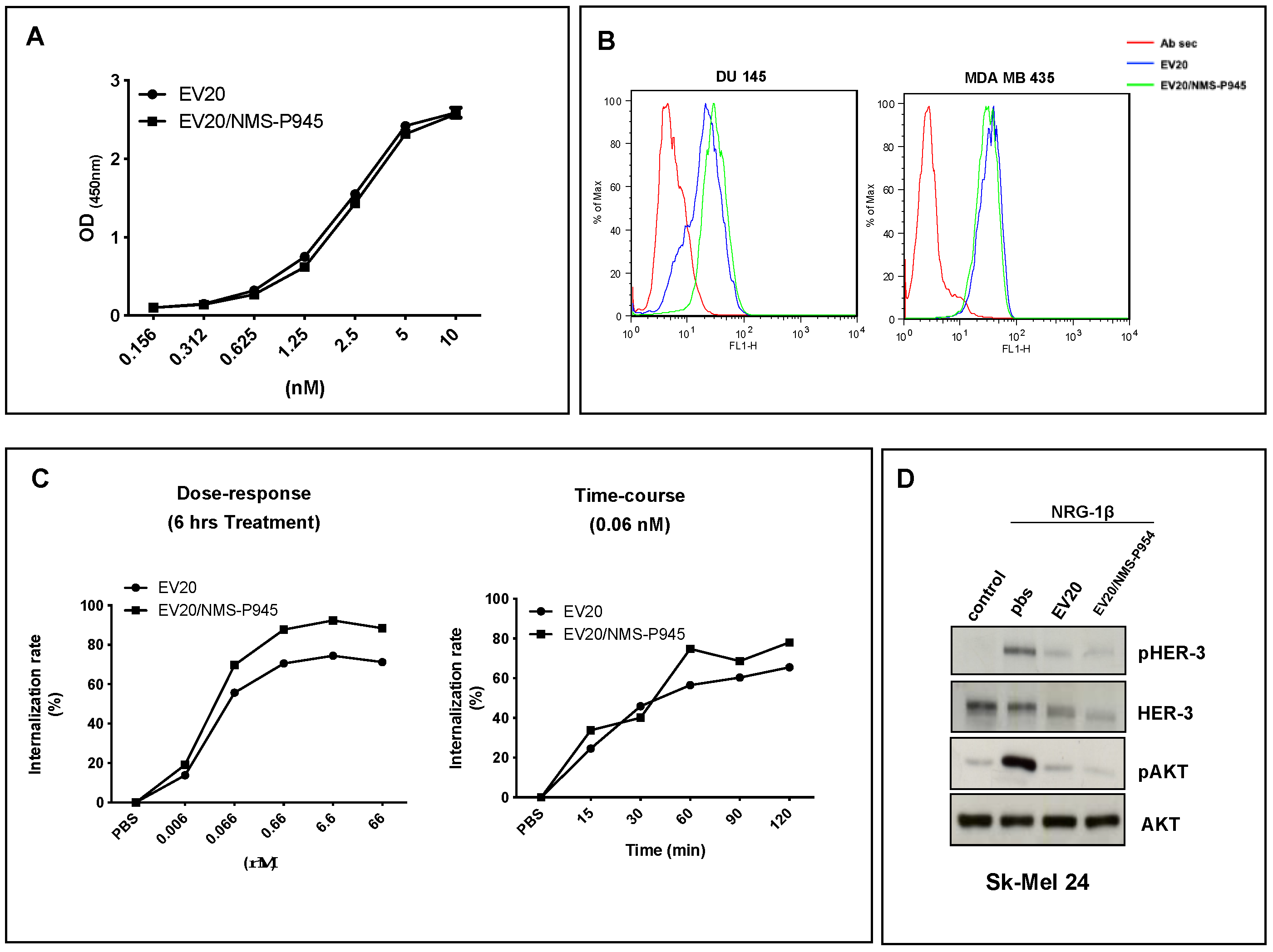
Recombinant HER-3 extracellular domain (ECD) (1 μg/mL) was pre-coated overnight at 4 °C on 96-well plates (NUNC Maxisorp modules). After blocking with 1% BSA in PBS for 1 h at room temperature, increasing concentrations (ranging between 0.15 nM and 10 nM) of naked EV20 or EV20/NMS-P945 were incubated for 1 h at room temperature. After several washes with PBS + 0.05% Tween-20, a goat anti-human IgG-HRP solution at a dilution of 1:5000 (A0170, Sigma-Aldrich Corporation) was added to each well and incubated for 1 h at room temperature. After washes, stabilized chromogen was added to each well for at least 10 min in the dark, then the reaction was stopped with the addition of H2SO4 1N and the absorbance was read at 450 nm with an ELISA reader.
4.7. Internalization Assays
For flow cytometric analysis of internalization, A375M cells were plated in 12-well plates and grown in DMEM containing 10% FBS for 24 h. Cells were then incubated at 37 °C with increasing doses of EV20 or EV20/NMS-P945 for 6 h in a dose-dependent assay, and with 0.06 nM of EV20 or EV20/NMS-P945 for the indicated times in a time-dependent assay. In order to quantify surface HER-3 internalization, the cells were detached and stained with 10 μg/mL of EV20 as a primary antibody for 30 min on ice. After washing, cells were labeled with PE-conjugate goat anti-Human Fc at a dilution of 1:300 (H10104, Molecular Probes, Life Technologies, Carlsbad, CA, USA) for 30 min on ice. Analysis was performed using a FACSCantoII cytometer (BD Pharmingen, Franklin Lakes, NJ, USA). Data were analyzed with FlowJo software V10.7 (FlowJo, LLC, BD Pharmingen, Franklin Lakes, NJ, USA).
4.8. Flow Cytometry
For the cell binding assay, DU145 and MDA MB 435 cells were detached and labeled with 10 μg/mL of EV20 and EV20/NMS-P945 for 30 min on ice, followed by staining of 30 min on ice with PE-conjugate goat anti-Human Fc as a secondary antibody at a dilution of 1:300 (H10104, Molecular Probes; Life Technologies; Thermo Fisher Scientific, Inc.). For HER-3 surface expression analysis, Sk-mel 24 and Sk-mel 28 cells were harvested and labeled with 10 μg/mL of EV20 as a primary antibody for 30 min on ice. After washing, cells were labelled with PE-conjugate goat anti-Human Fc as a secondary antibody at a dilution of 1:300 (H10104, Molecular Probes; Life Technologies; Thermo Fisher Scientific, Inc.) for 30 min on ice. For evaluation of HER-3 copy number on the surface of a panel of cell lines, QIFIKIT (Quantitative Analysis Kit, Dako Agilent, Santa Clara, CA, USA) was used following the manufacturer’s instructions. Analysis was performed using a FACSCantoII cytometer (BD Biosciences).
4.9. Cell Cycle Analysis
After 48 h of treatment with 0.6 nM of EV20/NMS-P945, Sk-mel 24 and Sk-mel 28 cells were harvested and centrifuged for 5 min. Then, PBS was added to the cells, followed by 70% ethanol to bring the final volume to 0.5 mL. The cells were stored overnight at 4 °C. Cells were centrifuged and washed twice with PBS, before resuspending and staining them at 37 °C for 30 min with 50 µL of RNAse solution (10 mg/mL, R6513, Sigma-Aldrich Corporation) and then with 200 µL of propidium iodide solution (1 mg/mL, P4170, Sigma-Aldrich Corporation). Cell cycle analysis was performed using a FACSCantoII cytometer (Beckton Dickinson, Buccinasco, Italy). Data were analyzed using FlowJo software V10.7.
4.10. Cytotoxicity Assays
Cell proliferation was assessed by an MTT [3-(4,5-dimethyldiazol-2-yl)-2,5-diphenyl tetrazolium bromide] assay (Sigma-Aldrich), Trypan Blue staining (Sigma-Aldrich), and cell count. For the MTT assay, cell lines were seeded into 24-well plates at a density of 5 × 103 cells/well in 500 µL of complete medium; they were treated with increasing doses of EV20/NMS-P945 or EV20/MMAF (ranging between 0.06 and 6.6 nM) and further incubated for 120 h. At the end of the incubation period, cells were incubated with 200 µL of MTT solution (medium serum-free with 0.5 mg/mL of MTT) for a further 2 h. After removal of the MTT solution, 200 µL of dimethyl sulfoxide (DMSO) was added to the wells for 10 min, and the absorption value at 570 nm was measured using a multi-plate reader. All experiments were performed in triplicate and the IC50 (Inhibit Cellular Proliferation by 50%) values were calculated using GraphPad Prism 8.0 software (GraphPad Software, Inc., San Diego, CA, USA).
For Trypan blue staining, after treatment with 0.6 nM of EV20/NMS-P945 for 24–48-72–96 h, MDA MB 435 cells were stained with 0.4% Trypan blue and were counted at various fields in the hemocytometer. The level of cytotoxicity was calculated as % of dead cells.
For cell count assay, MDA MB 435 cells were seeded in 6-well plates and exposed to naked EV20 and EV20/NMS-P945 at the doses of 0.6 and 6.6 nM for 120 h in a complete medium; then cells were trypsinized and counted using a hemocytometer. The average of cell counts from the triplicate was obtained, and the SD value was calculated statistically.
4.11. Western Blotting
Lysates from Sk-mel 24 cells exposed to the indicated treatments were prepared by washing cells twice in cold PBS followed by lysis with RIPA Buffer (50 mM Tris-HCl, 1% NP40, 0.1% SDS, 150 mM NaCl) supplemented with protease and phosphatase inhibitors (Sigma-Aldrich: Merck KGaA) for 10 min at 4 °C. Insoluble materials were removed by centrifugation (16,000× g for 10 min at 4 °C) and protein concentration was assessed using a Bradford assay. Equal amounts of protein (30 μg) were separated by SDS/PAGE on 10% polyacrylamide gel and transferred to nitrocellulose membrane. Membranes were blocked with 5% non-fat dry milk in PBS containing 0.1% Tween-20 for 1 h at room temperature and incubated with the indicated primary antibodies at the dilution of 1:1000 in PBS containing 0.1% Tween-20 overnight at 4 °C. After washing, membranes were hybridized for 1 h at room temperature with HRP-conjugated Goat anti-Rabbit IgG (STAR208P; Bio-rad Laboratories Inc., Berkeley, CA, USA) at a dilution of 1:20,000 in PBS containing 0.1% Tween-20. Detection was performed with a Plus-ECL chemiluminescence kit (Bio-Rad Laboratories, Inc.).
4.12. In Vivo Tumor Growth
Athymic CD-1 nu/nu female mice (5 or 7 weeks old) were purchased from Charles River Laboratories (Calco, Italy) and maintained at 22–24 °C and relative humidity, under pathogen-limiting conditions as required. Cages, bedding and food were autoclaved before use. Mice were provided with a standard diet and water ad libitum and acclimatized for 2 weeks before the start of the experiments. Housing and all procedures involving the mice were performed according to the protocol approved by the Institutional Animal Care and Use Committee of the Italian Ministry of Health (Authorization no. 292/2017-PR).
Xenografts were generated by subcutaneous injection into the right flank of mice of a range between 2 × 106 and 5 × 106 of cells (OVCAR-8, DU-145, SNU638, Fadu, MDA-MB-435 and BxPC-3) in 200 μL of PBS. When xenografts became palpable (approximately 100 mm3), animals were divided into groups to provide a similar range of tumor sizes for each group. The treated group received intravenous injections of EV20 or EV20/NMS-P945 in PBS at the indicated schedules and doses, whereas the control group received PBS only. The tumor volume was monitored weekly by a caliper and calculated using the following formula: tumor volume (mm3) = (length × width2)/2.
For the hematological and biochemical analysis, blood from mice treated with 40 mg/kg of EV20/NMS-P945 was collected after 48 h from administration. Samples were centrifuged at 2500× g for 10 min at 4 °C, and levels of alanine transaminase (ALT) and aspartate transaminase (AST) were quantified. Moreover, hematological analyses were performed including counts of red blood cells (RBC) and white blood cells (WBC), counts of platelets (PLT), levels of haemoglobin (HGB), haematocrit (HCT), mean corpuscular volume (MCV) and mean corpuscular haemoglobin (MCH).
4.13. Safety and Pharmacokinetic Study on Monkeys
EV20/NMS-P945 was administered to cynomolgus monkeys (one animal/sex/group) as a single IV dose at 4 or 8 mg/kg. One additional group of animals was administered with the naked antibody (EV20) at the dose of 8 mg/kg and one group with the vehicle with the same regimen. Animals were monitored for 4 weeks after administration; they were observed daily for mortality and clinical signs; qualitative food intake was recorded daily, and body weight was recorded weekly.
Hematology and clinical chemistry parameters were investigated pre-test and on days 3, 8, 15, 22 and 29 of the study; animals were sampled for 4 weeks for pharmacokinetics. The format of the ELISA was solid-phase and non-competitive, where the total antibody was extracted from the monkey matrix by means of a goat polyclonal anti-human IgG (Bethyl Lab. Cat n A80-319A) antibody bound to the surface of a 96-well high-binding microtiter plate. On the other hand, conjugated antibody was detected with an anti-NMS-P945 (custom made) antibody bound to the surface of a 96-well ELISA plate. After washing, blocking with 200 µL/well of TBS pH 8.0 containing 1% BSA, the bound analyte was tagged with a goat anti-Human IgG-h + l HRP Conjugated Antibody (Bethyl Lab. Cat n A80-319P). After an additional washing procedure, the HRP substrate TMB was added to allow the peroxidase reaction with the consequent substrate conversion to the colored product. The enzymatic reaction was stopped by the addition of 100 µL/well of Stop solution and finally the optical density (OD) at 450 nm was recorded. Conversion of the OD readings of samples into analyte concentrations was performed by interpolation with a calibration curve constructed by plotting the OD readings of calibration standards (CS), prepared by spiking known amounts of analyte into pooled blank monkey plasma, which was then diluted at the optimal MRD (Minimum Required Dilution). The most suitable mathematical model was used to fit the calibration standards results best and convert the OD reading values registered for the quality control (QC) samples into analyte concentrations. Finally, free NMS-P528 in monkey plasma was determined by LC/MS-MS analysis.
4.14. Statistical Analysis
For in vivo xenograft growth curves and cytotoxicity assays, p-values were calculated by two-way ANOVA, followed by Bonferroni’s post hoc test. (* p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001). Statistical analysis of the cell cycle was performed using two-way ANOVA, followed by Sidak’s multiple comparison test. Experimental sample numbers (n) are indicated in the figure legends. All statistical analysis was performed with GraphPad Prism 8.0 software.


 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}