1. Introduction
Nowadays, one of the main challenges in pharmaceutical technology is dealing with poorly water-soluble drugs. The number of Biopharmaceutics Classification System (BCS) Class II and Class IV drugs is estimated to make up ~40% of currently marketed drugs and ∼90% of compounds currently under development [
1,
2,
3]. One of the BCS Class II/IV drugs is ciprofloxacin (CIP), which is a worldwide-used, broad-spectrum, second-generation fluoroquinolone antibiotic. Fluoroquinolones inhibit DNA replication in bacteria because their mechanism of action is the inhibition of bacterial DNA gyrase and topoisomerase IV. The lack of cross-resistance between fluoroquinolones and other classes of antibiotics makes this antibiotic family important. CIP and its derivatives can effectively treat antibiotic-resistant infections (e.g., nosocomial pneumonia), so they are usually reserved as “drugs of last resort” [
4]. Since CIP is active against many Gram-negative and Gram-positive bacteria, it is directed to treat several bacterial infections. CIP is indicated for a broad spectrum of diseases, from bacterial conjunctivitis through bone and joint infections to complicated urinary tract infections. Accordingly, the available dosage forms on the market are ophthalmic and otic solutions, film-coated tablets, oral suspensions, and solutions for infusions. Thus, various routes for drug administration, both local and systemic, are applied. Despite common use, CIP has only 56–77% bioavailability [
4]. The solubility of the drug is pH-dependent, as CIP is highly soluble below pH 5 and above pH 10 but almost insoluble around neutral pH level [
5]. According to the BCS, CIP belongs to Class IV, with low water solubility (0.067 mg/mL at 25 °C, pH 7.5; [
6]) and poor permeability [
4].
In most cases, nanocarrier-based drug delivery systems can offer a solution to the low solubility problem of a drug. Nanostructures are innovative formulations with nanometer-scale in at least one dimension and a large surface-area-to-volume ratio. The latter ensures that a large amount of the drug can come into contact with the surrounding medium. In this way, the dissolution rate and even solubility can increase [
7,
8,
9]. Furthermore, numerous drug delivery systems contain the active pharmaceutical ingredient (API) in an amorphous state and therefore cause increased solubility of the drug [
10,
11]. With the increase of solubility and dissolution rate, the bioavailability of the API increases [
12,
13,
14]. Currently, intensive research is underway to formulate CIP into different nanocarriers to treat several diseases. Some examples are collected in
Table 1.
Polymeric nanofibers are considered solid dispersions with a large specific surface area that can stabilize drugs in their amorphous state. Thus, polymer-based nanofibers provide an attractive approach for the development of dosage forms due to the enhanced solubility and dissolution rate [
25,
26]. Nanofibers have a variety of uses currently under investigation in different fields of science. In pharmaceutical and medical fields, at least four applications have to be mentioned: wound dressings, filtration, drug delivery systems, and tissue engineering scaffolds [
27,
28,
29,
30,
31]. Electrospinning is an effective and inexpensive method for the fabrication of polymeric nanofibers; hence, it is the most widely used technique for nanofiber production in the industry. During the electrospinning procedure, which is generated by a high voltage power supply, the ejection and travel of the polymer-drug fluid jet are induced by the large potential difference between a needle and a collector [
32]. As the jet travels towards the collector, the solvent evaporates, and the jet solidifies into nano-sized fibers. It is possible to control the mean fiber diameter and morphology by changing the polymer-drug solution, the applied voltage, the needle–collector distance, the flow rate, the collector speed, or the environmental temperature and humidity [
33,
34].
Poly(vinyl pyrrolidone) (PVP) is a water-soluble and biocompatible polymer, which is commonly used as a pharmaceutical excipient and food additive [
35]. PVP is also widely used as a carrier polymer that enables electrospinning [
23,
36,
37,
38,
39,
40,
41,
42,
43,
44,
45].
Ciprofloxacin-loaded PVP nanofibers as potential wound dressings have been recently investigated [
23,
41,
44]. However, to the best of our knowledge, the development of a potential per os formulation has not been published yet.
In this article, the preformulation studies of PVP-based nanofiber mats loaded with CIP for per os administration are investigated. The aim of the present study was to increase the water solubility and diffusion of the API and study the in vitro drug release and its kinetics. Additionally, the study aimed to investigate the effect of the polymer–drug solution and the flow rate on the fiber diameter and morphology. Special attention was paid to structural characterization, drug entrapment efficiency, solubility, in vitro dissolution, and in vitro diffusion of the CIP-loaded nanofibrous samples when producing immediate-release nanofibrous formulation, and therefore achieving effective antibiotic therapy.
3. Results and Discussion
3.1. Optimization of the Electrospinning Parameters
Reviewing the works focusing on the electrospinning of drug-loaded PVP nanofibers, the solution flow rate was found to range between 0.2 and 2 mL/h [
23,
36,
37,
38,
39,
40,
41,
42,
43,
44,
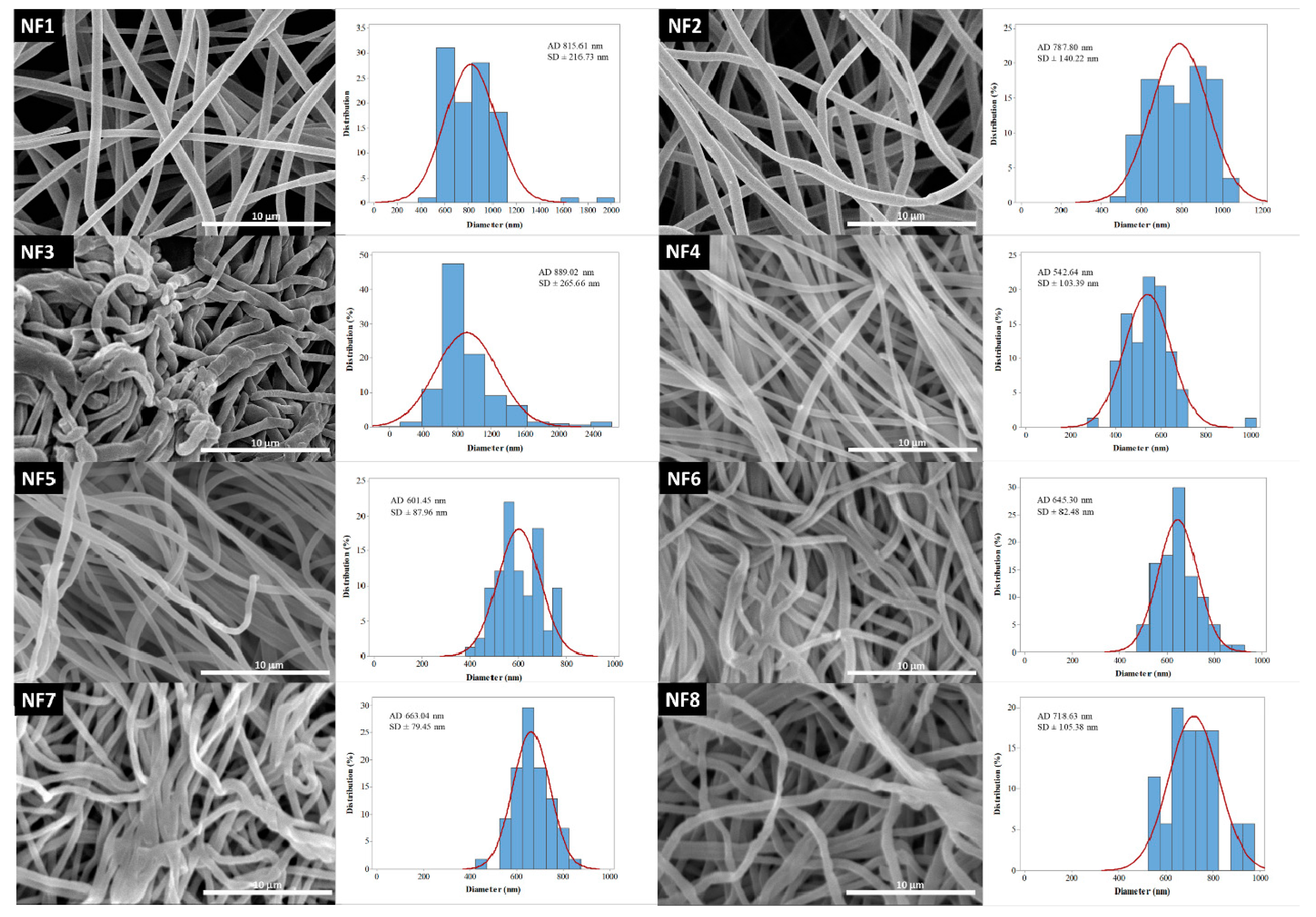
45]. The average fiber diameter of the different formulations was largely varying. One of the aims of this research was to investigate the effect of flow rate on the diameter of PVP nanofibers, keeping the other preparation parameters constant. Additionally, nanofibers with different PVP:CIP volume ratios were prepared and studied. SEM was used to visualize the nanofibers, and then their morphology was observed, and the average fiber diameter was measured. The narrowest nanofiber formulation with the finest morphology was targeted during the optimization.
The morphology of drug-free and various drug-loaded nanofibers are represented in
Table 3 and
Figure 2. Continuous, smooth-surfaced nanofibers were successfully prepared from all the solutions studied except for sample NF3. PVP:CIP 1:2 volume ratio eventuated discontinuous, worm-like nanofibers with largely varying diameters (889 ± 265 nm). The average diameter of the pure PVP fibers was 815 ± 216 nm. Considering the continuous fibers, the addition of the CIP decreased the fiber diameter. The decrease was significant except for sample NF2 (
Table 4). This is expected to be related to the increase in the conductivity of the electrospinning solutions by adding the CIP [
47]. Higher solution conductivity could facilitate the elongation of the jet and generate thinner fibers [
48]. Additionally, the PVP nanofibers had some narrowing that slightly resembled the formation of beads, as shown in
Figure 2. This was probably due to the low PVP concentration [
49]. As the flow rate was increased, the average fiber diameter increased, which was expected from the literature [
49]. However, the difference was not significant in every case (
Table 4). Besides, the 3 mL/h flow rate (NF7) caused incomplete solvent evaporation and merged fibers, while the sample produced by 4 mL/h flow rate (NF8) showed some bead-like structures on top of the merged fibers (
Figure 2). According to the results of this study, two formulations—namely, NF5 and NF6—had the most optimal morphology and diameter distribution. Thus, the 1:1 ratio between the PVP and CIP was found optimal, similar to our previous study [
50]. The higher flow rate is desirable for the preparation of samples because of the larger number of nanofibers produced during the same period of time. Since the applied flow rate of NF6 was 2 times higher than the flow rate of NF5, finally, NF6 was selected for further studies.
3.2. Structural Characterization
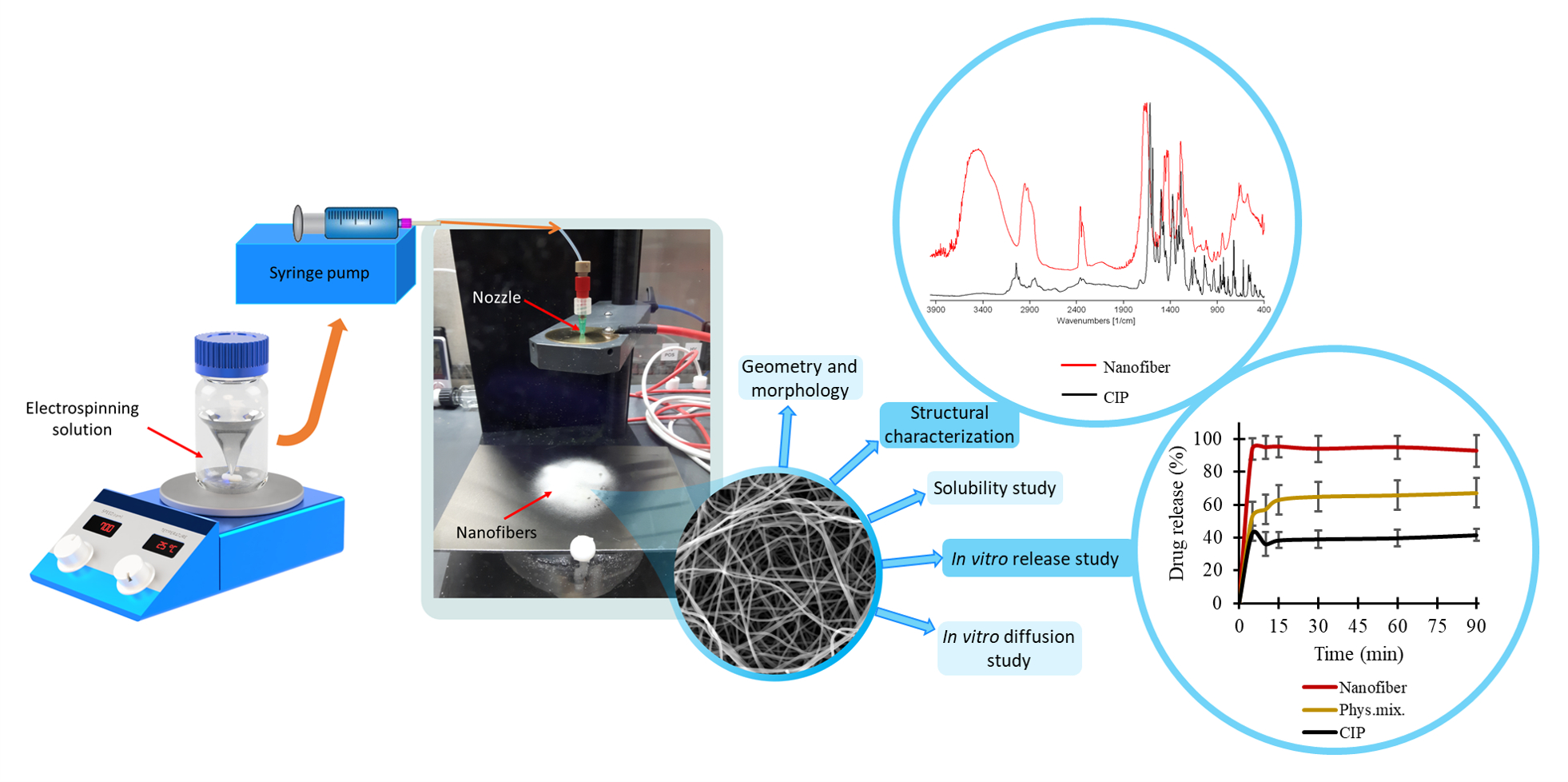
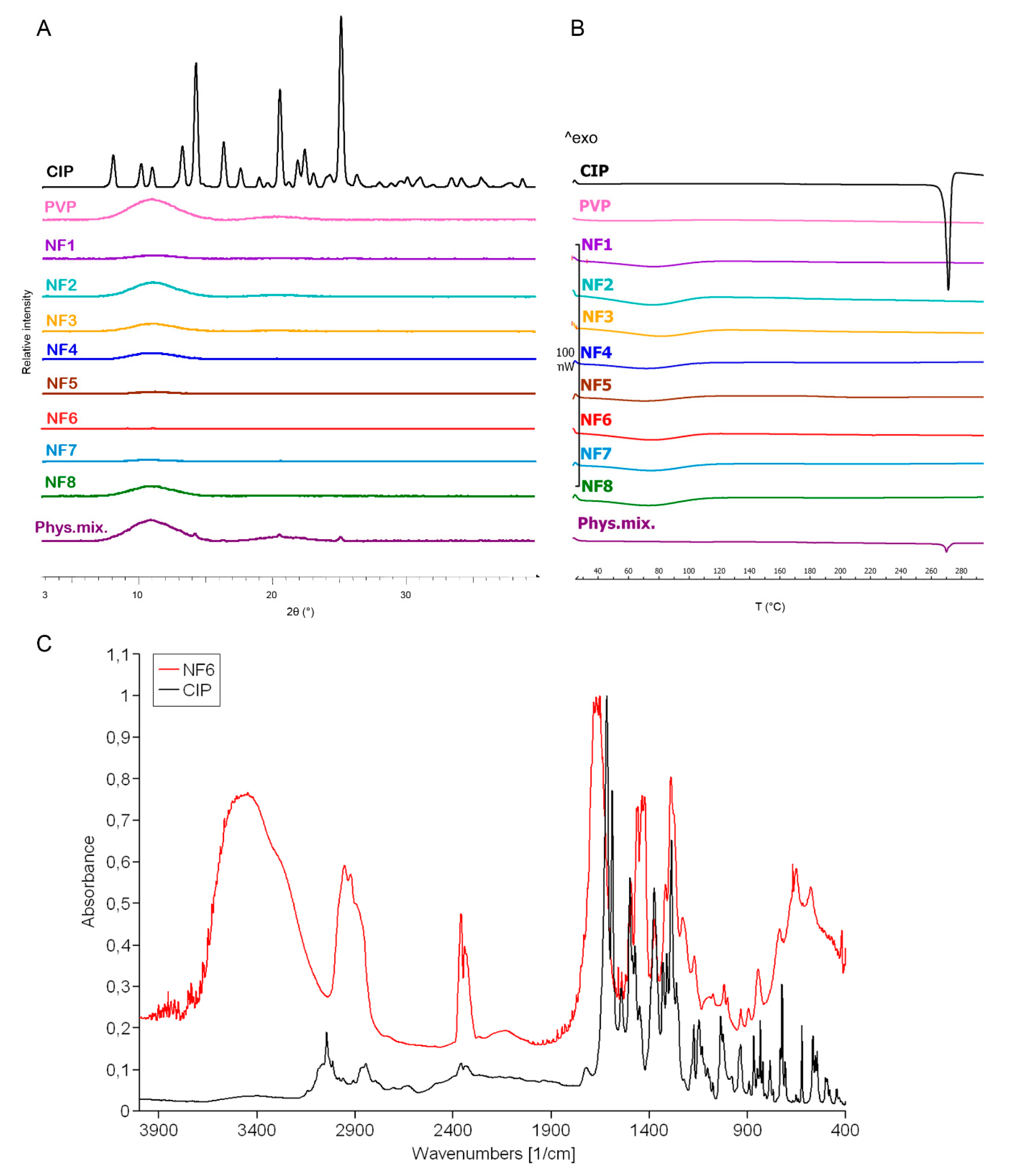
The change in the crystallinity of CIP was studied by XRPD and DSC (
Figure 3A,B). The XRPD of the CIP powder showed high crystallinity, represented by several smaller and three longer sharp peaks at around 2-Theta = 14.5, 20.9, and 25.4°. The diffractogram of the physical mixture also had the same peaks along with a broad peak between 2-Theta = 7–15°. The broad peak that appeared in the diffractogram of PVP, physical mixture, and the half of nanofibrous samples prove the polymer’s amorphous nature. The three characteristic peaks of the CIP were missing from the spectra of the nanofibrous samples, which indicates physicochemical interactions between the drug and the polymer matrix and the amorphous form of CIP created by the fast evaporation of the solvent during electrospinning.
Figure 3B presents the DSC thermograms of CIP, PVP, physical mixture, and all the samples listed in
Table 2. The melting point of the pure drug is well-defined by a large endothermic peak at 275 °C, also appearing on the thermogram of the physical mixture. However, the flat PVP and NF1-NF8 thermograms suggest amorphous components. It can be concluded from both results that the drug was incorporated in amorphous form into the nanofibers.
Main drug–polymer interactions were observed by FTIR, and the spectra are given in
Figure 3C. One prominent characteristic CIP peak at 1732 cm
−1 (νC=O) shifted toward the higher wavenumbers and stretched due to the interaction with the hydroxyl groups of the PVP [
51]. The wide band between 3050–3750 cm
−1 (νC-OH) was enlarged by the hydrogen bonding between the CIP and PVP in the nanofibers. Moreover, the bands between 3050–2750 cm
−1 (alkene and aromatic νC-H), between 2380–2275 cm
−1 (νC-N), at 1500 cm
−1 (quinolone), and at 1290 cm
−1 (νC-O) appeared wider in case of the nanofiber [
52]. These shifts and widenings confirm the successful incorporation of the CIP into the polymeric fibers.
3.3. Drug Loading and Drug Entrapment Efficiency
Nanofibers, in general, exhibit very high entrapment efficiency because the electrospinning procedure can be considered as an in situ solidification of a polymer solution. Since both CIP and PVP are non-volatile in nature, high entrapment efficiency was expected under the consideration of complete miscibility.
Entrapment efficiency, as well as drug loading, was calculated by UV spectrophotometry. Solvent with low pH (0.1 M HCl) was used to earn complete dissolution. The NF6 nanofibrous sample met our expectations with 92 ± 8% entrapment efficiency. Furthermore, the theoretical drug loading of the NF6 formulation was 0.99%, while the calculated drug loading was 0.92 ± 0.08%. These data are consistent with the high entrapment efficiency.
3.4. Solubility Test
The optimization study revealed that the sample NF6 had the most promising size and morphology for the solubility and dissolution studies. Solubility tests of the raw CIP powder, the physical mixture, and the drug-loaded nanofibrous samples were carried out in distilled water and PBS, as shown in
Table 5. CIP has a pH-dependent, U-shaped solubility profile showing high solubility at pH < 5 and pH > 10 and poor solubility around neutral pH level as its isoelectric point is 7.42 [
5]. Thus, distilled water and pH 7.4 PBS were chosen as solvents to observe the effect of the polymer nanocarrier on the solubility. The pH was measured at the beginning and the end of the tests. The pH values of the solvents before adding the CIP were 6.3 and 7.4 in the case of the distilled water and PBS, respectively. During the 24 h solubility study, the pH of the CIP solution in water increased to 7.1 because CIP as a weak base slightly alkalized the solution. With the increase of the pH level, the solubility of the drug decreases, which reduces the dissolution of the remaining solid CIP in the system. The buffer capacity of the PBS was sufficient to prevent a notable pH shift.
Studies with both solvents suggested that the incorporation of CIP into nanofibers caused a significant increase (p < 0.01 in the case of water and p < 0.05 in the case of PBS) in the drug solubility. In distilled water, the final CIP concentration showed a 12-fold increase in the case of nanofibers compared with the raw CIP, while in the PBS, the nanofibers were approx. 6.4 times more soluble. Furthermore, the solubility of the drug in the physical mixture was not significantly higher in either solvent. Thus, it can be seen that only the presence of PVP could enhance solubility by increasing wettability. However, this enhancement was much lower than in the case of nanofibers, where solid molecular dispersion of CIP formed during the electrospinning procedure. Solid molecular dispersions can guarantee increased solubility by decreasing the particle size and improving wettability. This finding may be correlated with the characterization results because inside the nanofiber the CIP is in its amorphous form.
3.5. In Vitro Drug Release
The in vitro release profiles of electrospun nanofibers and raw CIP powder were investigated both in single medium dissolution and two-stage biorelevant release tests. On the one hand, pH 7.4 PBS was chosen as a medium, while in this pH level, CIP has a minimum solubility value, and the pH of the terminal ileum [
53], as well as the blood, is around 7.4. The pH-dependent solubility of a drug can cause incomplete dissolution or precipitation, which leads to suboptimal bioavailability. This might generate a problem with CIP because it occurs mostly at high administered doses [
4]. On the other hand, any pH shift that occurs during the transfer from the stomach to the small intestine may affect the solubility and the bioavailability of CIP [
54]. To investigate this effect, conditions similar to in vivo (biosimilar media, change in pH after 30 min, body temperature) were used by a two-stage release study [
46].
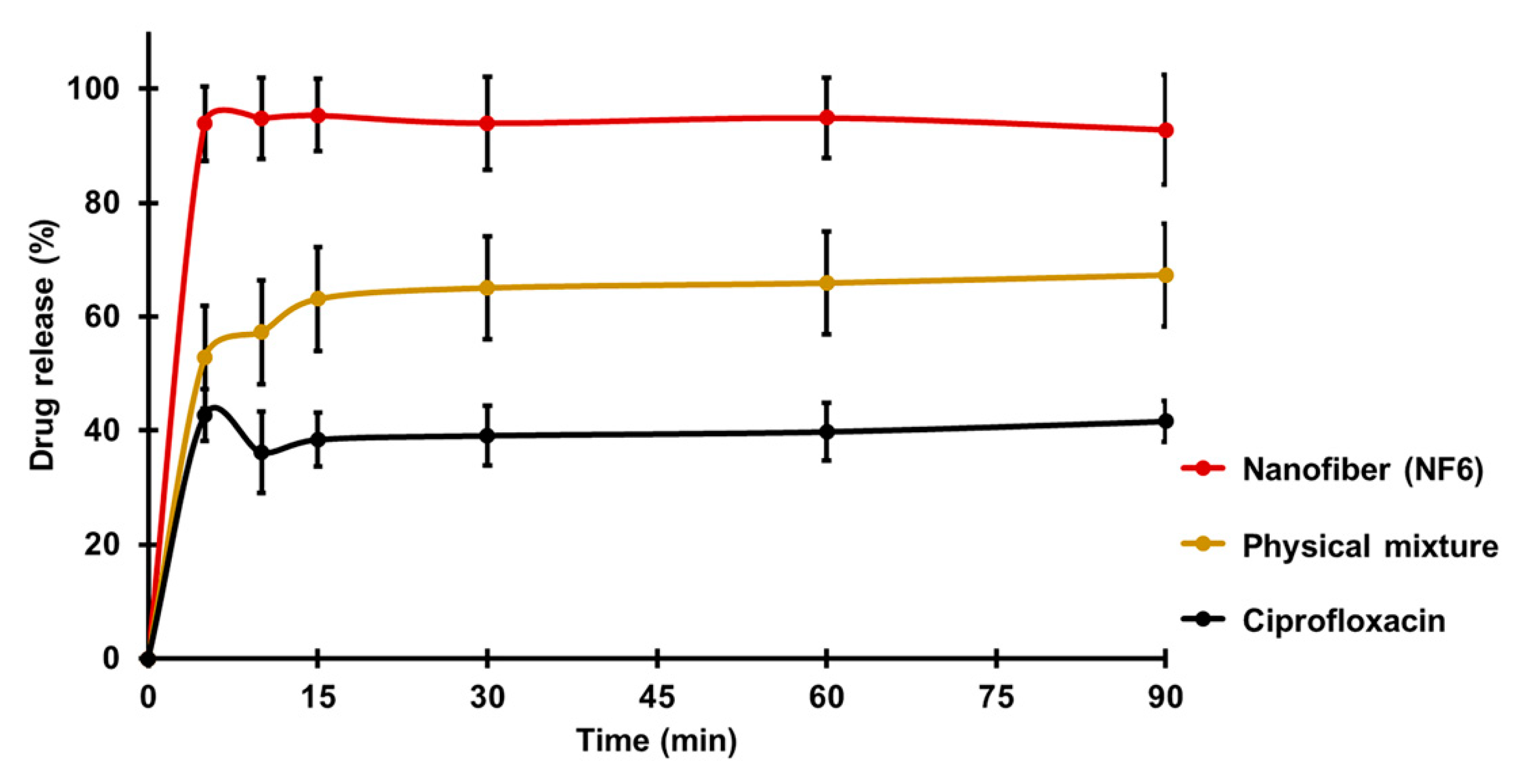
Figure 4 presents the cumulative drug release vs. time curves of CIP powder, physical mixture, and electrospun sample (NF6) measured in PBS (pH 7.4). In the case of the CIP powder, the dissolution was not complete within 90 min, most probably due to the poor solubility of the drug at this pH. Until 90 min, only 41 ± 3% of the drug was liberated. Similarly, the drug release from the physical mixture was 67 ± 12% at the end of the measurement (90 min). However, NF6 showed a significantly higher dissolution rate than CIP powder (
p < 0.001) at every measured point. Additionally, the nanofibrous sample demonstrated significantly higher drug release than the physical mixture (
p < 0.05) in time points at 5 and 10 min. Besides, while all the samples showed fast dissolution behavior, the release was the fastest from the nanofibers. The improved dissolution rate resulted in 94 ± 6% dissolved CIP within only 5 min. The distinct difference between the raw CIP and the nanofibers could be caused by the high surface-to-volume ratio, the high wettability, and the amorphous drug inside the nanofibers.
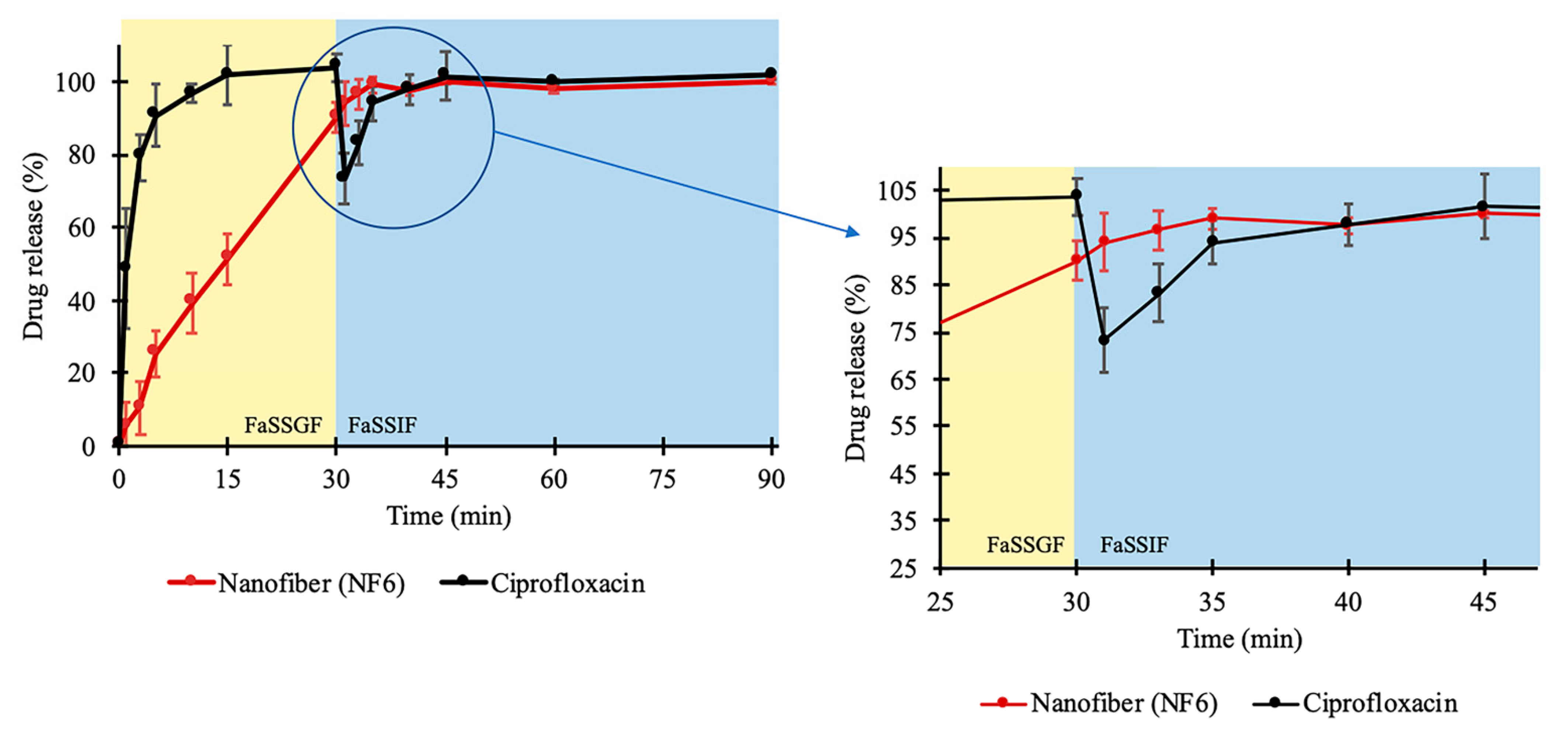
In addition, a two-stage biorelevant release study was executed to mimic the in vivo conditions since the solubility of CIP is sensitive to changes in pH of the gastrointestinal environment. The raw CIP powder and the NF6 nanofibrous sample were placed into 25 mL of FaSSGF medium, and the drug release was studied for 30 min. From the raw CIP, the release was fast and complete as the whole amount of the added powder was dissolved within 10 min (
Figure 5). This was expected since Hansmann et al. published a similar result with Ciprobay
® 500 mg IR tablet [
54], and the 24 h solubility test in FaSSGF (described in
Section 2.2.7) showed 7.794 ± 0.675 mg/mL average CIP concentration. However, in the case of the electrospun sample, the drug release was slower. It barely reached 90% until the last sampling of the FaSSGF medium. The difference could have been caused by the strong gelation of the high molecular weight PVP (Mw = 1,300,000) in low pH levels forming a viscous matrix around the CIP, which could remarkably slow down its diffusion to the dissolution medium.
As the second phase of the two-stage study, at 30 min, FaSSIF Concentrate was added to the FaSSGF dissolution medium to create a 1:1 volume ratio mixture (FaSSIF medium). With this step, the emptying of the stomach into the small intestine is modeled easily [
54,
55]. Due to the change of pH, the solubility of CIP decreased. As a result of the 24 h solubility test, 0.205 ± 0.002 mg/mL drug concentration was detected in FaSSIF (described in
Section 2.2.7). In the case of the raw CIP samples, this 38-fold decrease caused the precipitation of the pre-dissolved CIP, which was visible in the vessels. Additionally, a slope can be observed at the 30 min time point in
Figure 5. Interestingly, the curve of the nanofibrous sample has no slope, while the dissolution of the drug continued even after the change of the medium. Controlled release without precipitation was observed in the case of the electrospun sample. From the time point at 40 min, the two curves run together at around 100%. The time range between 25 and 45 min is magnified in
Figure 5.
3.6. Drug-Release Kinetics and Mechanism
To describe the release kinetics from nanofibers, five different mathematical models are usually used—namely, zero order, first order, Hixson–Crowell model, Higuchi model, and Korsmeyer–Peppas model. The regression coefficient (
R2) values of the different drug release models are listed in
Table 6.
In the case of the single medium (pH 7.4 PBS) dissolution study, the Korsmeyer–Peppas model showed superiority over the other models studied for describing the release kinetic of the raw CIP, the physical mixture, and the NF6 electrospun sample. However, the Korsmeyer–Peppas model could not be fitted very well to the curves of CIP powder (
R2 = 0.8967) and physical mixture (
R2 = 0.8684), while the drug release of nanofibers was almost perfectly described by the model (
R2 = 0.9993). This is reasonable since the Korsmeyer–Peppas model describes drug release from a polymeric system. It takes into account several mechanisms simultaneously, such as the diffusion of water into the polymer matrix, the swelling, and the dissolution of the polymer [
52,
56].
Furthermore, the Korsmeyer–Peppas model showed a high R2 value both in FaSSGF (R2 = 0.9794) and FaSSIF (R2 = 0.9229), considering the release kinetics of the two-stage biorelevant dissolution study. However, in the case of the latter, the first order kinetics could be fitted to the curve even more precisely (R2 = 0.9268). First order was found to be the release kinetics of the raw CIP in FaSSIF medium as well, which means that the dissolution rate of CIP from the PVP matrix was dependent on the drug concentration. This can be explained by the change of pH caused by the change of the medium at 30 min. The solution was supersaturated, and the higher pH caused the decrease of the solubility of the drug. Near to its solubility limit, the concentration of the CIP could affect the release kinetics.
In terms of the other medium, the release of the CIP powder followed the Higuchi model (
R2 = 0.9502) while the highest
R2 values of the nanofibrous sample were related to zero order (
R2 = 0.9810) and the Hixson–Crowell model (
R2 = 0.9873). The difference in the course of the curves can also be seen in
Figure 5. The Higuchi model describes drug release from different matrix systems that contain water-soluble drugs [
52]. The FaSSGF is a good solvent of the CIP and without any polymer in the system, it could dissolve freely. On the other hand, the PVP formed a viscous hemisphere gel at the bottom of the vessel, causing the Hixson–Crowell model. This release model considers the dissolution of a tablet or polymer matrix but with the maintenance of their geometrical characteristics.
3.7. In Vitro Diffusion Study
An in vitro diffusion study was executed to compare the capacity of CIP from different samples (NF6 nanofiber, physical mixture, CIP powder) for crossing biological barriers, e.g., the small intestine cells (
Figure 6). In the literature, it is suggested that the CIP is absorbed from the duodenum and the proximal jejunum [
57]. Thus, during the in vitro diffusion study, pH 6.8 PBS was used for the donor phase. The acceptor phase (pH 7.4 PBS) modeled the intracellular pH (“set-point” pHi 7.35) of intestinal absorptive cells [
58]. Since the absorption of ciprofloxacin seems to be mainly mediated not by active but by passive diffusion [
54], a synthetic membrane was used to separate the two phases.
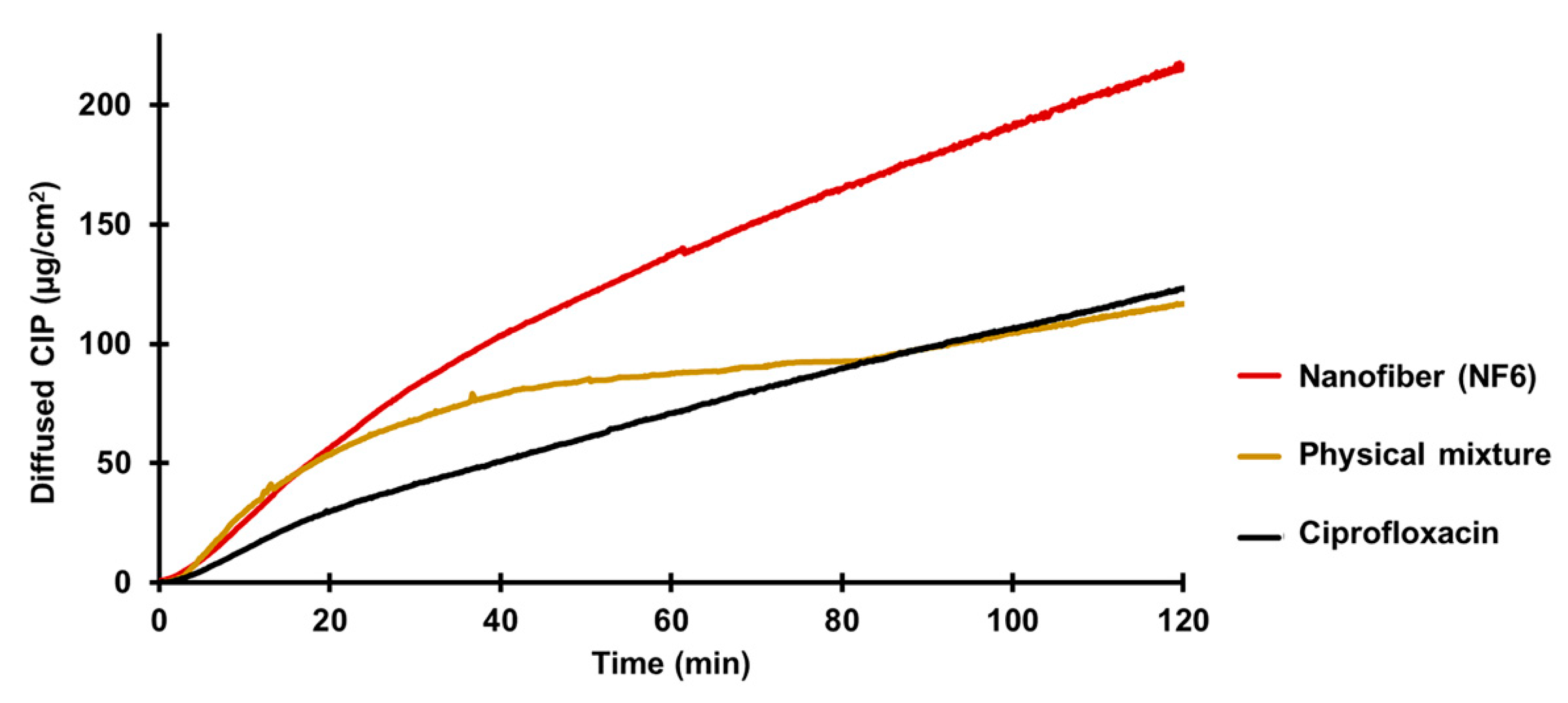
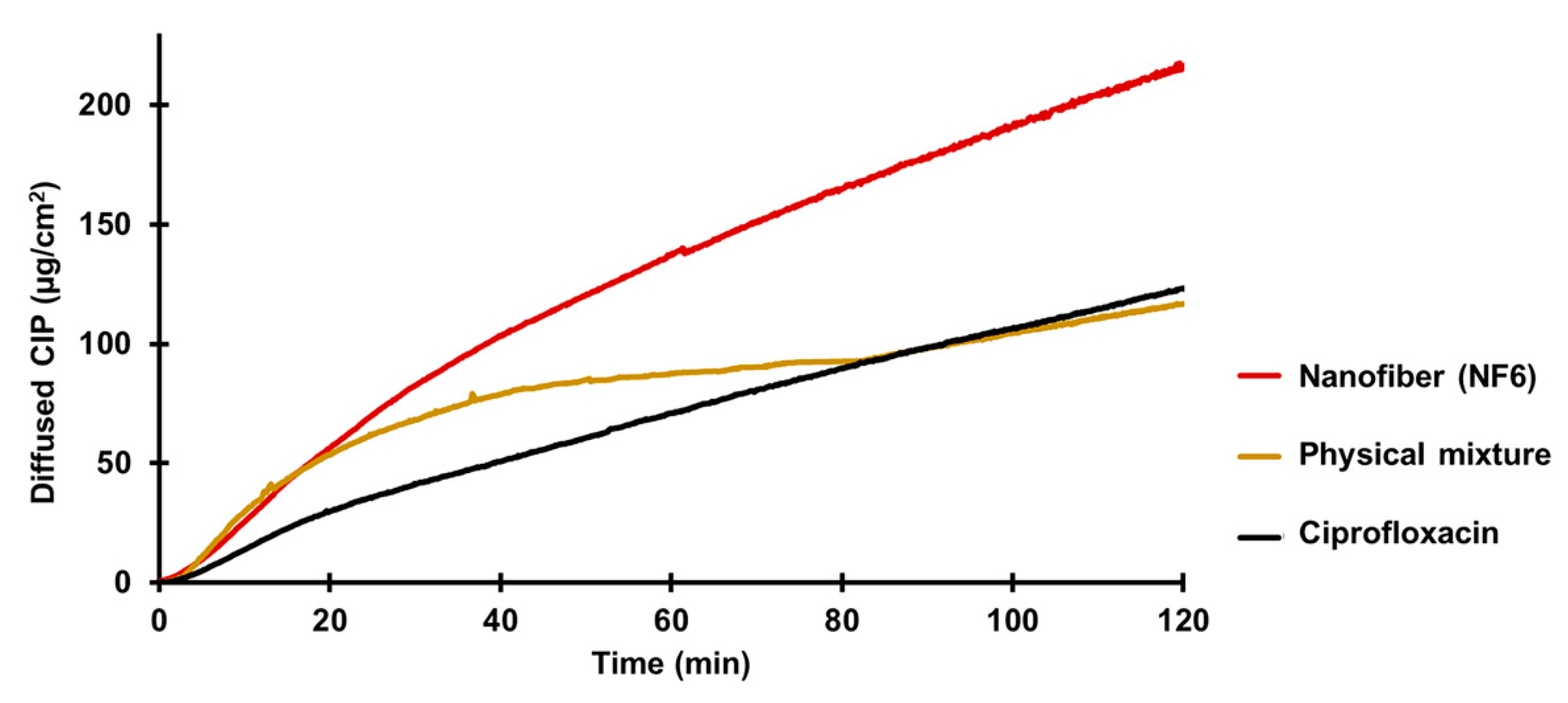
According to the results, the diffusion of the CIP was remarkably higher from the nanofibrous sample (215 µg/cm
2) than the raw CIP (122 µg/cm
2) or the physical mixture (118 µg/cm
2).
Figure 6 shows the diffusion profiles of CIP from the samples. The diffused CIP over time curve of the physical mixture interestingly runs with the curve of the nanofibrous sample for the first 20 min, but after that it becomes flatter as the CIP diffusion slows down. Finally, at the time range between 80 and 120 min, it overlaps with the curve of the pure CIP powder. This fascinating behavior could be caused by the presence of PVP. As written previously, PVP is a wetting agent that could increase the solubility of a drug. More solute CIP means a higher concentration gradient, which results in faster diffusion. However, the effect of this increase has a limit, and beyond that, the solution and diffusion of CIP slow down.
The calculated flux (
J) and permeability coefficient (
Kp) values are shown in
Table 7. The diffused CIP amount was 1.7 times higher from the NF6 sample than from the nonfibrous samples. The presence of the PVP did not affect the diffusion since the results of the physical mixture were similar to the CIP powders. Moreover, the permeability coefficient was increased 1.9 times by the nanofiber formulation.
4. Conclusions
Since CIP is a BSC Class IV drug, various pharmaceutical technological approaches are desirable for improving its bioavailability through the improvement in solubility, dissolution, and permeability. One such approach is the formulation of nanofibers via electrospinning, which is a simple and cost-effective production technique. Electrospun nanofibers, besides solid nanoparticles, are considered as amorphous solid dispersions and promising nanocarriers. Nanofibers are favorable drug carriers because of their high specific surface area, the wide variety of polymers and APIs that are spinnable, the ease of material combination, and the capability for mass production.
In the present study, CIP-loaded nanofibers were successfully fabricated by electrospinning. With the optimized process parameters, the nanofibers had a small, uniform fiber diameter with smooth surface morphology. The flow rates used in PVP-based nanofiber fabrication were compared. It was found that a higher flow rate produced thicker fibers, but in the case of too high a flow rate, the fibers were merged because the solvent evaporation was incomplete. According to the results of the XRPD and the DSC measurements, the CIP lost its crystallinity during the electrospinning procedure, and an amorphous form was produced. This form of the drug, along with the increased surface area, is the reason for the significantly higher solubility and in vitro dissolution rate in pH 7.4 PBS achieved with the nanofibrous samples. In the single medium release study (pH 7.4 PBS), the nanofibrous formulation demonstrated fast dissolution, and the release kinetics followed the Korsmeyer–Peppas model. In contrast, the raw CIP showed incomplete dissolution due to its poor solubility at this pH level. To mimic more precisely the in vivo conditions, a two-stage biorelevant release study was executed. Since the CIP is more soluble at low pH levels, a supersaturated solution was formed with FaSSGF medium. Then, with the change of the medium to FaSSIF, the solubility of the drug changed, and precipitation occurred. The precipitation could be prevented by nanofibers, since the PVP formed a viscous matrix around the CIP and released it with the dissolution kinetics described the best by Hixson–Crowell model and zero order kinetics. This study predicts a controlled release in the stomach for other API-loaded PVP-based nanofibers that might be useful in the development of local drug delivery systems. Moreover, incorporation of CIP into nanofibers could provide noticeably higher in vitro diffusion through the membrane. Therefore, our results show that CIP-loaded PVP-nanofibers could be considered as fast-dissolving formulations with improved physicochemical properties that may be suitable for further studies to keep in mind when developing an oral dosage form.


 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
