
A COVID-19 Drug Repurposing Strategy through Quantitative Homological Similarities Using a Topological Data Analysis-Based Framework

 , ,
, ,  ,
,  and
and
Abstract
:1. Introduction
2. Results
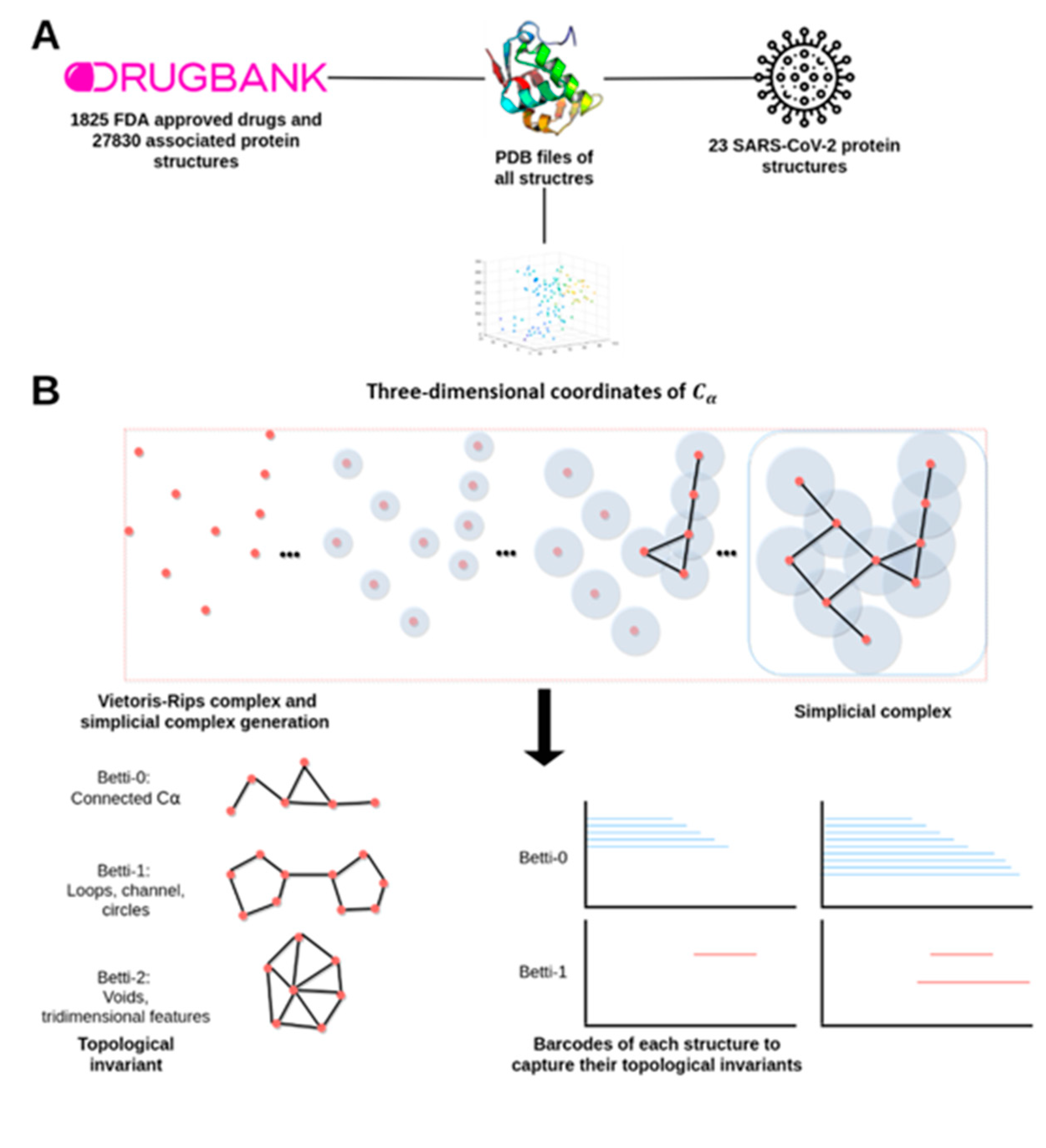
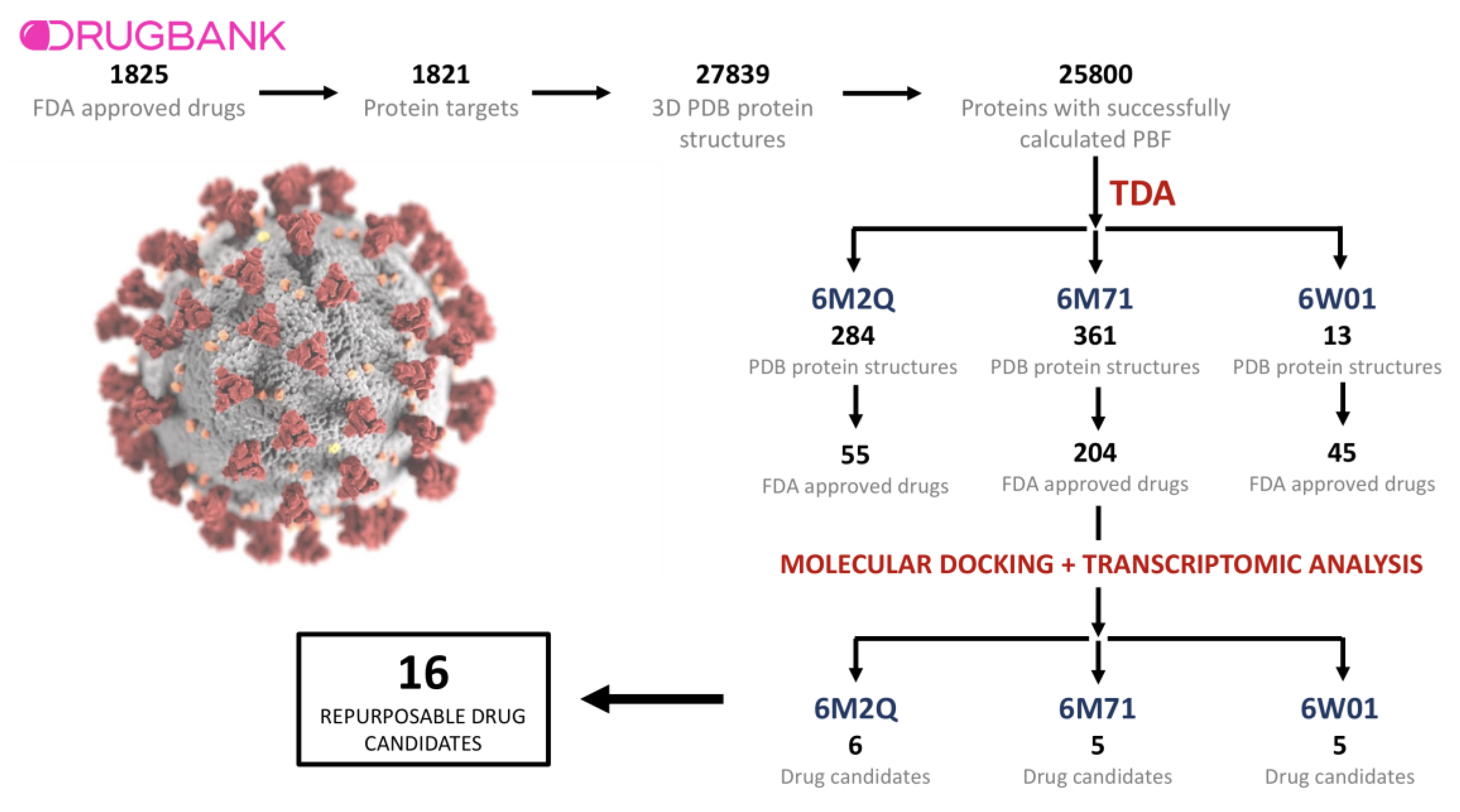
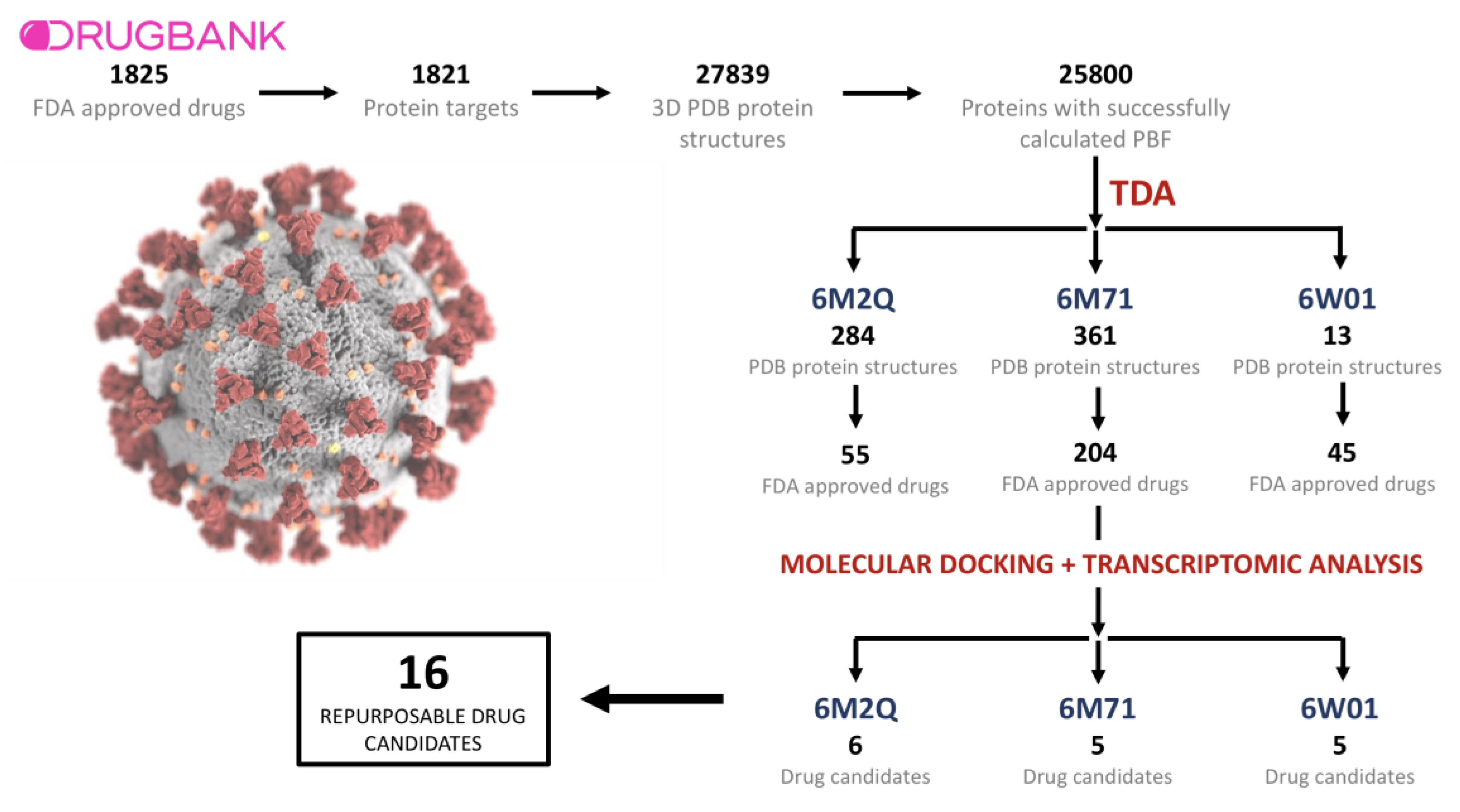
2.1. Drugs, Protein Targets, and PDB Structures Included in This Study
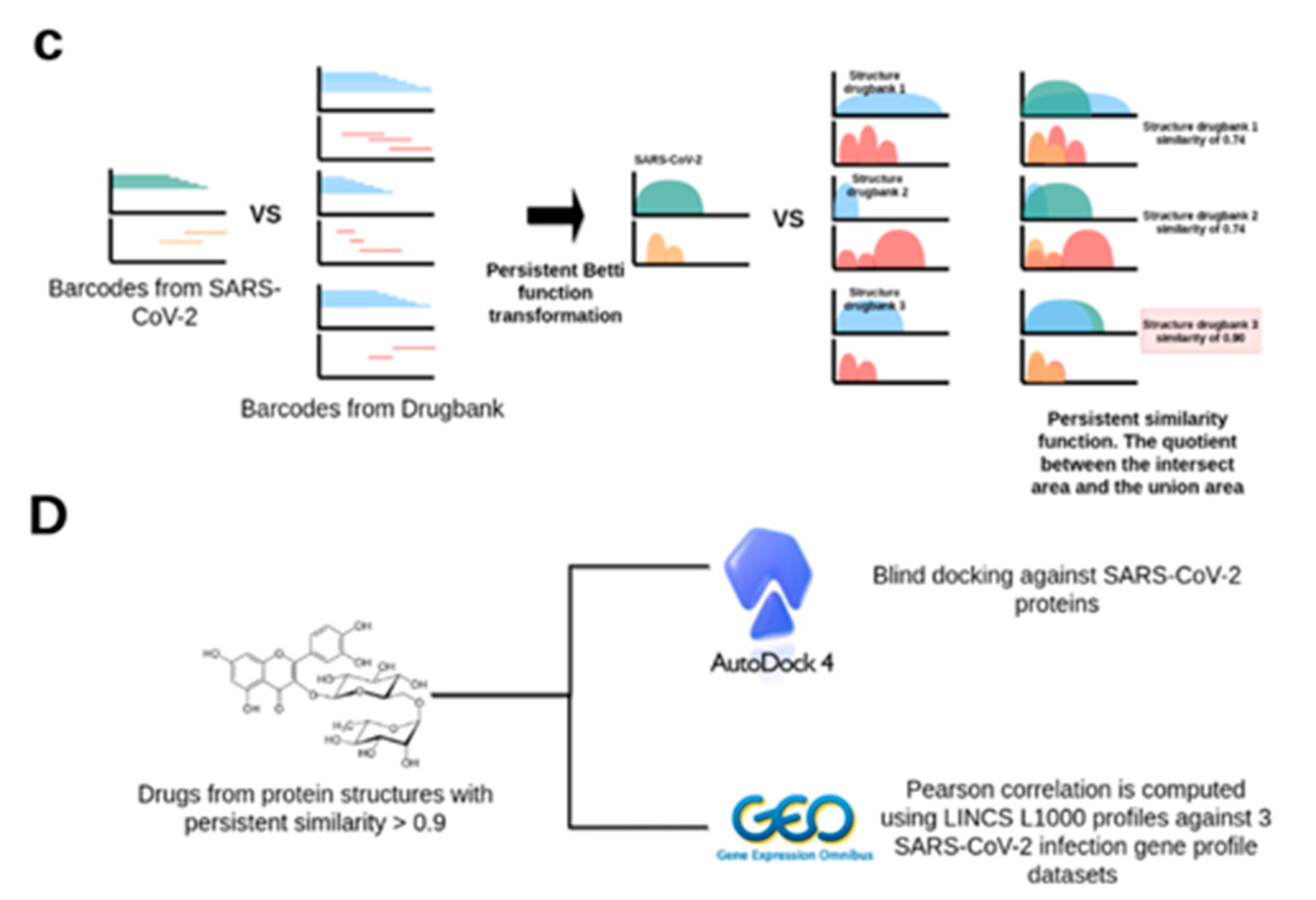
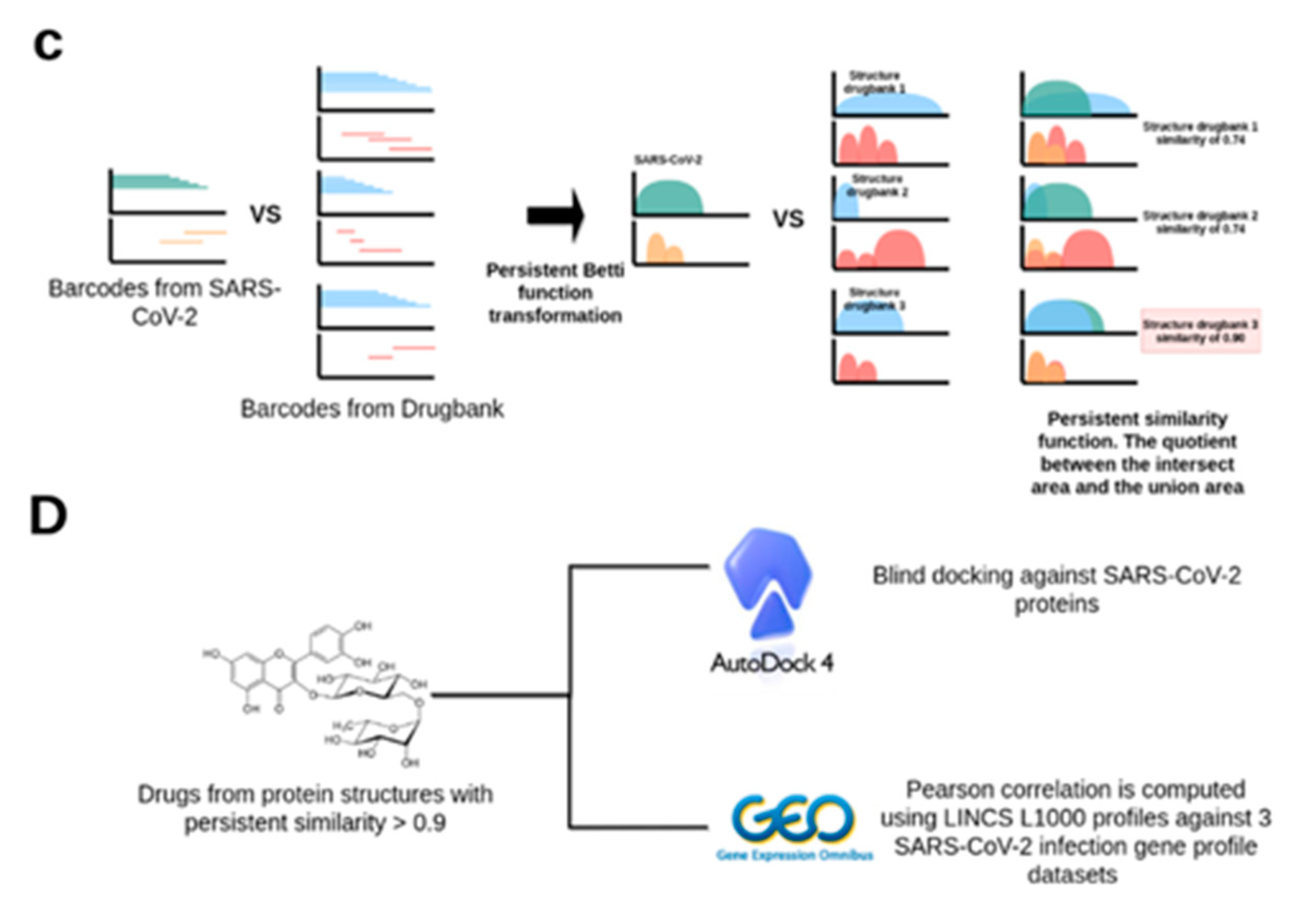
2.2. TDA Results, Viral Proteins Showing Mean Persistent Similarities above 0.9 with Structures Targeted by Known FDA-Approved Drugs
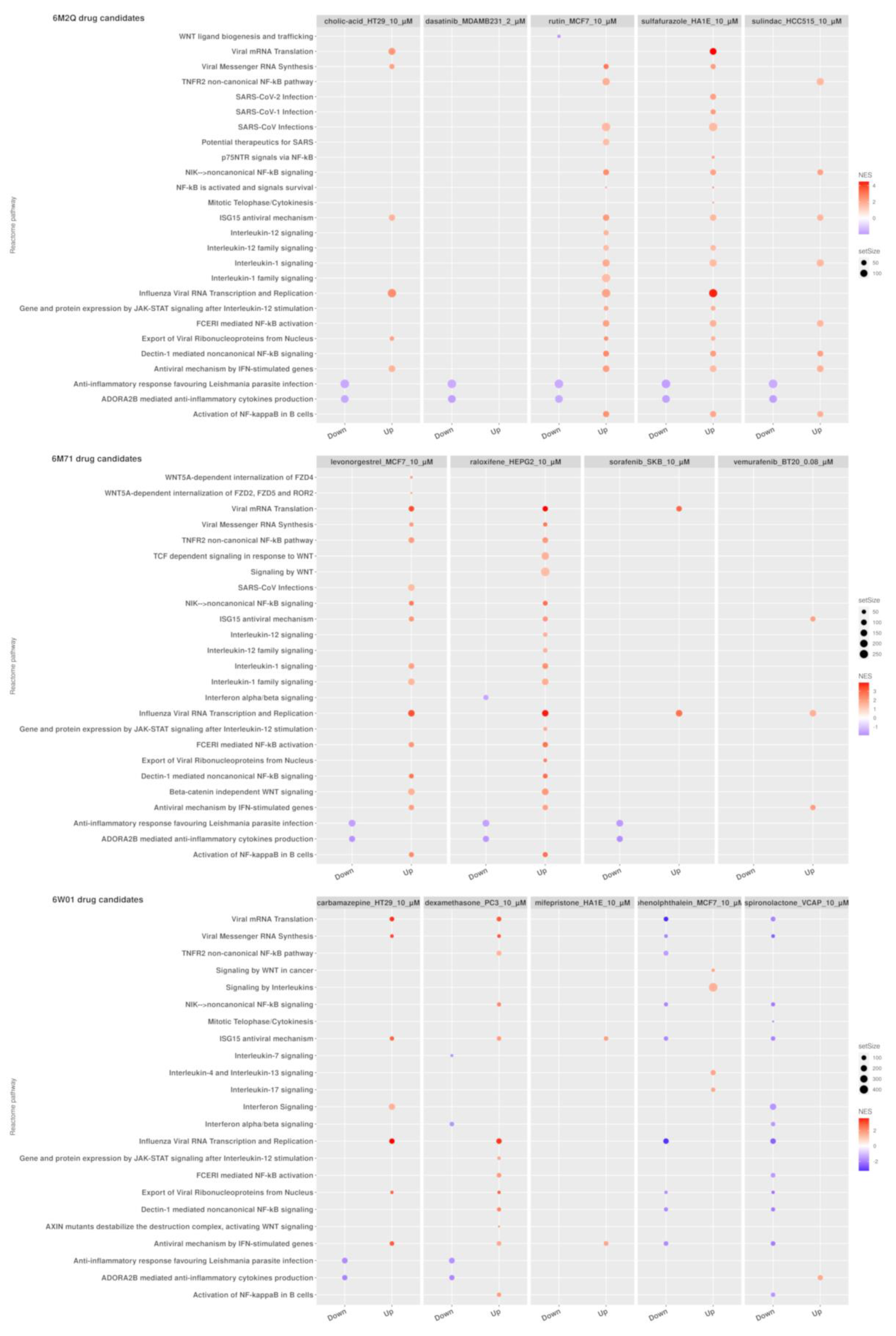
2.3. Transcriptomic Data Analysis Results
2.4. GSEA Analysis of the Repurposing Candidates
3. Discussion
4. Materials and Methods
4.1. Data Acquisition
4.2. A Topological Data Analysis Based Formalism to Compare, at Quantitative Level, the Homological Similarities of Pairwise Three-Dimensional Molecules Considered as Surfaces
4.3. Data Preprocessing and Persistent Similarity Measures Computation
4.4. Protein–Ligand Binding with AutoDock 4.2
4.5. Differential Gene Expression Analyses of SARS-CoV-2 Infected Human Samples and Cell Lines and Uninfected Controls
4.6. Identification of LINCS 1000 Signatures Negatively Correlated with the SARS-CoV-2 Differential Gene Expression Profiles
4.7. Gene Set Enrichment Analysis (GSEA)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Al-Mandhari, A.; Samhouri, D.; Abubakar, A.; Brennan, R. Coronavirus Disease 2019 outbreak: Preparedness and readiness of countries in the Eastern Mediterranean Region. East. Mediterr. Health J. 2020, 26, 136–137. [Google Scholar] [CrossRef]
- WHO Solidarity Trial Consortium; Faust, S.; Horby, P.; Lim, W.S.; Emberson, J.; Mafaham, M.; Bell, J.; Linsell, L.; Staplin, N.; Brightling, C.; et al. Repurposed Antiviral Drugs for Covid-19—Interim WHO Solidarity Trial Results. N. Eng. J. Med. 2020, NEJMoa2023184. [Google Scholar] [CrossRef]
- Mohs, R.C.; Greig, N.H. Drug discovery and development: Role of basic biological research. Alzheimers Dement. 2017, 3, 651–657. [Google Scholar] [CrossRef]
- DiMasi, J.A.; Feldman, L.; Seckler, A.; Wilson, A. Trends in risks associated with new drug development: Success rates for investigational drugs. Clin. Pharmacol. Ther. 2010, 87, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S. Covid-19: A drug repurposing and biomarker identification by using comprehensive gene-disease associations through protein-protein interaction network analysis. Preprints 2020. [Google Scholar] [CrossRef]
- Griffith, M.; Griffith, O.L.; Coffman, A.C.; Weible, J.V.; McMichael, J.F.; Spies, N.C.; Koval, J.; Das, I.; Callaway, M.B.; Eldred, J.M.; et al. DGIdb: Mining the druggable genome. Nat. Methods 2013, 10, 1209–1210. [Google Scholar] [CrossRef]
- Freshour, S.L.; Kiwala, S.; Cotto, K.C.; Coffman, A.C.; McMichael, J.F.; Song, J.J.; Griffith, M.; Griffith, O.L.; Wagner, A.H. Integration of the drug-gene interaction database (dgidb) with open crowdsource efforts. Nucleic Acids Res. 2021, 49, D1144–D1151. [Google Scholar] [CrossRef]
- Zhou, Y.; Hou, Y.; Shen, J.; Huang, Y.; Martin, W.; Cheng, F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020, 6, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keretsu, S.; Bhujbal, S.P.; Cho, S.J. Rational approach toward COVID-19 main protease inhibitors via molecular docking, molecular dynamics simulation and free energy calculation. Sci. Rep. 2020, 10, 17716. [Google Scholar] [CrossRef]
- Xia, K.; Wei, G.W. Persistent homology analysis of protein structure, flexibility, and folding. Int. J. Numer. Methods Biomed. Eng. 2014, 30, 814–844. [Google Scholar] [CrossRef] [PubMed]
- Javed, H.; Khan, M.M.; Ahmad, A.; Vaibhav, K.; Ahmad, M.E.; Khan, A.; Ashafaq, M.; Islam, F.; Siddiqui, M.S.; Safhi, M.M.; et al. Rutin prevents cognitive impairments by ameliorating oxidative stress and neuroinflammation in rat model of sporadic dementia of Alzheimer type. Neuroscience 2012, 210, 340–352. [Google Scholar] [CrossRef] [PubMed]
- Richetti, S.K.; Blank, M.; Capiotti, K.M.; Piato, A.L.; Bogo, M.R.; Vianna, M.R.; Bonan, C.D. Quercetin and rutin prevent scopolamine-induced memory impairment in zebrafish. Behav. Brain Res. 2011, 217, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Lucas, S. The Pharmacology of Indomethacin. Headache 2016, 56, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Munjal, A.; Wadhwa, R. Sulindac; [Updated 2020 November 27] in StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, January 2021. Available online: https://www.ncbi.nlm.nih.gov/books/NBK556107/ (accessed on 1 April 2021).
- Keskin, D.; Sadri, S.; Eskazan, A.E. Dasatinib for the treatment of chronic myeloid leukemia: Patient selection and special considerations. Drug Des. Dev. Ther. 2016, 10, 3355–3361. [Google Scholar] [CrossRef] [Green Version]
- Shefrin, A.E.; Goldman, R.D. Use of dexamethasone and prednisone in acute asthma exacerbations in pediatric patients. Can. Fam. Physician 2009, 55, 704–706. [Google Scholar] [PubMed]
- Nakano, S.; Kobayashi, N.; Yoshida, K.; Ohno, T.; Matsuoka, H. Cardioprotective mechanisms of spironolactone associated with the angiotensin-converting enzyme/epidermal growth factor receptor/extracellular signal-regulated kinases, nad(p)h oxidase/lectin-like oxidized low-density lipoprotein receptor-1, and rho-kinase pathways in aldosterone/salt-induced hypertensive rats. Hypertens. Res. 2005, 28, 925–936. [Google Scholar] [CrossRef] [Green Version]
- National Center for Biotechnology Information. “PubChem Compound Summary for CID 4764, Phenolphthalein” PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Phenolphthalein (accessed on 1 April 2021).
- Díaz-Castro, F.; Monsalves-Álvarez, M.; Rojo, L.E.; del Campo, A.; Troncoso, R. Mifepristone for treatment of metabolic syndrome: Beyond cushing’s syndrome. Front. Pharmacol. 2020, 11, 429. [Google Scholar] [CrossRef]
- Silvestre, L.; Dubois, C.; Renault, M.; Rezvani, Y. Voluntary interruption of pregnancy with mifepristone (ru 486) and a prostaglandin analogue. N. Eng. J. Med. 1990, 322, 645–648. [Google Scholar] [CrossRef]
- Al-Quliti, K.W. Update on neuropathic pain treatment for trigeminal neuralgia. The pharmacological and surgical options. Neurosciences 2015, 20, 107–114. [Google Scholar] [CrossRef] [Green Version]
- National Center for Biotechnology Information. “PubChem Compound Summary for CID 42611257, Vemurafenib” PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Vemurafenib (accessed on 1 April 2021).
- Sosman, J.A.; Kim, K.B.; Schuchter, L.; Gonzalez, R.; Pavlick, A.C.; Weber, J.S.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N. Engl. J. Med. 2012, 366, 707–714. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Cao, Y.; Chen, C.; Zhang, X.; McNabola, A.; Wilkie, D.; Wilhelm, S.; Lynch, M.; Carter, C. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res. 2006, 66, 11851–11858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Center for Biotechnology Information. “PubChem Compound Summary for CID 5035, Raloxifene” PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Raloxifene (accessed on 1 April 2021).
- Cang, Z.; Mu, L.; Wu, K.; Opron, K.; Xia, K.; Wei, G.W. A topological approach for protein classification. Mol. Based Math. Biol. 2015, 3, 140–162. [Google Scholar] [CrossRef]
- Dey, T.K.; Mandal, S. Protein classification with improved topological data analysis. DROPS 2018, 113, 6:1–6:13. [Google Scholar] [CrossRef]
- Holm, L. Dali and the persistence of protein shape. Protein Sci. 2020, 29, 128–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baby, K.; Maity, S.; Mehta, C.H.; Suresh, A.; Nayak, U.Y.; Nayak, Y. Targeting SARS-CoV-2 RNA- dependent RNA polymerase: An in silico drug repurposing for COVID-19. F1000Res 2020, 9, 1166. [Google Scholar] [CrossRef]
- Acharya, A.; Agarwal, R.; Baker, M.B.; Baudry, J.; Bhowmik, D.; Boehm, S.; Byler, K.G.; Chen, S.Y.; Coates, L.; Cooper, C.J.; et al. Supercomputer-Based Ensemble Docking Drug Discovery Pipeline with Application to Covid-19. J. Chem. Inf. Model. 2020, 60, 5832–5852. [Google Scholar] [CrossRef] [PubMed]
- Marak, B.N.; Dowarah, J.; Khiangte, L.; Singh, V.P. Step toward repurposing drug discovery for COVID- 19 therapeutics through in silico approach. Drug Dev. Res. 2020. [Google Scholar] [CrossRef]
- Trezza, A.; Iovinelli, D.; Santucci, A.; Prischi, F.; Spiga, O. An integrated drug repurposing strategy for the rapid identification of potential SARS-CoV-2 viral inhibitors. Sci. Rep. 2020, 10, 13866. [Google Scholar] [CrossRef]
- Jia, Z.; Song, X.; Shi, J.; Wang, W.; He, K. Transcriptome-based drug repositioning for coronavirus disease 2019 (COVID-19). Pathog. Dis. 2020, 78, ftaa036. [Google Scholar] [CrossRef]
- Kumar, Y.; Singh, H.; Patel, C.N. In silico prediction of potential inhibitors for the main protease of SARS-CoV-2 using molecular docking and dynamics simulation based drug-repurposing. J. Infect. Public Health 2020, 13, 1210–1223. [Google Scholar] [CrossRef]
- Elmezayen, A.D.; Al-Obaidi, A.; Sahin, A.T.; Yelekci, K. Drug repurposing for coronavirus (COVID-19): In silico screening of known drugs against coronavirus 3CL hydrolase and protease enzymes. J. Biomol. Struct. Dyn. 2020, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kneller, D.W.; Phillips, G.; O’Neill, H.M.; Jedrzejczak, R.; Stols, L.; Langan, P.; Joachimiak, A.; Coates, L.; Kovalevsky, A. Structural plasticity of SARS-CoV-2 3CL Mpro active site cavity revealed by room temperature X-ray crystallography. Nat. Commun. 2020, 11, 3202. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Yan, L.; Huang, Y.; Liu, F.; Zhao, Y.; Cao, L.; Wang, T.; Sun, Q.; Ming, Z.; Zhang, L.; et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science 2020, 368, 779–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhardwaj, K.; Liu, P.; Leibowitz, J.L.; Kao, C.C. The coronavirus endoribonuclease Nsp15 interacts with retinoblastoma tumor suppressor protein. J. Virol. 2020, 86, 4294–4304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, X.; Hackbart, M.; Mettelman, R.C.; O’Brien, A.; Mielech, A.M.; Yi, G.; Kao, C.C.; Baker, S.C. Coronavirus nonstructural protein 15 mediates evasion of dsRNA sensors and limits apoptosis in macrophages. Proc. Natl. Acad. Sci. USA 2017, 114, E4251–E4260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niclosamide in COVID-19; Identifier: Nct04542434. 29 February 2000. Available online: https://clinicaltrials.gov/ct2/show/NCT04542434 (accessed on 1 April 2021).
- Jeon, S.; Ko, M.; Lee, J.; Choi, I.; Byun, S.Y.; Park, S.; Shum, D.; Kim, S. Identification of Antiviral Drug Candidates against SARS-CoV-2 from FDA-Approved Drugs. Antimicrob. Agents Chemother. 2020, 64, e00819-20. [Google Scholar] [CrossRef]
- Wu, C.J.; Jan, J.T.; Chen, C.M.; Hsieh, H.P.; Hwang, D.R.; Liu, H.W.; Liu, C.Y.; Huang, H.W.; Chen, S.C.; Hong, C.F.; et al. Inhibition of severe acute respiratory syndrome coronavirus replication by niclosamide. Antimicrob. Agents Chemother. 2004, 48, 2693–2696. [Google Scholar] [CrossRef] [Green Version]
- Gassen, N.C.; Niemeyer, D.; Muth, D.; Corman, V.M.; Martinelli, S.; Gassen, A.; Hafner, K.; Papies, J.; Mösbauer, K.; Zellner, A.; et al. Skp2 attenuates autophagy through beclin1- ubiquitination and its inhibition reduces mers-coronavirus infection. Nat. Commun. 2019, 10, 5770. [Google Scholar] [CrossRef]
- Wang, X.; Guan, Y. COVID-19 drug repurposing: A review of computational screening methods, clinical trials, and protein interaction assays. Med. Res. Rev. 2021, 41, 5–28. [Google Scholar] [CrossRef]
- Riva, L.; Yuan, S.; Yin, X.; Martin-Sancho, L.; Matsunaga, N.; Pache, L.; Burgstaller-Muehlbacher, S.; de Jesus, P.D.; Teriete, P.; Hull, M.V. Discovery of SARS-CoV-2 antiviral drugs through large-scale compound repurposing. Nature 2020, 586, 113–119. [Google Scholar] [CrossRef]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the Treatment of Covid-19—Final Report. N. Engl. J. Med. 2020, 383, 1813–1826. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, D.; Du, G.; Du, R.; Zhao, J.; Jin, Y.; Fu, S.; Gao, L.; Cheng, Z.; Lu, Q.; et al. Remdesivir in adults with severe COVID-19: A randomised, double-blind, placebo-controlled, multicentre trial. Lancet 2020, 395, 1569–1578. [Google Scholar] [CrossRef]
- Cao, B.; Wang, Y.; Wen, D.; Liang, W.; Ou, C.; He, J.; Liu, L.; Shan, H.; Lei, C.; David, S.C.; et al. A Trial of Lopinavir-Ritonavir in Adults Hospitalized with Severe Covid-19. N. Engl. J. Med. 2020, 382, 1787–1799. [Google Scholar] [CrossRef]
- White, K.M.; Rosales, R.; Yildiz, S.; Kehrer, T.; Miorin, L.; Moreno, E.; Jangra, S.; Uccellini, M.B.; Rathnasinghe, R.; Coughlan, L.; et al. Plitidepsin has potent preclinical efficacy against SARS-CoV-2 by targeting the host protein eEF1A. Science 2021, 371, 926–931. [Google Scholar] [CrossRef]
- Sharma, S.; Ali, A.; Ali, J.; Sahni, J.K.; Baboota, S. Rutin: Therapeutic potential and recent advances in drug delivery. Expert Opin. Investig. Drugs 2013, 22, 1063–1079. [Google Scholar] [CrossRef]
- Perng, Y.C.; Lenschow, D.J. ISG15 in antiviral immunity and beyond. Nat. Rev. Microbiol. 2018, 16, 423–439. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Brown, H.M.; Hwang, S. Direct Antiviral Mechanisms of Interferon-Gamma. Immune Netw. 2018, 18, e33. [Google Scholar] [CrossRef]
- Hu, X.; Cai, X.; Song, X.; Li, C.; Zhao, J.; Luo, W.; Zhang, Q.; Ekumi, I.O.; He, Z. Possible SARS-coronavirus 2 inhibitor revealed by simulated molecular docking to viral main protease and host toll-like receptor. Future Virol. 2020, 15, 359–368. [Google Scholar] [CrossRef]
- RECOVERY Collaborative Group; Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; et al. Dexamethasone in hospitalized patients with covid-19—Preliminary report. N. Engl. J. Med. 2020, NEJMoa2021436. [Google Scholar] [CrossRef]
- Sarkar, I.; Sen, A. In silico screening predicts common cold drug Dextromethorphan along with Prednisolone and Dexamethasone can be effective against novel Coronavirus disease (COVID-19). J. Biomol. Struct. Dyn. 2020, 1–5. [Google Scholar] [CrossRef]
- Matsuyama, S.; Kawase, M.; Nao, N.; Shirato, K.; Ujike, M.; Kamitani, W.; Shimojima, M.; Fukushi, S. The inhaled steroid ciclesonide blocks sars-cov-2 rna replication by targeting the viral replication- transcription complex in cultured cells. J. Virol. 2020, 95, e01648-20. [Google Scholar] [CrossRef] [PubMed]
- Ribaudo, G.; Ongaro, A.; Oselladore, E.; Zagotto, G.; Memo, M.; Gianoncelli, A. A computational approach to drug repurposing against SARS-CoV-2 RNA dependent RNA polymerase (RdRp). J. Biomol. Struct. Dyn. 2020, 1–8. [Google Scholar] [CrossRef]
- Parvez, M.S.A.; Karim, M.A.; Hasan, M.; Jaman, J.; Karim, Z.; Tahsin, T.; Hasan, N.; Hosen, M.J. Prediction of potential inhibitors for RNA-dependent RNA polymerase of SARS-CoV-2 using comprehensive drug repurposing and molecular docking approach. Int. J. Biol. Macromol. 2020, 163, 1787–1797. [Google Scholar] [CrossRef]
- Ahmad, J.; Ikram, S.; Ahmad, F.; Rehman, I.U.; Mushtaq, M. SARS-CoV-2 RNA Dependent RNA polymerase (RdRp)—A drug repurposing study. Heliyon 2020, 6, e04502. [Google Scholar] [CrossRef] [PubMed]
- Pokhrel, R.; Chapagain, P.; Siltberg-Liberles, J. Potential RNA-dependent RNA polymerase inhibitors as prospective therapeutics against SARS-CoV-2. J. Med. Microbiol. 2020, 69, 864–873. [Google Scholar] [CrossRef] [PubMed]
- Pleschka, S.; Wolff, T.; Ehrhardt, C.; Hobom, G.; Planz, O.; Rapp, U.R.; Ludwig, S. Influenza virus propagation is impaired by inhibition of the Raf/MEK/ERK signalling cascade. Nat. Cell Biol. 2001, 3, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Adnane, L.; Trail, P.A.; Taylor, I.; Wilhelm, S.M. Sorafenib (BAY 43-9006, Nexavar), a dual-action inhibitor that targets RAF/MEK/ERK pathway in tumor cells and tyrosine kinases VEGFR/PDGFR in tumor vasculature. Methods Enzymol. 2006, 407, 597–612. [Google Scholar] [CrossRef]
- Pleschka, S. RNA viruses and the mitogenic Raf/MEK/ERK signal transduction cascade. Biol. Chem. 2008, 389, 1273–1282. [Google Scholar] [CrossRef]
- Cai, Y.; Liu, Y.; Zhang, X. Suppression of coronavirus replication by inhibition of the MEK signaling pathway. J. Virol. 2007, 81, 446–456. [Google Scholar] [CrossRef] [Green Version]
- Ghasemnejad-Berenji, M.; Pashapour, S. SARS-CoV-2 and the Possible Role of Raf/MEK/ERK Pathway in Viral Survival: Is This a Potential Therapeutic Strategy for COVID-19? Pharmacology 2021, 106, 119–122. [Google Scholar] [CrossRef]
- Wishart, D.S. Drugbank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.; Ezzat, A. DrugBank Database XML Parser. Dainanahan, R Package Version 1.2.0. 2020. Available online: https://CRAN.R-project.org/package=dbparser (accessed on 21 March 2020).
- Munkres, J. Elements of Algebraic Topology; Perseus Publishing: Cambridge, MA, USA, 1984. [Google Scholar]
- Robins, V. Towards computing homology from finite approximations. Topol. Proc. 1999, 24, 503–532. [Google Scholar]
- Pérez-Moraga, R.; Forés-Martors, J.; Suay-García, B.; Duval, J.L.; Falcó, A.; Climent, J. A COVID-19 Drug Repurposing Strategy Through Quantitative Homological Similarities by using a Topological Data Analysis Based Formalism. Preprints 2020, 2020120281. [Google Scholar] [CrossRef]
- Grant, B.J.; Rodrigues, A.P.C.; ElSawy, K.M.; McCammon, J.A.; Caves, L.S.D. Bio3d: An R package for the comparative analysis of protein structures. Bioinformatics 2006, 22, 2695–2696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wadhwa, R.R.; Williamson, D.F.; Dhawan, A.; Scott, J.G. Tdastats: R pipeline for computing persistent homology in topological data analysis. J. Open Source Softw. 2018, 3, 860. [Google Scholar] [CrossRef] [PubMed]
- Bauer, U. Ripser: Efficient computation of Vietoris-Rips persistence barcodes. arXiv 2019, arXiv:1908.02518. [Google Scholar]
- Jain, B.J.; Lappe, M. Joining softassign and dynamic programming for the contact map overlap problem. BIRD 2007, 4414, 410–423. [Google Scholar] [CrossRef]
- Lancia, G.; Carr, R.; Walenz, B.; Istrail, S. 101 optimal pdb structure alignments: A branch-and-cut algorithm for the maximum contact map overlap problem. In Proceedings of the Fifth Annual International Conference on Computational Biology, Montreal, QC, Canada, 22–25 April 2001; pp. 193–202. [Google Scholar] [CrossRef]
- Fox, N.K.; Brenner, S.E.; Chandonia, J.M. SCOPe: Structural Classification of Proteins–extended, integrating SCOP and ASTRAL data and classification of new structures. Nucleic Acids Res. 2014, 42, D304–D309. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Forli, S.; Huey, R.; Pique, M.; Sanner, M.; Goodsell, D.; Olson, A. Computational protein-ligand docking and virtual drug screening with the autodock suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef] [Green Version]
- Desai, N.; Neyaz, A.; Szabolcs, A.; Shih, A.R.; Chen, J.H.; Thapar, V.; Nieman, L.T.; Solovyov, A.; Mehta, A.; Lieb, D.J.; et al. Temporal and spatial heterogeneity of host response to sars-cov-2 pulmonary infection. Nat. Commun. 2020, 11, 6319. [Google Scholar] [CrossRef]
- Xiong, Y.; Liu, Y.; Cao, L.; Wang, D.; Guo, M.; Jiang, A.; Guo, D.; Hu, W.; Yang, J.; Tang, Z.; et al. Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in covid-19 patients. Emerg. Microbes Infect. 2020, 9, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. The R package rsubread is easier, faster, cheaper and better for alignment and quantification of rna sequencing reads. Nucleic Acids Res. 2019, 47, e47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.C.; Møller, R.; Panis, M.; Sachs, D.; Albrecht, R.A.; tenOever, B.R.; Uhl, S.; Hoagland, D.; et al. Imbalanced host response to sars-cov-2 drives development of covid-19. Cell 2020, 181, 1036–1045. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K.; et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 2017, 171, 1437–1452. [Google Scholar] [CrossRef]
- Korotkevich, G.; Sukhov, V.; Sergushichev, A. Fast gene set enrichment analysis. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. ClusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry ID | Structure Title | Macromolecule Name | Chain ID |
|---|---|---|---|
| 6LVN | 2019-nCoV HR2 Domain | Spike protein S2 | A, B, C, D |
| 6YI3 | The N-terminal RNA-binding domain of the SARS-CoV-2 nucleocapsid phosphoprotein | Nucleoprotein | A |
| 6M3M | SARS-CoV-2 nucleocapsid protein N-terminal RNA binding domain | SARS-CoV-2 nucleocapsid protein | A, B, C, D |
| 6VYO | RNA binding domain of nucleocapsid phosphoprotein from SARS coronavirus 2 | Nucleoprotein | A, B, C, D |
| 6WJI | C-terminal Dimerization Domain of Nucleocapsid Phosphoprotein from SARS-CoV-2 | SARS-CoV-2 nucleocapsid protein | A, B, C, D, E, F |
| 6LXT | Structure of post fusion core of 2019-nCoV S2 subunit | Spike protein S2 | A, B, C, D, E, F |
| 6VSB | Prefusion 2019-nCoV spike glycoprotein with a single receptor-binding domain up | SARS-CoV-2 spike glycoprotein | A, B, C |
| 6VYB | SARS-CoV-2 spike ectodomain structure (open state) | Spike glycoprotein | A, B, C |
| 6W41 | Crystal structure of SARS-CoV-2 receptor binding domain in complex with human antibody CR3022 | CR3022 Fab heavy chain | H |
| CR3022 Fab light chain | L | ||
| Spike protein S1 | C | ||
| 6YLA | Crystal structure of the SARS-CoV-2 receptor binding domain in complex with CR3022 Fab | Spike glycoprotein | A, E |
| Heavy Chain | B, H | ||
| Light chain | C, L | ||
| 6M0J | Crystal structure of SARS-CoV-2 spike receptor-binding domain bound with ACE2 | Angiotensin converting enzyme 2 | A |
| Spike receptor binding domain | E | ||
| 6M17 | 2019-nCoV RBD/ACE2-B0AT1 complex | Sodium-dependent neutral amino acid transporter B(0)AT1 | A, C |
| Angiotensin converting enzyme 2 | B, D | ||
| SARS-coV-2 Receptor Binding Domain | E, F | ||
| 6M2Q | SARS-CoV-2 3CL protease (3CL pro) apo structure (space group C21) | SARS-CoV-2 3CL protease | A |
| 6W4B | Crystal structure of Nsp9 RNA binding protein of SARS CoV-2 | Non-structural protein 9 | A, B |
| 6W9Q | Peptide-bound SARS-CoV-2 Nsp9 RNA replicase | 3C-like proteinase peptide, Nonstructural protein 9 fusion | A |
| 6VXS | Crystal Structure of ADP ribose phosphatase of NSP3 from SARS CoV-2 | Non-structural protein 3 | A, B |
| 6W9C | Crystal structure of papain-like protease of SARS CoV-2 | Papain-like proteinase | A, B, C |
| 6WCF | Crystal Structure of ADP ribose phosphatase of NSP3 from SARS-CoV-2 in complex with MES | Non-structural protein 3 | A |
| 6WEN | Crystal Structure of ADP ribose phosphatase of NSP3 from SARS-CoV-2 in the apo form | Non-structural protein 3 | A |
| 6WIQ | Crystal structure of the co-factor complex of NSP7 and the C-terminal domain of NSP8 from SARS CoV-2 | SARS-CoV-2 NSP7 | A |
| SARS-CoV-2 NSP8 | B | ||
| 6M71 | SARS-Cov-2 RNA-dependent RNA polymerase in complex with cofactors | SARS-Cov-2 NSP 12 | A |
| SARS-Cov-2 NSP 8 | C | ||
| SARS-Cov-2 NSP 7 | B, D | ||
| 6W01 | 1.9 A Crystal Structure of NSP15 Endoribonuclease from SARS CoV-2 in the Complex with a Citrate | Uridylate-specific endoribonuclease | A, B |
| 6VWW | Crystal Structure of NSP15 Endoribonuclease from SARS CoV-2 | Uridylate-specific endoribonuclease | A, B |
| 6M2Q (SARS-CoV-2 3CL Protease) | ||||||
|---|---|---|---|---|---|---|
| Drug Name | Drug ID | PC DS1 (GSE150316) | PC DS2 (CRA002390) | PC DS3 (GSE147507) | AutoDock LE (kcal/mol) | AutoDock Cluster |
| CholicAcid | DB02659 | −0.09 | −0.11 | −0.08 | −15.06 | 74 |
| Rutin | DB01698 | −0.07 | −0.18 | −0.1 | −14.52 | 149 |
| Indomethacin | DB00328 | −0.07 | −0.12 | −0.05 | −13.31 | 146 |
| Sulindac | DB00605 | −0.07 | −0.12 | −0.07 | −13.14 | 73 |
| Sulfisoxazole | DB00263 | −0.05 | −0.13 | −0.09 | −11.59 | 77 |
| Dasatinib | DB01254 | −0.04 | −0.15 | −0.09 | −10.94 | 43 |
| 6W01 (NSP15 Endoribonuclease) | ||||||
| Dexamethasone | DB01234 | −0.07 | −0.15 | −0.08 | −11.42 | 49 |
| Phenolphthalein | DB04824 | −0.13 | −0.1 | −0.04 | −11.15 | 101 |
| Spironolactone | DB00421 | −0.12 | −0.1 | −0.09 | −10.99 | 110 |
| Mifepristone | DB00834 | −0.13 | −0.14 | −0.06 | −10.04 | 28 |
| Carbamazepine | DB00564 | −0.08 | −0.14 | −0.07 | −9.66 | 86 |
| 6M71 (NSP12 RNA-dependent RNA polymerase) | ||||||
| Vemurafenib | DB08881 | −0.09 | −0.16 | −0.08 | −8.09 | 13 |
| Sorafenib | DB00398 | −0.11 | −0.15 | −0.05 | −7.34 | 30 |
| Levonorgestrel | DB00367 | −0.08 | −0.14 | −0.08 | −7.21 | 89 |
| Naloxone | DB01183 | −0.06 | −0.12 | −0.09 | −7.07 | 69 |
| Raloxifene | DB00481 | −0.13 | −0.17 | −0.07 | −7.05 | 6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pérez-Moraga, R.; Forés-Martos, J.; Suay-García, B.; Duval, J.-L.; Falcó, A.; Climent, J. A COVID-19 Drug Repurposing Strategy through Quantitative Homological Similarities Using a Topological Data Analysis-Based Framework. Pharmaceutics 2021, 13, 488. https://doi.org/10.3390/pharmaceutics13040488
Pérez-Moraga R, Forés-Martos J, Suay-García B, Duval J-L, Falcó A, Climent J. A COVID-19 Drug Repurposing Strategy through Quantitative Homological Similarities Using a Topological Data Analysis-Based Framework. Pharmaceutics. 2021; 13(4):488. https://doi.org/10.3390/pharmaceutics13040488
Chicago/Turabian StylePérez-Moraga, Raul, Jaume Forés-Martos, Beatriz Suay-García, Jean-Louis Duval, Antonio Falcó, and Joan Climent. 2021. "A COVID-19 Drug Repurposing Strategy through Quantitative Homological Similarities Using a Topological Data Analysis-Based Framework" Pharmaceutics 13, no. 4: 488. https://doi.org/10.3390/pharmaceutics13040488
APA StylePérez-Moraga, R., Forés-Martos, J., Suay-García, B., Duval, J.-L., Falcó, A., & Climent, J. (2021). A COVID-19 Drug Repurposing Strategy through Quantitative Homological Similarities Using a Topological Data Analysis-Based Framework. Pharmaceutics, 13(4), 488. https://doi.org/10.3390/pharmaceutics13040488