
Importance of Nanoparticles for the Delivery of Antiparkinsonian Drugs



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction of Parkinson’s Disease
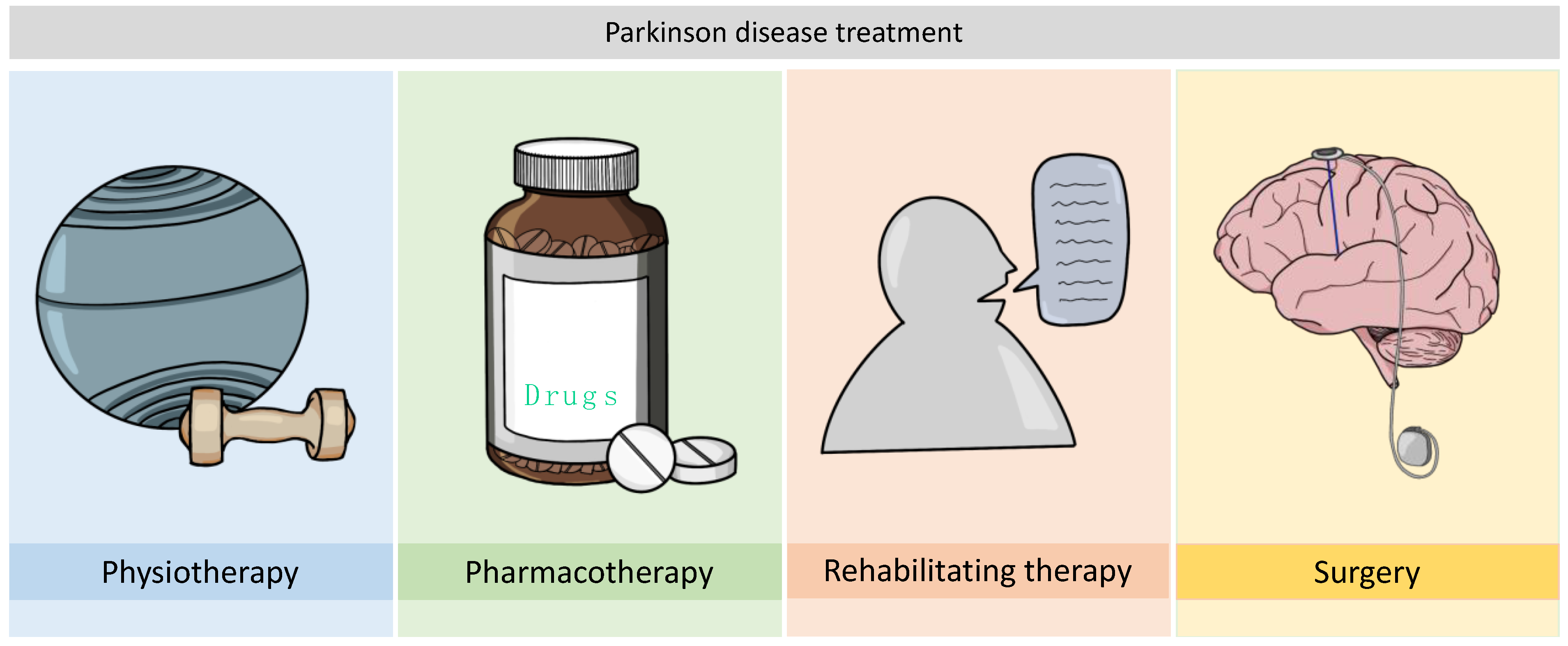
2. Considerations about PD Treatment
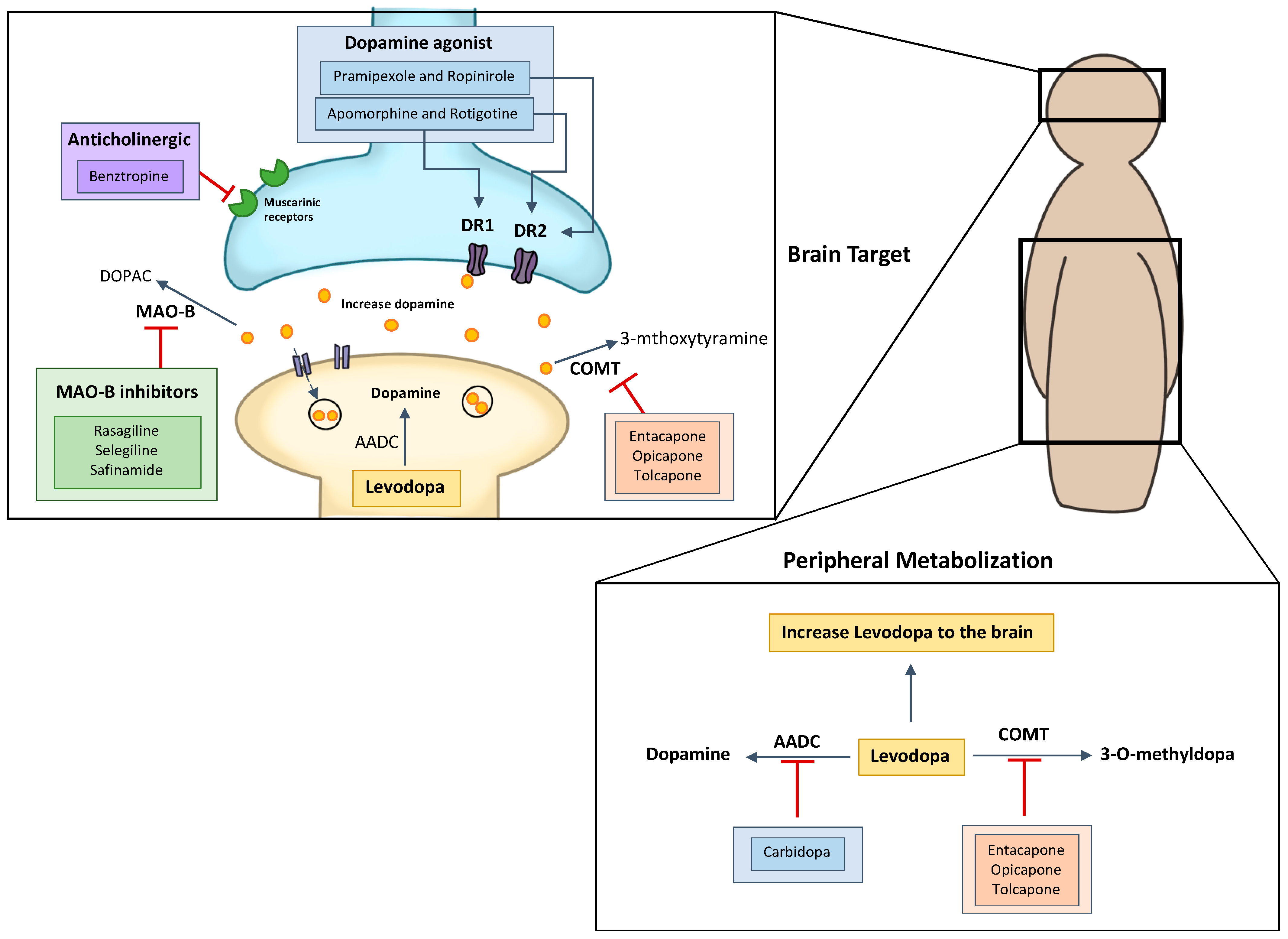
2.1. Levodopa and Carbidopa
2.2. Dopamine Agonists
2.2.1. Apomorphine
2.2.2. Pramipexole
2.2.3. Ropinirole
2.2.4. Rotigotine
2.3. MAO-B Inhibitors
2.4. Catechol O-Methyltransferase Inhibitors
2.5. Anticholinergic
2.5.1. Trihexyphenidyl
2.5.2. Benztropine
2.6. Amantadine
2.7. Neurosurgical Treatments
3. Nanoparticles as Drug Delivery System Repurposing in PD
3.1. Parenteral/Intravenous Delivery
3.2. Transdermal Delivery
3.3. Intranasal Delivery
3.4. Oral Delivery
3.5. Buccal Delivery
4. Nanotheranostics in PD
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Erkkinen, M.G.; Kim, M.-O.; Geschwind, M.D. Clinical Neurology and Epidemiology of the Major Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2018, 10, a033118. [Google Scholar] [CrossRef] [Green Version]
- Beitz, J.M. Parkinson s disease a review. Front. Biosci. 2014, S6, S415. [Google Scholar] [CrossRef]
- Hayes, M.T. Parkinson’s Disease and Parkinsonism. Am. J. Med. 2019, 132, 802–807. [Google Scholar] [CrossRef]
- Dorsey, E.R.; Elbaz, A.; Nichols, E.; Abd-Allah, F.; Abdelalim, A.; Adsuar, J.C.; Ansha, M.G.; Brayne, C.; Choi, J.-Y.J.; Collado-Mateo, D.; et al. Global, regional, and national burden of Parkinson’s disease, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 939–953. [Google Scholar] [CrossRef] [Green Version]
- Samii, A.; Nutt, J.G.; Ransom, B.R. Parkinson’s disease. Lancet 2004, 363, 1782–1793. [Google Scholar] [CrossRef] [Green Version]
- Sherer, T.B.; Chowdhury, S.; Peabody, K.; Brooks, D.W. Overcoming obstacles in Parkinson’s disease. Mov. Disord. 2012, 27, 1606–1611. [Google Scholar] [CrossRef] [PubMed]
- Tolosa, E.; Wenning, G.; Poewe, W. The diagnosis of Parkinson’s disease. Lancet Neurol. 2006, 5, 75–86. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Okun, M.S. Diagnosis and Treatment of Parkinson Disease: A Review. JAMA-J. Am. Med. Assoc. 2020, 323, 548–560. [Google Scholar] [CrossRef]
- Cavaliere, F.; Cerf, L.; Dehay, B.; Ramos-Gonzalez, P.; De Giorgi, F.; Bourdenx, M.; Bessede, A.; Obeso, J.A.; Matute, C.; Ichas, F.; et al. In vitro α-synuclein neurotoxicity and spreading among neurons and astrocytes using Lewy body extracts from Parkinson disease brains. Neurobiol. Dis. 2017, 103, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Antony, P.M.A.; Diederich, N.J.; Krüger, R.; Balling, R. The hallmarks of Parkinson’s disease. FEBS J. 2013, 280, 5981–5993. [Google Scholar] [CrossRef] [Green Version]
- Ceccatelli, S. Mechanisms of neurotoxicity and implications for neurological disorders. J. Intern. Med. 2013, 273, 426–428. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.-E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Prim. 2017, 3, 17013. [Google Scholar] [CrossRef]
- Kam, T.-I.; Hinkle, J.T.; Dawson, T.M.; Dawson, V.L. Microglia and astrocyte dysfunction in parkinson’s disease. Neurobiol. Dis. 2020, 144, 105028. [Google Scholar] [CrossRef]
- Dehay, B.; Martinez-Vicente, M.; Ramirez, A.; Perier, C.; Klein, C.; Vila, M.; Bezard, E. Lysosomal dysfunction in Parkinson disease. Autophagy 2012, 8, 1389–1391. [Google Scholar] [CrossRef] [PubMed]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Ascherio, A.; Schwarzschild, M.A. The epidemiology of Parkinson’s disease: Risk factors and prevention. Lancet Neurol. 2016, 15, 1257–1272. [Google Scholar] [CrossRef]
- Deng, H.; Wang, P.; Jankovic, J. The genetics of Parkinson disease. Ageing Res. Rev. 2018, 42, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Rui, Q.; Ni, H.; Li, D.; Gao, R.; Chen, G. The Role of LRRK2 in Neurodegeneration of Parkinson Disease. Curr. Neuropharmacol. 2018, 16, 1348–1357. [Google Scholar] [CrossRef] [PubMed]
- Reich, S.G.; Savitt, J.M. Parkinson’s Disease. Med. Clin. North Am. 2019, 103, 337–350. [Google Scholar] [CrossRef]
- Gazewood, J.D.; Richards, D.R.; Clebak, K. Parkinson disease: An update. Am. Fam. Physician 2013, 87, 267–273. [Google Scholar] [PubMed]
- Markham, C.H.; Diamond, S.G.; Treciokas, L.J. Carbidopa in Parkinson Disease and in Nausea and Vomiting of Levodopa. Arch. Neurol. 1974, 31, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Haddad, F.; Sawalha, M.; Khawaja, Y.; Najjar, A.; Karaman, R. Dopamine and levodopa prodrugs for the treatment of Parkinson’s disease. Molecules 2018, 23, 40. [Google Scholar] [CrossRef] [Green Version]
- Whitfield, A.C.; Moore, B.T.; Daniels, R.N. Classics in chemical neuroscience: Levodopa. ACS Chem. Neurosci. 2014, 5, 1192–1197. [Google Scholar] [CrossRef]
- Fahn, S. Levodopa in the treatment of Parkinson’s disease. J. Neural Transm. Suppl. 2006, 1–15. [Google Scholar] [CrossRef]
- Jenner, P.; Katzenschlager, R. Apomorphine-pharmacological properties and clinical trials in Parkinson’s disease. Park. Relat. Disord. 2016, 33, S13–S21. [Google Scholar] [CrossRef]
- Auffret, M.; Drapier, S.; Vérin, M. Pharmacological Insights into the Use of Apomorphine in Parkinson’s Disease: Clinical Relevance. Clin. Drug Investig. 2018, 38, 287–312. [Google Scholar] [CrossRef] [PubMed]
- Subramony, J.A. Apomorphine in dopaminergic therapy. Mol. Pharm. 2006, 3, 380–385. [Google Scholar] [CrossRef]
- Antonini, A.; Barone, P.; Ceravolo, R.; Fabbrini, G.; Tinazzi, M.; Abbruzzese, G. Role of pramipexole in the management of Parkinsons disease. CNS Drugs 2010, 24, 829–841. [Google Scholar] [CrossRef]
- Hametner, E.M.; Seppi, K.; Poewe, W. Pramipexole extended release in Parkinson’s disease. Expert Rev. Neurother. 2011, 11, 1229–1234. [Google Scholar] [CrossRef]
- Lloret, S.P.; Rey, M.V.; Ratti, L.; Rascol, O. Pramipexole for the treatment of early Parkinsons disease. Expert Rev. Neurother. 2011, 11, 925–935. [Google Scholar] [CrossRef]
- Frampton, J.E. Rotigotine Transdermal Patch: A Review in Parkinson’s Disease. CNS Drugs 2019, 33, 707–718. [Google Scholar] [CrossRef]
- Pahwa, R.; Lyons, K.E.; Hauser, R.A. Ropinirole therapy for Parkinson’s disease. Expert Rev. Neurother. 2004, 4, 581–588. [Google Scholar] [CrossRef]
- Reynolds, N.A.; Wellington, K.; Easthope, S.E. Rotigotine. CNS Drugs 2005, 19, 973–981. [Google Scholar] [CrossRef]
- Morgan, J.C.; Sethi, K.D. Rotigotine for the treatment of Parkinson’s disease. Expert Rev. Neurother. 2006, 6, 1275–1282. [Google Scholar] [CrossRef] [PubMed]
- Ebadi, M.; Sharma, S.; Shavali, S.; El Refaey, H. Neuroprotective actions of selegiline. J. Neurosci. Res. 2002, 67, 285–289. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, M.A.A.; Plosker, G.L.M. Rasagiline. Drugs Aging 2005, 22, 83–91. [Google Scholar] [CrossRef]
- Schrag, A. Entacapone in the treatment of Parkinson’s disease. Lancet Neurol. 2005, 4, 366–370. [Google Scholar] [CrossRef]
- Chong, B.S.; Mersfelder, T.L. Entacapone. Ann. Pharmacother. 2000, 34, 1056–1065. [Google Scholar] [CrossRef]
- Keating, G.M.; Lyseng-Williamson, K.A. Tolcapone. CNS Drugs 2005, 19, 165–184. [Google Scholar] [CrossRef] [PubMed]
- Shear, N. TRIHEXYPHENIDYL. In Litt’s Drug Eruptions & Reactions Manual; CRC Press: Boca Raton, FL, USA, 2010; pp. 603–610. [Google Scholar]
- Katzenschlager, R.; Sampaio, C.; Costa, J.; Lees, A. Anticholinergics for symptomatic management of Parkinson´s disease. Cochrane Database Syst. Rev. 2002. [Google Scholar] [CrossRef] [PubMed]
- Rascol, O.; Lozano, A.; Stern, M.; Poewe, W. Milestones in Parkinson’s disease therapeutics. Mov. Disord. 2011, 26, 1072–1082. [Google Scholar] [CrossRef]
- Chang, C.; Ramphul, K. Amantadine; Pfeiffer, R.F., Wszolek, Z.K., Ebadi, M., Eds.; CRC Press: Boca Raton, FL, USA, 2020; ISBN 9780429149344. [Google Scholar]
- Groiss, S.J.; Wojtecki, L.; Südmeyer, M.; Schnitzler, A. Review: Deep brain stimulation in Parkinson’s disease. Ther. Adv. Neurol. Disord. 2009, 2, 379–391. [Google Scholar] [CrossRef] [Green Version]
- Okun, M.S. Deep-Brain Stimulation for Parkinson’s Disease. N. Engl. J. Med. 2012, 367, 1529–1538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olanow, C.W.; Schapira, A.H.V. Therapeutic prospects for Parkinson disease. Ann. Neurol. 2013, 74, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Alonso, M.; Couvreur, P. Chapter 1:Historical View of the Design and Development of Nanocarriers for Overcoming Biological Barriers. In Nanostructured Biomaterials for Overcoming Biological Barriers; The Royal Society of Chemistry Cambridge: Cambridge, UK, 2012. [Google Scholar]
- Singh, R.; Lillard, J.W. Nanoparticle-based targeted drug delivery. Exp. Mol. Pathol. 2009, 86, 215–223. [Google Scholar] [CrossRef] [Green Version]
- Lewinski, N.; Colvin, V.; Drezek, R. Cytotoxicity of nanopartides. Small 2008, 4, 26–49. [Google Scholar] [CrossRef]
- Yohan, D.; Chithrani, B.D. Applications of nanoparticles in nanomedicine. J. Biomed. Nanotechnol. 2014, 10, 2371–2392. [Google Scholar] [CrossRef] [PubMed]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef]
- Gaspar, D.P.; Faria, V.; Almeida, J.P.Q.; Quintas, J.P.; Almeida, A.J. Targeted Delivery of Lipid Nanoparticles by Means of Surface Chemical Modification. Curr. Org. Chem. 2017, 21, 2360–2375. [Google Scholar] [CrossRef]
- Garud, A.; Singh, D.; Garud, N. Solid Lipid Nanoparticles (SLN): Method, Characterization and Applications. Int. Curr. Pharm. J. 2012, 1, 384–393. [Google Scholar] [CrossRef] [Green Version]
- Gaucher, G.; Marchessault, R.H.; Leroux, J.-C. Polyester-based micelles and nanoparticles for the parenteral delivery of taxanes. J. Control. Release 2010, 143, 2–12. [Google Scholar] [CrossRef]
- Hwang, T.-L.; Lin, Y.-K.; Chi, C.-H.; Huang, T.-H.; Fang, J.-Y. Development and Evaluation of Perfluorocarbon Nanobubbles for Apomorphine Delivery. J. Pharm. Sci. 2009, 98, 3735–3747. [Google Scholar] [CrossRef]
- Barcia, E.; Boeva, L.; García-García, L.; Slowing, K.; Fernández-Carballido, A.; Casanova, Y.; Negro, S. Nanotechnology-based drug delivery of ropinirole for Parkinson’s disease. Drug Deliv. 2017, 24, 1112–1123. [Google Scholar] [CrossRef] [Green Version]
- Somasundaram, I. Formulation of Polymeric Nanosuspension Containing Pramipexole Dihydrochloride and Hesperidin for Improved Treatment of Parkinson’s Diseases. Asian J. Pharm. Clin. Res. 2018, 11, 133–140. [Google Scholar] [CrossRef] [Green Version]
- Ray, S.; Sinha, P.; Laha, B.; Maiti, S.; Bhattacharyya, U.K.; Nayak, A.K. Polysorbate 80 coated crosslinked chitosan nanoparticles of ropinirole hydrochloride for brain targeting. J. Drug Deliv. Sci. Technol. 2018, 48, 21–29. [Google Scholar] [CrossRef]
- Bahari Javan, N.; Jafary Omid, N.; Moosavi Hasab, N.; Rezaie Shirmard, L.; Rafiee-Tehrani, M.; Dorkoosh, F. Preparation, statistical optimization and in vitro evaluation of pramipexole prolonged delivery system based on poly (3-hydroxybutyrate-co-3-hydroxyvalerate) nanoparticles. J. Drug Deliv. Sci. Technol. 2018, 44, 82–90. [Google Scholar] [CrossRef]
- Jawanjal, P.M.; Patil, P.B.; Patil, J.; Waghulde, M.; Naik, J.B. Development of Graphene Oxide-Trihexyphenidyl Hydrochloride Nanohybrid and Release behavior. Curr. Environ. Eng. 2019, 6, 134–140. [Google Scholar] [CrossRef]
- Zhou, Y.Z.; Alany, R.G.; Chuang, V.; Wen, J. Optimization of PLGA nanoparticles formulation containing L-DOPA by applying the central composite design. Drug Dev. Ind. Pharm. 2013, 39, 321–330. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, B.H.; Singh, G.; Hak, S.; Bandyopadhyay, S.; Augestad, I.L.; Peddis, D.; Sandvig, I.; Sandvig, A.; Glomm, W.R. L-DOPA-Coated Manganese Oxide Nanoparticles as Dual MRI Contrast Agents and Drug-Delivery Vehicles. Small 2016, 12, 301–306. [Google Scholar] [CrossRef] [Green Version]
- Cao, X.; Hou, D.; Wang, L.; Li, S.; Sun, S.; Ping, Q.; Xu, Y. Effects and molecular mechanism of chitosan-coated levodopa nanoliposomes on behavior of dyskinesia rats. Biol. Res. 2016, 49, 32. [Google Scholar] [CrossRef] [Green Version]
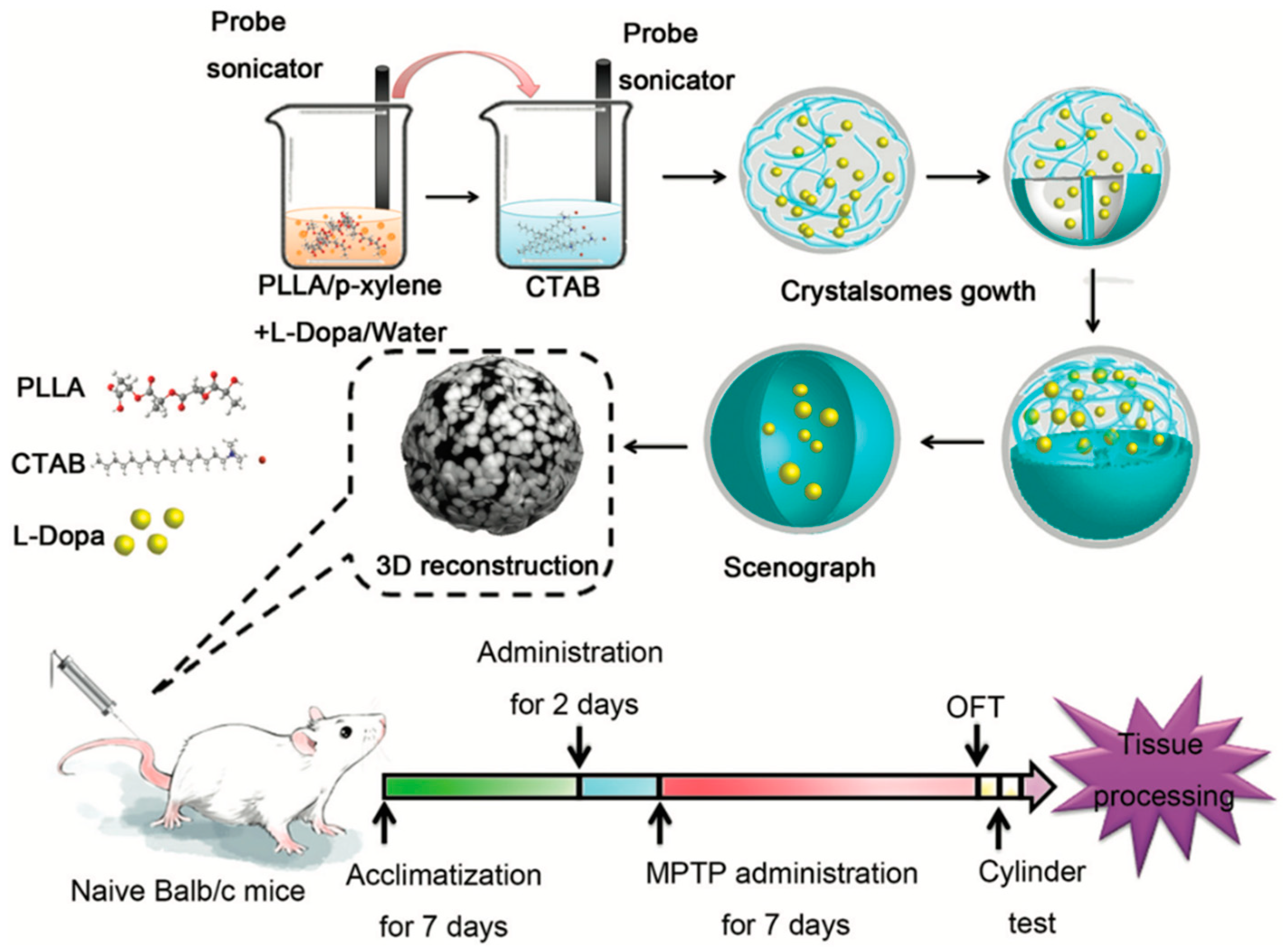
- Wang, W.; Qi, H.; Zhou, T.; Mei, S.; Han, L.; Higuchi, T.; Jinnai, H.; Li, C.Y. Highly robust crystalsome via directed polymer crystallization at curved liquid/liquid interface. Nat. Commun. 2016, 7, 10599. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Liu, Q.; Zhu, D.; Che, Y.; Feng, X. Preparation of levodopa-loaded crystalsomes through thermally induced crystallization reverses functional deficits in Parkinsonian mice. Biomater. Sci. 2019, 7, 1623–1631. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, K.; Kunasekaran, V. Multi criteria decision making to select the best method for the preparation of solid lipid nanoparticles of rasagiline mesylate using analytic hierarchy process. J. Adv. Pharm. Technol. Res. 2014, 5, 115. [Google Scholar] [CrossRef] [PubMed]
- Kunasekaran, V.; Krishnamoorthy, K. Experimental Design for the Optimization of Nanoscale Solid Lipid Particles Containing Rasagiline Mesylate. J. Young Pharm. 2015, 7, 285–295. [Google Scholar] [CrossRef] [Green Version]
- Kunasekaran, V.; Krishnamoorthy, K. Kinetic modeling of Rasagiline mesylate from nanoscale solid lipid particles. Int. J. Pharm. Pharm. Sci. 2015, 7, 300–305. [Google Scholar]
- Kunasekaran, V.; Krishnamoorthy, K. Formulation and Evaluation of Nanoscale Solid Lipid Particles Containing a Hydrophilic Drug-Rasagiline Mesylate. J. Appl. Pharm. Sci. 2016, 6, 044–050. [Google Scholar] [CrossRef] [Green Version]
- Kannan, K.; Viveksarathi, K. Effect of the moist-heat sterilization on fabricated nanoscale solid lipid particles containing rasagiline mesylate. Int. J. Pharm. Investig. 2015, 5, 87. [Google Scholar] [CrossRef] [Green Version]
- Hsu, S.H.; Wen, C.J.; Al-Suwayeh, S.A.; Chang, H.W.; Yen, T.C.; Fang, J.Y. Physicochemical characterization and in vivo bioluminescence imaging of nanostructured lipid carriers for targeting the brain: Apomorphine as a model drug. Nanotechnology 2010, 21. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.-S.; Wen, C.-J.; Yen, T.-C.; Sung, K.C.; Ku, M.-C.; Wang, J.-J.; Fang, J.-Y. Combined strategies of apomorphine diester prodrugs and nanostructured lipid carriers for efficient brain targeting. Nanotechnology 2012, 23, 095103. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.-H.; Al-Suwayeh, S.A.; Chen, C.-C.; Chi, C.-H.; Fang, J.-Y. PEGylated Liposomes Incorporated with Nonionic Surfactants as an Apomorphine Delivery System Targeting the Brain: In Vitro Release and In Vivo Real-time Imaging. Curr. Nanosci. 2011, 7, 191–199. [Google Scholar] [CrossRef]
- Fang, J.-Y.; Wen, C.-J.; Zhang, L.-W.; Al-Suwayeh, S.A.; Yen, T.-C. Theranostic liposomes loaded with quantum dots and apomorphine for brain targeting and bioimaging. Int. J. Nanomed. 2012, 7, 1599. [Google Scholar] [CrossRef] [Green Version]
- Gunay, M.S.; Ozer, A.Y.; Erdogan, S.; Bodard, S.; Baysal, I.; Gulhan, Z.; Guilloteau, D.; Chalon, S. Development of nanosized, pramipexole-encapsulated liposomes and niosomes for the treatment of Parkinson’s disease. J. Nanosci. Nanotechnol. 2017, 17, 5155–5167. [Google Scholar] [CrossRef]
- Shingade, G.M. Review on: Recent Trend on Transdermal Drug Delivery System. J. Drug Deliv. Ther. 2012, 2. [Google Scholar] [CrossRef]
- Fleischhack, G.; Garnett, M.; Beccaria, K. Drug Delivery. In Brain and Spinal Tumors of Childhood; CRC Press: Boca Raton, FL, USA, 2020; Volume 197, pp. 150–168. ISBN 9781420008333. [Google Scholar]
- Ferini-Strambi, L.; Marelli, S.; Galbiati, A. Clinical pharmacology and efficacy of rotigotine (Neupro® patch) in the treatment of restless leg syndrome. Expert Opin. Drug Metab. Toxicol. 2016, 12, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Jessen, L.; Kovalick, L.J.; Azzaro, A.J. The selegiline transdermal system (emsam): A therapeutic option for the treatment of major depressive disorder. P T 2008, 33, 212–246. [Google Scholar]
- Davis, S.S.; Illum, L. Polymeric microspheres as drug carriers. Biomaterials 1988, 9, 111–115. [Google Scholar] [CrossRef]
- Cheng, Y.; Xu, Z.; Ma, M.; Xu, T. Dendrimers as Drug Carriers: Applications in Different Routes of Drug Administration. J. Pharm. Sci. 2008, 97, 123–143. [Google Scholar] [CrossRef] [PubMed]
- Trucillo, P. Drug Carriers: Classification, Administration, Release Profiles, and Industrial Approach. Processes 2021, 9, 470. [Google Scholar] [CrossRef]
- Fang, J.Y.; Hung, C.F.; Chi, C.H.; Chen, C.C. Transdermal permeation of selegiline from hydrogel-membrane drug delivery systems. Int. J. Pharm. 2009, 380, 33–39. [Google Scholar] [CrossRef]
- Azeem, A.; Rizwan, M.; Ahmad, F.J.; Iqbal, Z.; Khar, R.K.; Aqil, M.; Talegaonkar, S. Nanoemulsion components screening and selection: A technical note. AAPS PharmSciTech 2009. [Google Scholar] [CrossRef] [PubMed]
- Azeem, A.; Rizwan, M.; Ahmad, F.; Khar, R.; Iqbal, Z.; Talegaonkar, S. Components Screening and Influence of Surfactant and Cosurfactant on Nanoemulsion Formation. Curr. Nanosci. 2009. [Google Scholar] [CrossRef]
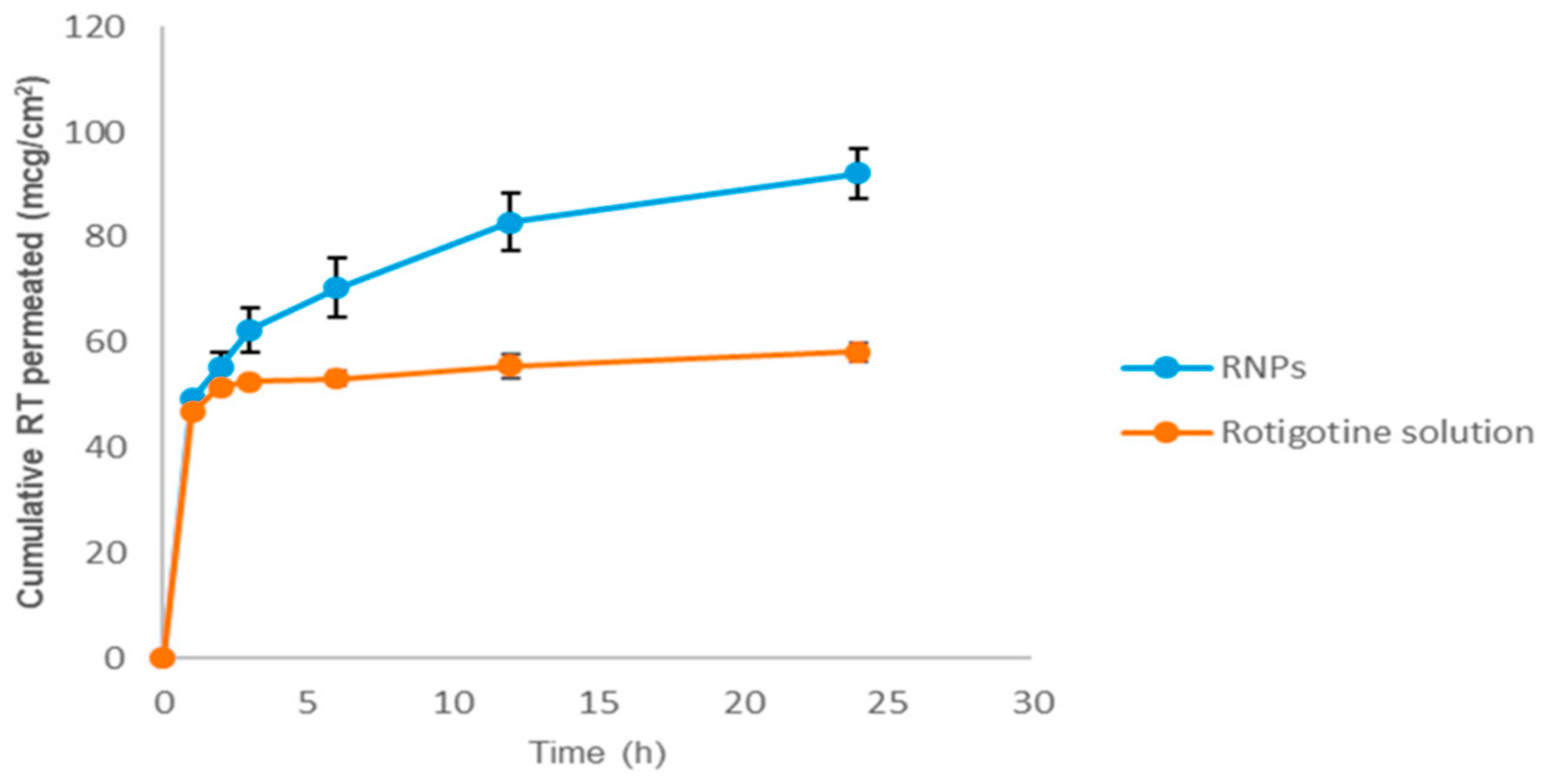
- Azeem, A.; Talegaonkar, S.; Negi, L.M.; Ahmad, F.J.; Khar, R.K.; Iqbal, Z. Oil based nanocarrier system for transdermal delivery of ropinirole: A mechanistic, pharmacokinetic and biochemical investigation. Int. J. Pharm. 2012, 422, 436–444. [Google Scholar] [CrossRef]
- Chen, C.C.; Fang, C.L.; Al-Suwayeh, S.A.; Leu, Y.L.; Fang, J.Y. Transdermal delivery of selegiline from alginate-Pluronic composite thermogels. Int. J. Pharm. 2011, 415, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.S.; Sung, K.C.; Al-Suwayeh, S.A.; Ku, M.C.; Chu, C.C.; Wang, J.J.; Fang, J.Y. Enhancement of transdermal apomorphine delivery with a diester prodrug strategy. Eur. J. Pharm. Biopharm. 2011, 78, 422–431. [Google Scholar] [CrossRef]
- Patel, P.; Pol, A.; More, S.; Kalaria, D.R.; Kalia, Y.N.; Patravale, V.B. Colloidal soft nanocarrier for transdermal delivery of dopamine agonist: Ex vivo and in vivo evaluation. J. Biomed. Nanotechnol. 2014, 10, 3291–3303. [Google Scholar] [CrossRef] [PubMed]
- Sintov, A.C.; Levy, H.V.; Greenberg, I. Continuous Transdermal Delivery of L-DOPA Based on a Self-Assembling Nanomicellar System. Pharm. Res. 2017, 34, 1459–1468. [Google Scholar] [CrossRef]
- Bali, N.R.; Salve, P.S. Selegiline nanoparticle embedded transdermal film: An alternative approach for brain targeting in Parkinson’s disease. J. Drug Deliv. Sci. Technol. 2019, 54, 101299. [Google Scholar] [CrossRef]
- Dudhipala, N.; Gorre, T. Neuroprotective effect of ropinirole lipid nanoparticles enriched hydrogel for parkinson’s disease: In vitro, ex vivo, pharmacokinetic and pharmacodynamic evaluation. Pharmaceutics 2020, 12, 448. [Google Scholar] [CrossRef] [PubMed]
- Tucker, C.; Tucker, L.; Brown, K. The Intranasal Route as an Alternative Method of Medication Administration. Crit. Care Nurse 2018, 38, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, L.; Panciani, P.P.; Muntoni, E.; Capucchio, M.T.; Biasibetti, E.; De Bonis, P.; Mioletti, S.; Fontanella, M.; Swaminathan, S. Lipid nanoparticles for intranasal administration: Application to nose-to-brain delivery. Expert Opin. Drug Deliv. 2018, 15, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Ali, J.; Ali, M.; Baboota, S.; Kaur Sahni, J.; Ramassamy, C.; Dao, L.; Bhavna. Potential of Nanoparticulate Drug Delivery Systems by Intranasal Administration. Curr. Pharm. Des. 2010, 16, 1644–1653. [Google Scholar] [CrossRef] [PubMed]
- Pardeshi, C.V.; Rajput, P.V.; Belgamwar, V.S.; Tekade, A.R.; Surana, S.J. Novel surface modified solid lipid nanoparticles as intranasal carriers for ropinirole hydrochloride: Application of factorial design approach. Drug Deliv. 2013, 20, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Pardeshi, C.V.; Belgamwar, V.S.; Tekade, A.R.; Surana, S.J. Novel surface modified polymer-lipid hybrid nanoparticles as intranasal carriers for ropinirole hydrochloride: In vitro, ex vivo and in vivo pharmacodynamic evaluation. J. Mater. Sci. Mater. Med. 2013, 24, 2101–2115. [Google Scholar] [CrossRef]
- Pardeshi, C.V.; Belgamwar, V.S. Ropinirole-dextran sulfate nanoplex for nasal administration against Parkinson’s disease: In silico molecular modeling and in vitro–ex vivo evaluation. Artif. Cells Nanomed. Biotechnol. 2017, 45, 635–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patil, G.B.; Surana, S.J. Fabrication and statistical optimization of surface engineered PLGA nanoparticles for naso-brain delivery of ropinirole hydrochloride: In-vitro–ex-vivo studies. J. Biomater. Sci. Polym. Ed. 2013, 24, 1740–1756. [Google Scholar] [CrossRef] [PubMed]
- Lungare, S.; Bowen, J.; Badhan, R. Development and Evaluation of a Novel Intranasal Spray for the Delivery of Amantadine. J. Pharm. Sci. 2016, 105, 1209–1220. [Google Scholar] [CrossRef]
- Kumar, S.; Dang, S.; Nigam, K.; Ali, J.; Baboota, S. Selegiline Nanoformulation in Attenuation of Oxidative Stress and Upregulation of Dopamine in the Brain for the Treatment of Parkinson’s Disease. Rejuvenation Res. 2018, 21, 464–476. [Google Scholar] [CrossRef]
- Salatin, S.; Alami-Milani, M.; Daneshgar, R.; Jelvehgari, M. Box–Behnken experimental design for preparation and optimization of the intranasal gels of selegiline hydrochloride. Drug Dev. Ind. Pharm. 2018, 44, 1613–1621. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Xu, L.; Bi, C.; Duan, D.; Chu, L.; Yu, X.; Wu, Z.; Wang, A.; Sun, K. Lactoferrin-modified rotigotine nanoparticles for enhanced nose-to-brain delivery: LESA-MS/MS-based drug biodistribution, pharmacodynamics, and neuroprotective effects. Int. J. Nanomed. 2018, 13, 273–281. [Google Scholar] [CrossRef] [Green Version]
- Tan, J.P.K.; Voo, Z.X.; Lim, S.; Venkataraman, S.; Ng, K.M.; Gao, S.; Hedrick, J.L.; Yang, Y.Y. Effective encapsulation of apomorphine into biodegradable polymeric nanoparticles through a reversible chemical bond for delivery across the blood–brain barrier. Nanomed. Nanotechnol. Biol. Med. 2019, 17, 236–245. [Google Scholar] [CrossRef]
- Arisoy, S.; Sayiner, O.; Comoglu, T.; Onal, D.; Atalay, O.; Pehlivanoglu, B. In vitro and in vivo evaluation of levodopa-loaded nanoparticles for nose to brain delivery. Pharm. Dev. Technol. 2020, 25, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Katual, M.K.; Singh, G.; Harikumar, S.L. Formulation Development, Characterization and in-vitro Evaluation of Nano-Lipid Carriers of Ropinirole HCl for Persistent Parkinsonian Management. Trends Drug Deliv. 2019, 6, 14–23. [Google Scholar] [CrossRef]
- Sharma, S.; Lohan, S.; Murthy, R.S.R. Formulation and characterization of intranasal mucoadhesive nanoparticulates and thermo-reversible gel of levodopa for brain delivery. Drug Dev. Ind. Pharm. 2014, 40, 869–878. [Google Scholar] [CrossRef]
- Gulati, N.; Nagaich, U.; Saraf, S. Fabrication and in vitro characterization of polymeric nanoparticles for Parkinson’s therapy: A novel approach. Braz. J. Pharm. Sci. 2014, 50, 869–876. [Google Scholar] [CrossRef] [Green Version]
- Jafarieh, O.; Md, S.; Ali, M.; Baboota, S.; Sahni, J.K.; Kumari, B.; Bhatnagar, A.; Ali, J. Design, characterization, and evaluation of intranasal delivery of ropinirole-loaded mucoadhesive nanoparticles for brain targeting. Drug Dev. Ind. Pharm. 2015, 41, 1674–1681. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, G.; Ahuja, A.; Al Rohaimi, A.H.; Muslim, S.; Hassan, A.A.; Baboota, S.; Ali, J. Nano-ropinirole for the management of Parkinsonism: Blood-brain pharmacokinetics and carrier localization. Expert Rev. Neurother. 2015, 15, 695–710. [Google Scholar] [CrossRef] [PubMed]
- Raj, R.; Wairkar, S.; Sridhar, V.; Gaud, R. Pramipexole dihydrochloride loaded chitosan nanoparticles for nose to brain delivery: Development, characterization and in vivo anti-Parkinson activity. Int. J. Biol. Macromol. 2018, 109, 27–35. [Google Scholar] [CrossRef]
- Sridhar, V.; Wairkar, S.; Gaud, R.; Bajaj, A.; Meshram, P. Brain targeted delivery of mucoadhesive thermosensitive nasal gel of selegiline hydrochloride for treatment of Parkinson’s disease. J. Drug Target. 2018, 26, 150–161. [Google Scholar] [CrossRef]
- Tzeyung, A.; Md, S.; Bhattamisra, S.; Madheswaran, T.; Alhakamy, N.; Aldawsari, H.; Radhakrishnan, A. Fabrication, Optimization, and Evaluation of Rotigotine-Loaded Chitosan Nanoparticles for Nose-To-Brain Delivery. Pharmaceutics 2019, 11, 26. [Google Scholar] [CrossRef] [Green Version]
- Bhattamisra, S.K.; Shak, A.T.; Xi, L.W.; Safian, N.H.; Choudhury, H.; Lim, W.M.; Shahzad, N.; Alhakamy, N.A.; Anwer, M.K.; Radhakrishnan, A.K.; et al. Nose to brain delivery of rotigotine loaded chitosan nanoparticles in human SH-SY5Y neuroblastoma cells and animal model of Parkinson’s disease. Int. J. Pharm. 2020, 579. [Google Scholar] [CrossRef]
- Choudhury, H.; Zakaria, N.F.B.; Tilang, P.A.B.; Tzeyung, A.S.; Pandey, M.; Chatterjee, B.; Alhakamy, N.A.; Bhattamishra, S.K.; Kesharwani, P.; Gorain, B.; et al. Formulation development and evaluation of rotigotine mucoadhesive nanoemulsion for intranasal delivery. J. Drug Deliv. Sci. Technol. 2019, 54, 101301. [Google Scholar] [CrossRef]
- Pridgen, E.M.; Alexis, F.; Farokhzad, O.C. Polymeric nanoparticle drug delivery technologies for oral delivery applications. Expert Opin. Drug Deliv. 2015, 12, 1459–1473. [Google Scholar] [CrossRef] [Green Version]
- Tsai, M.-J.; Huang, Y.-B.; Wu, P.-C.; Fu, Y.-S.; Kao, Y.-R.; Fang, J.-Y.; Tsai, Y.-H. Oral Apomorphine Delivery from Solid Lipid Nanoparticles with Different Monostearate Emulsifiers: Pharmacokinetic and Behavioral Evaluations. J. Pharm. Sci. 2011, 100, 547–557. [Google Scholar] [CrossRef]
- Borkar, N.; Holm, R.; Yang, M.; Müllertz, A.; Mu, H. In vivo evaluation of lipid-based formulations for oral delivery of apomorphine and its diester prodrugs. Int. J. Pharm. 2016, 513, 211–217. [Google Scholar] [CrossRef]
- Al-Dhubiab, B.E. Formulation and In Vitro Evaluation of Gelatin Nanospheres for the Oral Delivery of Selegiline. Curr. Nanosci. 2013, 9, 21–25. [Google Scholar] [CrossRef]
- Papadimitriou, S.; Bikiaris, D.; Avgoustakis, K.; Karavas, E.; Georgarakis, M. Chitosan nanoparticles loaded with dorzolamide and pramipexole. Carbohydr. Polym. 2008, 73, 44–54. [Google Scholar] [CrossRef]
- Tzankov, B.; Voycheva, C.; Yordanov, Y.; Aluani, D.; Spassova, I.; Kovacheva, D.; Lambov, N.; Tzankova, V. Development and in vitro safety evaluation of pramipexole-loaded hollow mesoporous silica (HMS) particles. Biotechnol. Biotechnol. Equip. 2019, 33, 1204–1215. [Google Scholar] [CrossRef] [Green Version]
- Tzankov, B.; Tzankova, V.; Aluani, D.; Yordanov, Y.; Spassova, I.; Kovacheva, D.; Avramova, K.; Valoti, M.; Yoncheva, K. Development of MCM-41 mesoporous silica nanoparticles as a platform for pramipexole delivery. J. Drug Deliv. Sci. Technol. 2019, 51, 26–35. [Google Scholar] [CrossRef]
- Ngwuluka, N.C.; Choonara, Y.E.; Kumar, P.; du Toit, L.C.; Modi, G.; Pillay, V. An optimized gastroretentive nanosystem for the delivery of levodopa. Int. J. Pharm. 2015, 494, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Nagai, T.; Machida, Y. Buccal delivery systems using hydrogels. Adv. Drug Deliv. Rev. 1993, 11, 179–191. [Google Scholar] [CrossRef]
- Tran, P.H.L.; Duan, W.; Tran, T.T.D. Recent developments of nanoparticle-delivered dosage forms for buccal delivery. Int. J. Pharm. 2019, 571, 118697. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Heng, P.W.S. Buccal Delivery Systems. Drug Dev. Ind. Pharm. 2003, 29, 821–832. [Google Scholar] [CrossRef] [PubMed]
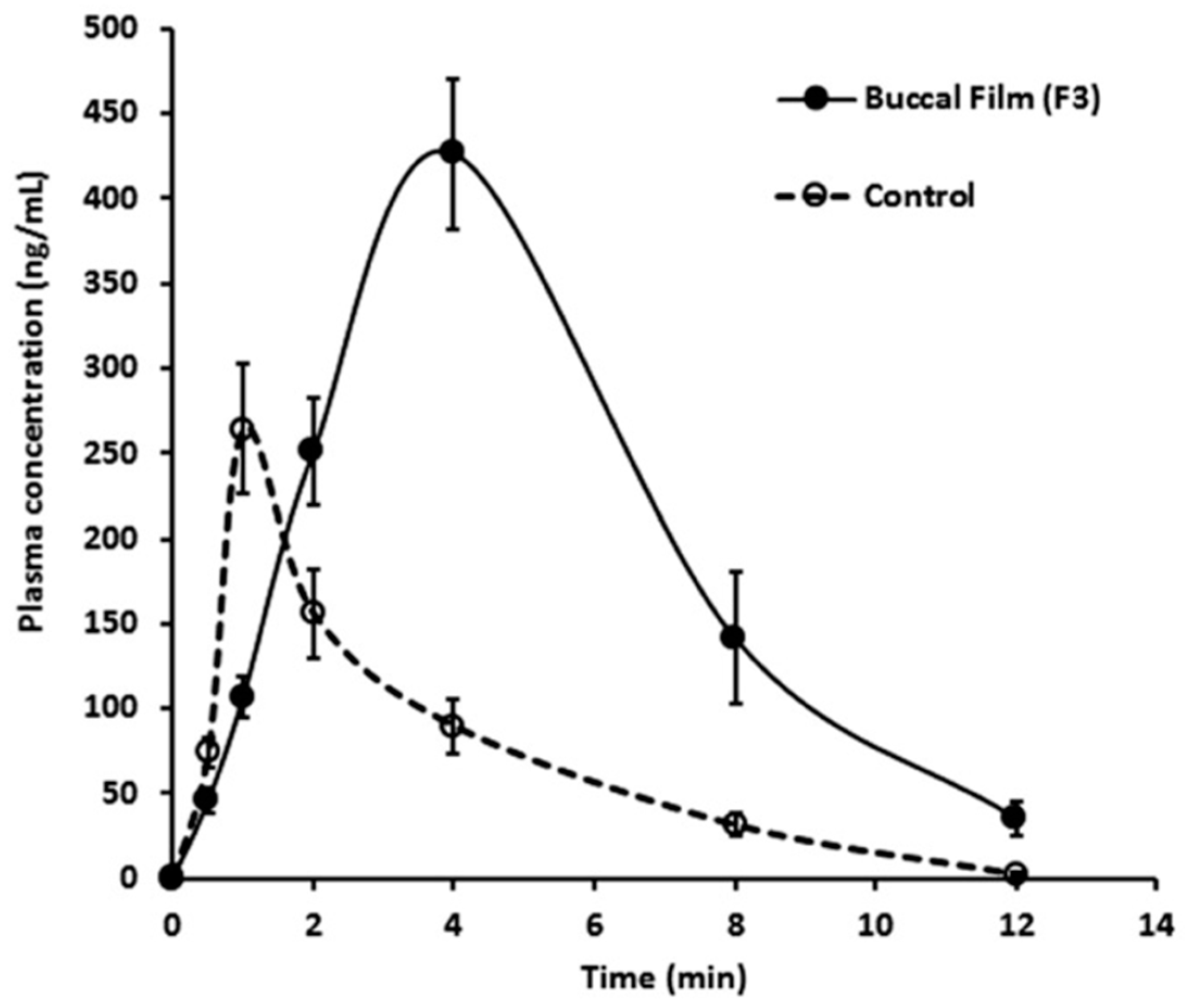
- Al-Dhubiab, B.E.; Nair, A.B.; Kumria, R.; Attimarad, M.; Harsha, S. Development and evaluation of buccal films impregnated with selegiline-loaded nanospheres. Drug Deliv. 2016, 23, 2154–2162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itin, C.; Komargodski, R.; Domb, A.J.; Hoffman, A. Controlled Delivery of Apomorphine Through Buccal Mucosa, Towards a Noninvasive Administration Method in Parkinson’s Disease: A Preclinical Mechanistic Study. J. Pharm. Sci. 2020. [Google Scholar] [CrossRef]
- Aulić, S.; Bolognesi, M.L.; Legname, G. Small-Molecule Theranostic Probes: A Promising Future in Neurodegenerative Diseases. Int. J. Cell Biol. 2013, 2013, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Liu, R.; Ji, W.; Li, Y.; Liu, L.; Zhang, X. Delivery systems for theranostics in neurodegenerative diseases. Nano Res. 2018, 11, 5535–5555. [Google Scholar] [CrossRef]
- Silindir Gunay, M.; Yekta Ozer, A.; Chalon, S. Drug Delivery Systems for Imaging and Therapy of Parkinson’;s Disease. Curr. Neuropharmacol. 2016, 14, 376–391. [Google Scholar] [CrossRef]
- Mi, L.; Wang, P.; Yan, J.; Qian, J.; Lu, J.; Yu, J.; Wang, Y.; Liu, H.; Zhu, M.; Wan, Y.; et al. A novel photoelectrochemical immunosensor by integration of nanobody and TiO2 nanotubes for sensitive detection of serum cystatin C. Anal. Chim. Acta 2016. [Google Scholar] [CrossRef]
- Sanpui, P.; Paul, A.; Chattopadhyay, A. Theranostic potential of gold nanoparticle-protein agglomerates. Nanoscale 2015, 7, 18411–18423. [Google Scholar] [CrossRef]
- Yan, K.; Sedgwick, A.C.; Zang, Y.; Chen, G.; He, X.; Li, J.; Yoon, J.; James, T.D. Sensors, Imaging Agents, and Theranostics to Help Understand and Treat Reactive Oxygen Species Related Diseases. Small Methods 2019, 3, 1900013. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, W.; Yu, D.X.; Xiao, Z.; He, Z. Application of nanodiagnostics and nanotherapy to CNS diseases. Nanomedicine 2018, 13, 2341–2371. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.Y.; Chiu, M.J.; Lin, C.H.; Horng, H.E.; Yang, C.C.; Chieh, J.J.; Chen, H.H.; Liu, B.H. Development of an ultra-high sensitive immunoassay with plasma biomarker for differentiating Parkinson disease dementia from Parkinson disease using antibody functionalized magnetic nanoparticles. J. Nanobiotechnol. 2016, 14, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrmann, Y.; Bujnicki, T.; Zafiu, C.; Kulawik, A.; Kühbach, K.; Peters, L.; Fabig, J.; Willbold, J.; Bannach, O.; Willbold, D. Nanoparticle standards for immuno-based quantitation of α-synuclein oligomers in diagnostics of Parkinson’s disease and other synucleinopathies. Clin. Chim. Acta 2017, 466, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-W.; Yang, S.-Y.; Yang, C.-C.; Chang, C.-W.; Wu, Y.-R. Plasma and Serum Alpha-Synuclein as a Biomarker of Diagnosis in Patients With Parkinson’s Disease. Front. Neurol. 2020, 10, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Govindaraju, S.; Ankireddy, S.R.; Viswanath, B.; Kim, J.; Yun, K. Fluorescent Gold Nanoclusters for Selective Detection of Dopamine in Cerebrospinal fluid. Sci. Rep. 2017, 7, 40298. [Google Scholar] [CrossRef]
- Sonuç Karaboğa, M.N.; Sezgintürk, M.K. Cerebrospinal fluid levels of alpha-synuclein measured using a poly-glutamic acid-modified gold nanoparticle-doped disposable neuro-biosensor system. Analyst 2019, 144, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.G.; Lu, S.; Liu, D.Q.; Zhang, L.; Zhang, L.X.; Yu, X.L.; Liu, R.T. ScFv-conjugated superparamagnetic iron oxide nanoparticles for MRI-based diagnosis in transgenic mouse models of Parkinson’s and Huntington’s diseases. Brain Res. 2019, 1707, 141–153. [Google Scholar] [CrossRef]
- Joshi, N.; Basak, S.; Kundu, S.; De, G.; Mukhopadhyay, A.; Chattopadhyay, K. Attenuation of the early events of α-synuclein aggregation: A fluorescence correlation spectroscopy and laser scanning microscopy study in the presence of surface-coated Fe3O4 nanoparticles. Langmuir 2015, 31, 1469–1478. [Google Scholar] [CrossRef]
- Malvindi, M.A.; Di Corato, R.; Curcio, A.; Melisi, D.; Rimoli, M.G.; Tortiglione, C.; Tino, A.; George, C.; Brunetti, V.; Cingolani, R.; et al. Multiple functionalization of fluorescent nanoparticles for specific biolabeling and drug delivery of dopamine. Nanoscale 2011, 3, 5110–5119. [Google Scholar] [CrossRef]
- Ramos-Gómez, M.; Seiz, E.G.; Martínez-Serrano, A. Optimization of the magnetic labeling of human neural stem cells and MRI visualization in the hemiparkinsonian rat brain. J. Nanobiotechnol. 2015, 13, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Long, L.; Cai, X.; Guo, R.; Wang, P.; Wu, L.; Yin, T.; Liao, S.; Lu, Z. Treatment of Parkinson’s disease in rats by Nrf2 transfection using MRI-guided focused ultrasound delivery of nanomicrobubbles. Biochem. Biophys. Res. Commun. 2017, 482, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Kulisevsky, J.; Oliveira, L.; Fox, S.H. Update in therapeutic strategies for Parkinson’s disease. Curr. Opin. Neurol. 2018, 31, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Fariello, R.G. Safinamide. Neurotherapeutics 2007. [Google Scholar] [CrossRef] [Green Version]
- Bette, S.; Shpiner, D.S.; Singer, C.; Moore, H. Safinamide in the management of patients with Parkinson’s disease not stabilized on levodopa: A review of the current clinical evidence. Ther. Clin. Risk Manag. 2018, 14, 1737–1745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, J.J.; Lees, A.; Rocha, J.-F.; Poewe, W.; Rascol, O.; Soares-da-Silva, P. Opicapone as an adjunct to levodopa in patients with Parkinson’s disease and end-of-dose motor fluctuations: A randomised, double-blind, controlled trial. Lancet Neurol. 2016, 15, 154–165. [Google Scholar] [CrossRef]
- Scott, L.J. Opicapone: A Review in Parkinson’s Disease. Drugs 2016, 76, 1293–1300. [Google Scholar] [CrossRef]
- Fields, C.R.; Bengoa-Vergniory, N.; Wade-Martins, R. Targeting Alpha-Synuclein as a Therapy for Parkinson’s Disease. Front. Mol. Neurosci. 2019, 12. [Google Scholar] [CrossRef] [Green Version]
- Parmar, M.; Grealish, S.; Henchcliffe, C. The future of stem cell therapies for Parkinson disease. Nat. Rev. Neurosci. 2020, 21, 103–115. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, S.; Almeida, A.J.; Vale, N. Importance of Nanoparticles for the Delivery of Antiparkinsonian Drugs. Pharmaceutics 2021, 13, 508. https://doi.org/10.3390/pharmaceutics13040508
Silva S, Almeida AJ, Vale N. Importance of Nanoparticles for the Delivery of Antiparkinsonian Drugs. Pharmaceutics. 2021; 13(4):508. https://doi.org/10.3390/pharmaceutics13040508
Chicago/Turabian StyleSilva, Sara, António J. Almeida, and Nuno Vale. 2021. "Importance of Nanoparticles for the Delivery of Antiparkinsonian Drugs" Pharmaceutics 13, no. 4: 508. https://doi.org/10.3390/pharmaceutics13040508