1. Introduction
Therapeutic proteins such as monoclonal antibodies (mAbs) can degrade after administration to patients [
1,
2]. Once injected or infused, mAbs are subjected to sudden changes as human biological fluids/tissues differ fundamentally from drug product formulations. mAbs are subjected to a macromolecule-rich matrix and undergo changes in conditions such as temperature, pH, ionic composition, and osmolality [
1]. Moreover, the stabilizing excipients administered along with the mAb degrade/diffuse away. Such changes may impact protein stability and, in turn, can have ramifications on their efficacy and/or safety profile. In recent years, in vitro models appeared as a promising tool to evaluate the stability of potential lead candidates under physiological conditions. These invitro models allow one to predict the impact on critical quality attributes upon administration into a human body compartment [
3].
Different in vitro models and analytical approaches have been developed to evaluate the in vivo stability of therapeutic proteins [
4,
5,
6,
7]. However, simulating physiological conditions and detecting the protein of interest in biological fluids is accompanied by challenges [
8,
9]. Purification techniques and/or fluorescence labeling have been successfully used to detect the target protein [
10,
11]. However, both methods can be laborious and potentially impact the intrinsic protein properties and stability. Instead, biological fluids can be substituted with buffer systems that enable analysis in fluids directly yet deviate markedly from the conditions found in vivo (e.g., phosphate-buffered saline) [
6,
12,
13]. Alternatively, stripping the neat biological fluid of macromolecules can lead to higher resemblance of the physiological conditions encountered.
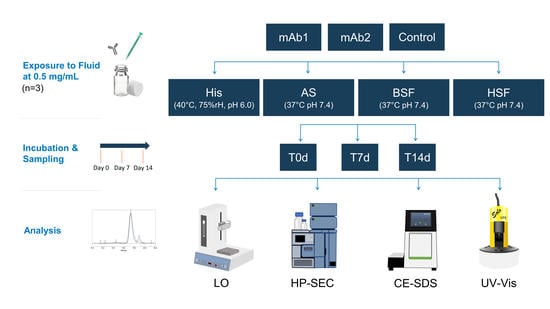
In this study, we used centrifugal ultrafiltration to strip human and bovine serum of macromolecules >3 kDa. We simulated intravenous (IV) administration and assessed the impact on protein stability by comparing human serum filtrate (HSF), bovine serum filtrate (BSF), artificial serum (AS), and histidine buffer under accelerated stability conditions. The stability of two mAbs was monitored over 14 days by light obscuration (LO), high performance-size exclusion chromatography (HP-SEC), and capillary electrophoresis–sodium dodecyl sulfate (CE-SDS).
2. Materials and Methods
2.1. Materials
Proprietary mAb1 (pI 9.1) and mAb2 (pI 8.1) were obtained from Lonza Biologics, Slough, UK and formulated at 10 mg/mL, 20 mM histidine buffer, pH 6.0. Buffers for analytical methods and in vitro models were prepared using alpha-D glucose, calcium chloride, histidine, L-histidine monohydrochloride monohydrate, potassium chloride, magnesium chloride hexahydrate, sodium bicarbonate, sodium chloride, sodium phosphate monobasic monohydrate, and sodium phosphate dibasic. All buffer components were purchased from Sigma-Aldrich (St. Louis, MO, USA). Fetal bovine serum (BS) and human serum (HS) were obtained from Gibco (ThermoFischer Scientific, Waltham, MA, USA) and Gemini Bio-Products (West Sacramento, CA, USA). Pierce™ Protein Concentrator PES, 3 kDa molecular weight (MW) cut-off, 5–20 mL was purchased from ThermoFischer Scientific. All samples were prepared and incubated in 10 mL sterile Nalgene cryoware (PETG media bottles, Sigma-Aldrich).
2.2. Preparation of Fluids
Four fluids were prepared; namely, 20 mM histidine buffer (pH 6.0), AS, BSF, and, HSF. AS and serum filtrates were prepared at pH 7.4. Sera were stripped of macromolecules by centrifugal ultrafiltration (3 h, 2000 rpm) with a 3 kDa protein concentrators. Protein concentrators were primed with 2.5 mL of MilliQ H
2O prior usage. AS contains electrolytes (127 mM Na
+, 111 mM Cl
−, 25 mM HCO
3−, 5 mM K
+, 3.75 mM Ca
2+) and 5 mM glucose to match the values provided by the vendor of human serum and the literature [
14]. CaCl
2 was added to AS after adjusting the pH to 7.4 with CO
2 to avoid Ca
3(PO
4)
2 and CaCO
3 precipitation [
15]. All fluids were filtered using a 0.22 µm PES filter (Merck Millipore, Darmstadt, Germany) before use.
2.3. Physiological In Vitro Model
Intravenous administration was simulated by spiking mAb1 and mAb2 in 4 mL of the aforementioned fluids to obtain a final concentration of 0.5 mg/mL, as used in a previous in vitro model [
16]. Blank controls were prepared by spiking each fluid with a 20 mM histidine buffer without a mAb. The 20 mM histidine buffer samples (pH 6.0) were incubated in a chamber under accelerated stability conditions, i.e., 40 °C and 75% relative humidity. AS, BSF, and, HSF samples were maintained at pH 7.4 by incubating at 37 °C and 6.5% CO
2. Samples were analyzed after 30 min (T0d), 7 days (T7d), and, 14 days (T14d) of incubation. The incubation period was selected based on previous studies [
17]. All samples and controls were characterized in regard to visible particles, pH, sub-visible particles (SbVP), protein aggregation, and, fragmentation. All samples and controls were prepared in triplicates. The values were compared to reference material (RM) of mAb1 and mAb2, stored in 20 mM histidine buffer (pH 6.0) at 5 °C.
2.4. pH, Conductivity and Osmolality
pH and conductivity were measured using SevenExcellence pH and conductivity meter (Mettler Toledo, Columbus, OH, USA). The osmolality of each fluid was determined using a freezing point OsmoPRO micro-osmometer (Advanced instruments, Norwood, MA, USA).
2.5. Visible Particles
Each sample and control were inspected for visible particles according to the method described in the European Pharmacopoeia (2.9.20).
2.6. Light Obscuration
SbVPs were analyzed using HIAC 9703+ equipped with a HRLD-400 detector (Beckman Coulter, Brea, CA, USA) and a 1 mL Hamilton syringe, at a flow rate of 10 mL/min. SbVPs ≥2, ≥5, ≥10, and, ≥25 µm in size were characterized using the LO method described in the European Pharmacopoeia (2.9.19); however, with a modified volume of 0.2 mL. Each sample was measured four times and the data from last three measurements were averaged and reported. Data was acquired using the PharmSpec software (Beckman Coulter, Brea, CA, USA). The instrument was calibrated using 5 µm COUNT-CAL Count Precision Size Standard beads (Thermo Fisher Scientific, Waltham, MA, USA).
2.7. Protein Content
The protein concentration of mAbs and fluids was determined using variable path spectroscopy. SoloVPE, C Technologies (Bridgewater Township, NJ, USA) mounted onto an Agilent Cary 60 UV–Vis spectrometer (Santa Clara, CA, USA). Samples were measured at 280 nm in fibrettes. Data was acquired by Cary WinUV software 5.0.0.1008 (Agilent Technologies, Santa Clara, CA, USA). The mAb concentration in BSF and HSF was determined by subtracting the absorbance of each blank control from the serum samples spiked with a mAb.
Protein content was also estimated using the Pierce™ Coomassie Plus (Bradford) Assay Kit (ThermoFischer Scientific, Waltham, MA, USA). For measurements, Coomassie Plus Reagent was mixed with samples and incubated for 10 min at room temperature. The absorbance of the samples was measured at 595 nm using Lico 690 spectrophotometer (Hach, Loveland, CO, USA). Protein concentration was determined using an albumin calibration curve according to the manufacturer.
2.8. High Performance Size Exclusion Chromatography
Protein monomer, high-molecular weight species (HMWS), and, low-molecular weight species (LMWS) were monitored by HP-SEC. Samples were analyzed using a TSKgel GS3000SWXL column (Tosoh Biosciences, Griesheim, Germany) and a Waters Alliance e2695 HPLC system equipped with a UV–Vis detector. The instrument was flushed with mobile phase consisting of 0.2 M sodium phosphate buffer (pH 7.0) at a flow rate of 0.5 mL/min. Samples (15 µL) were detected at 214 nm. The obtained chromatograms were processed with Empower3 software (Waters, Milford, MA, USA).
2.9. Capillary Electrophoresis–Sodium Dodecyl Sulfate
Samples were analyzed by CE-SDS using LabChip GXII (Caliper Life Sciences, Hopkinton, MA, USA). Samples were prepared using a Protein ClearTM HR Reagent kit (Caliper Life Sciences, Hopkinton, MA, USA). The reducing sample buffer was prepared by mixing 1 M dichlorodiphenyltrichloroethane with Protein Clear HR sample buffer, while the non-reducing buffer consisted of only Protein Clear HR sample buffer. Samples were mixed with reducing or non-reducing buffer and denatured at 70 °C for 10 min. Samples were centrifuged at 2000 rpm for 2 min and diluted with MilliQ H2O prior to analysis. The peaks between 16 to 250 kDa were processed using the Empower3 software (Waters).
4. Discussion
Serum/plasma is the liquid component of blood, containing thousands of proteins and low-molecular weight molecules such as peptides, electrolytes, and, amino acids [
18]. The protein concentration of 55 to 80 mg/mL in serum [
19] prevents one from monitoring the stability of protein of interest (e.g., spiked mAb). Analytical approaches to overcome this challenge include the enrichment of the protein of interest (e.g., immunoaffinity purification) [
20] or labeling (e.g., fluorescence dye) [
5,
11]. Alternatively, the biological fluid can be substituted with an appropriate surrogate buffer [
6,
12].
We present an alternative approach by stripping serum of endogenous proteins. We demonstrated that mAbs can be analyzed directly in serum filtrates without additional sample work-up procedures. The Bradford assay did not allow one to determine the protein content, presumably because molecules <3 kDa such as small peptides do not bind to the dye [
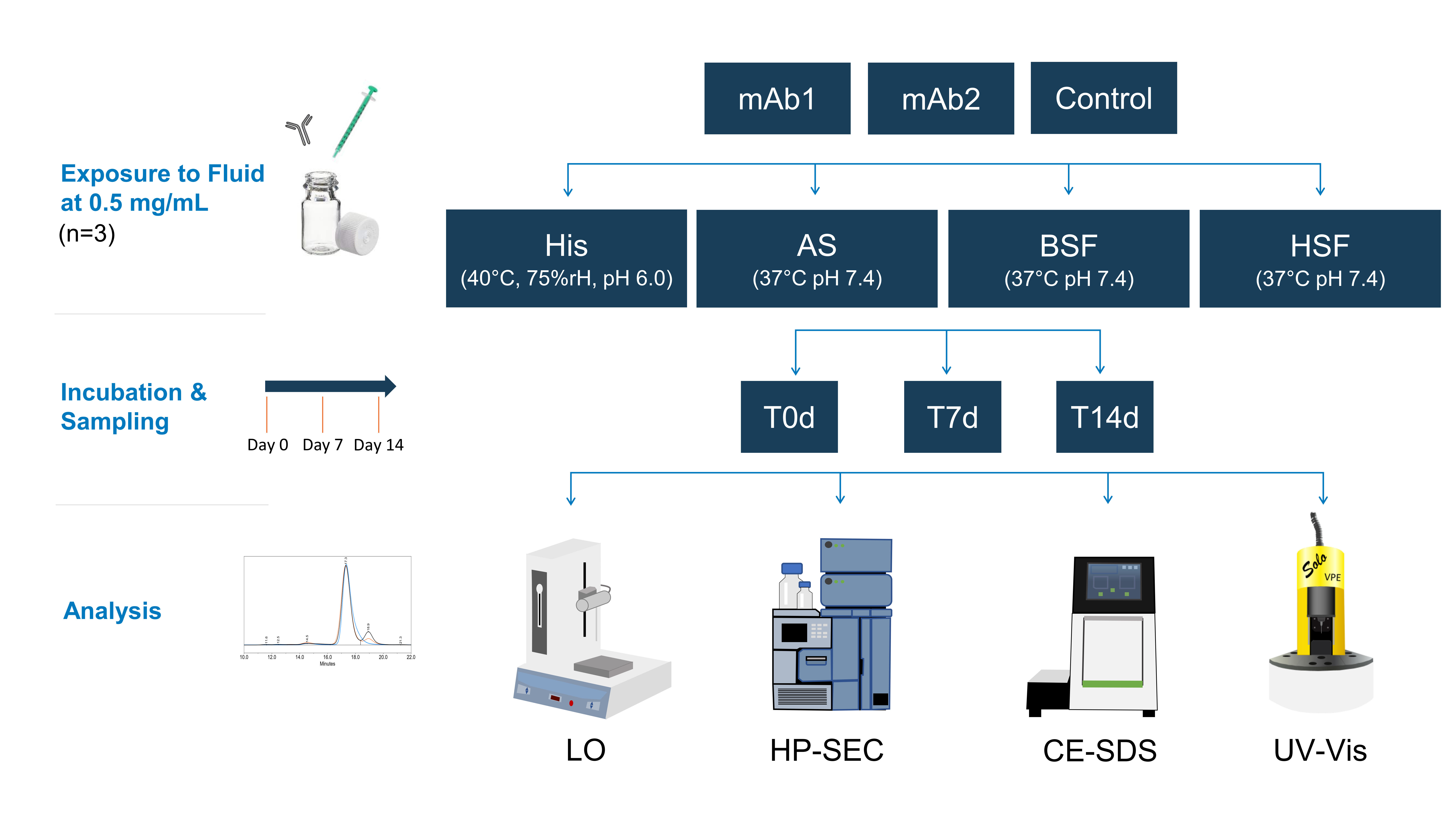
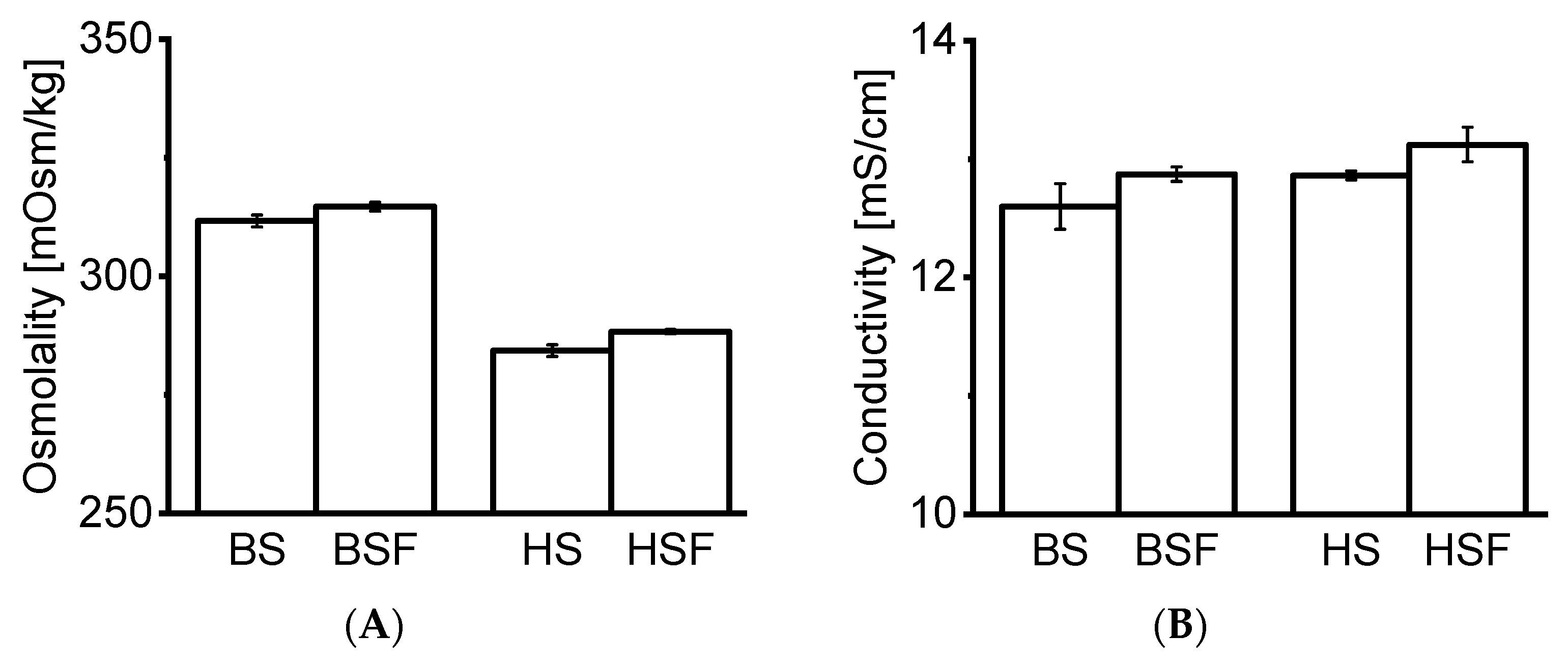
21]. Precise protein concentration measurement via spectroscopy was not possible as the extinction coefficient of the remaining macromolecules in the filtrate was unknown. HP-SEC data indicated the presence of molecules of approx. ≤ 1–3 kDa in serum blanks, most likely peptides and aromatic amino acids. This is in agreement with our CE-SDS data, as no matrix interference from serum blanks was observed. Osmolality and conductivity values of the serum filtrates were negligibly higher than in neat serum, indicating that the electrolyte concentration remained similar. The composition of the serum filtrates was not further characterized as it was out of the scope of this study.
Kinderman et al. showed that protein candidates with poor colloidal stability under physiological conditions may aggregate or precipitate in vivo and thus, particles (visible and sub-visible) should be assessed during pre-clinical development [
22]. During administration, a protein experiences sudden changes from the formulation conditions to those encountered in patients. This sudden transition can be simulated by pH jump assays in vitro [
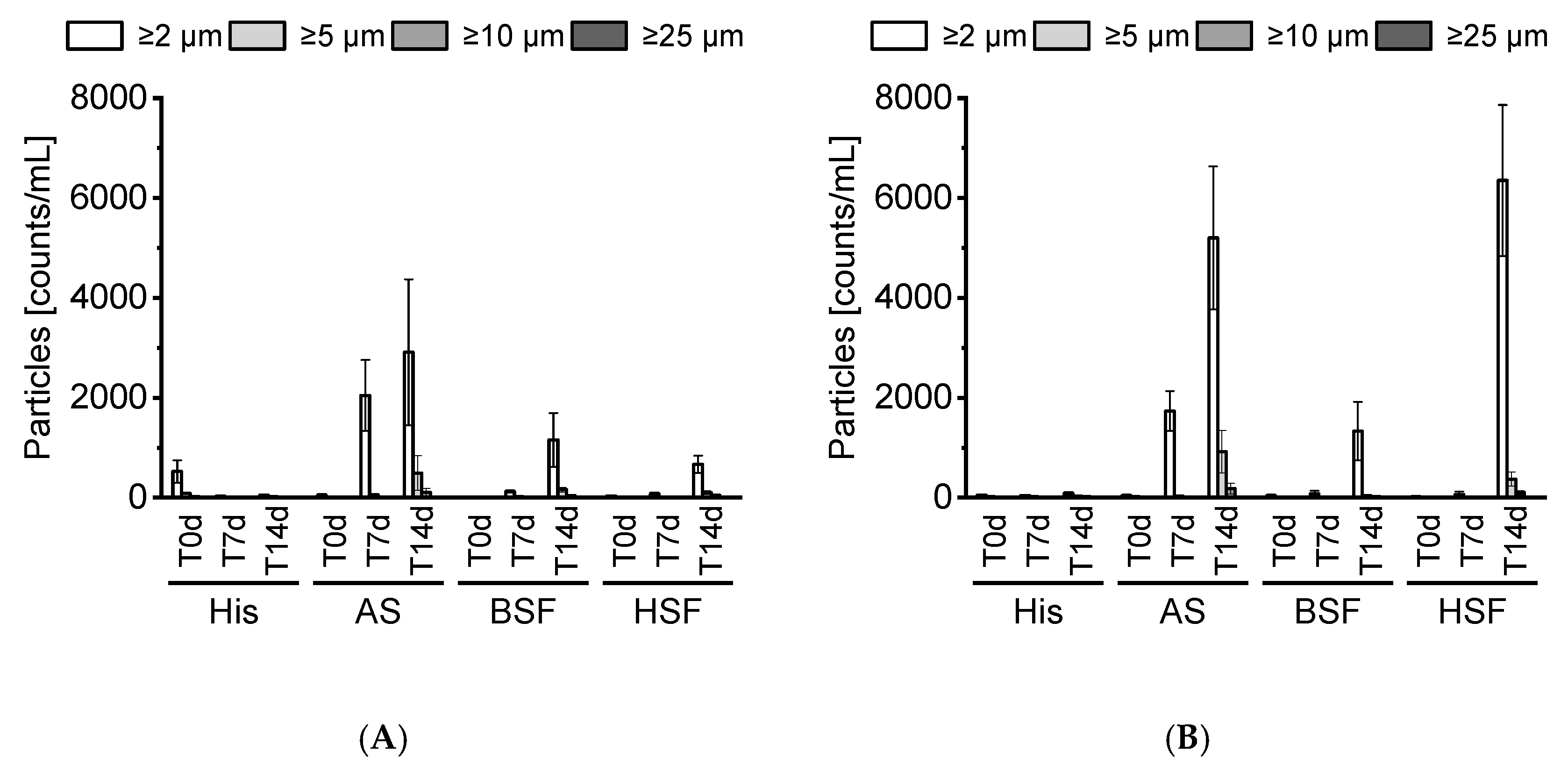
22]. In this study, the tested mAbs formed no visible particles. SbVPs were detected by LO under simulated physiological conditions. mAb2 appeared less stable compared to mAb1 and formed substantial SbVPs in HSF over time. The protein-free sera enabled the inspection of particles, which is typically not possible with biological fluids due to their poor stability in vitro [
5,
13,
23]. Our prepared protein-free sera remained stable over 14 days and displayed a negligible particle count.
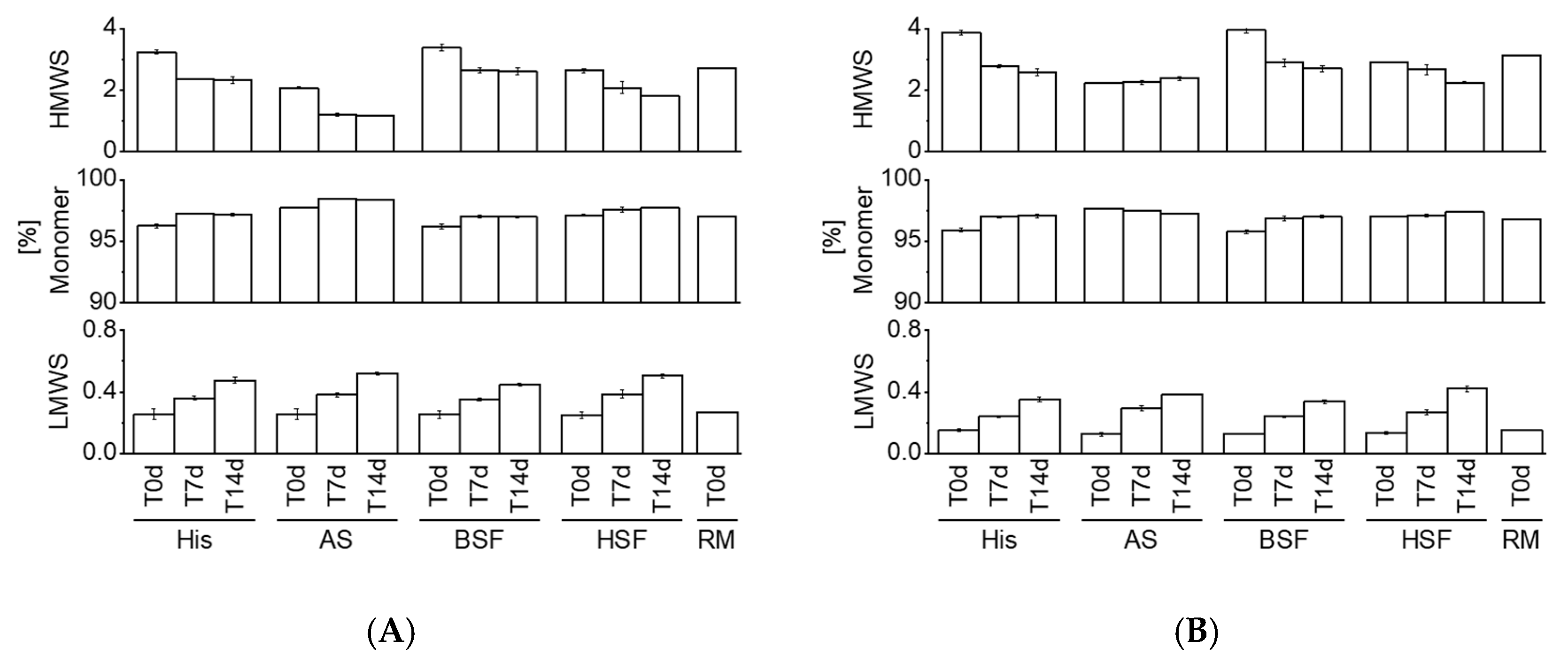
HP-SEC data showed a consistent increase in LMWS of both mAbs in all fluids over time. HMWS decreased consistently over time with the exception of mAb2 which remained within the same range in AS over 14 days. Previous studies reported an increase in HMWS under simulated physiological conditions [
5,
12]. Comparison to other studies is challenging due to differences in the analytical approach, protein concentration, fluids, sampling pull points, and, molecules. Our results suggest that certain protein aggregates may dissolve under physiological conditions. A variety of factors such as ionic composition, temperature, pH, etc. could contribute to the fate of protein aggregates in vivo [
1]. Knowledge on in vivo protein aggregation is generally limited as it remains unknown if administered particles (e.g., formed over the shelf-life duration or during clinical preparation) dissolve in vivo or persist. Future studies could aim at exploring if the nature of protein particles formed during manufacturing and/or clinical preparation impacts the fate in vivo. A previous study by Filipe et al. reported dissimilar protein aggregation behavior in serum by comparing particles generated under different stress conditions [
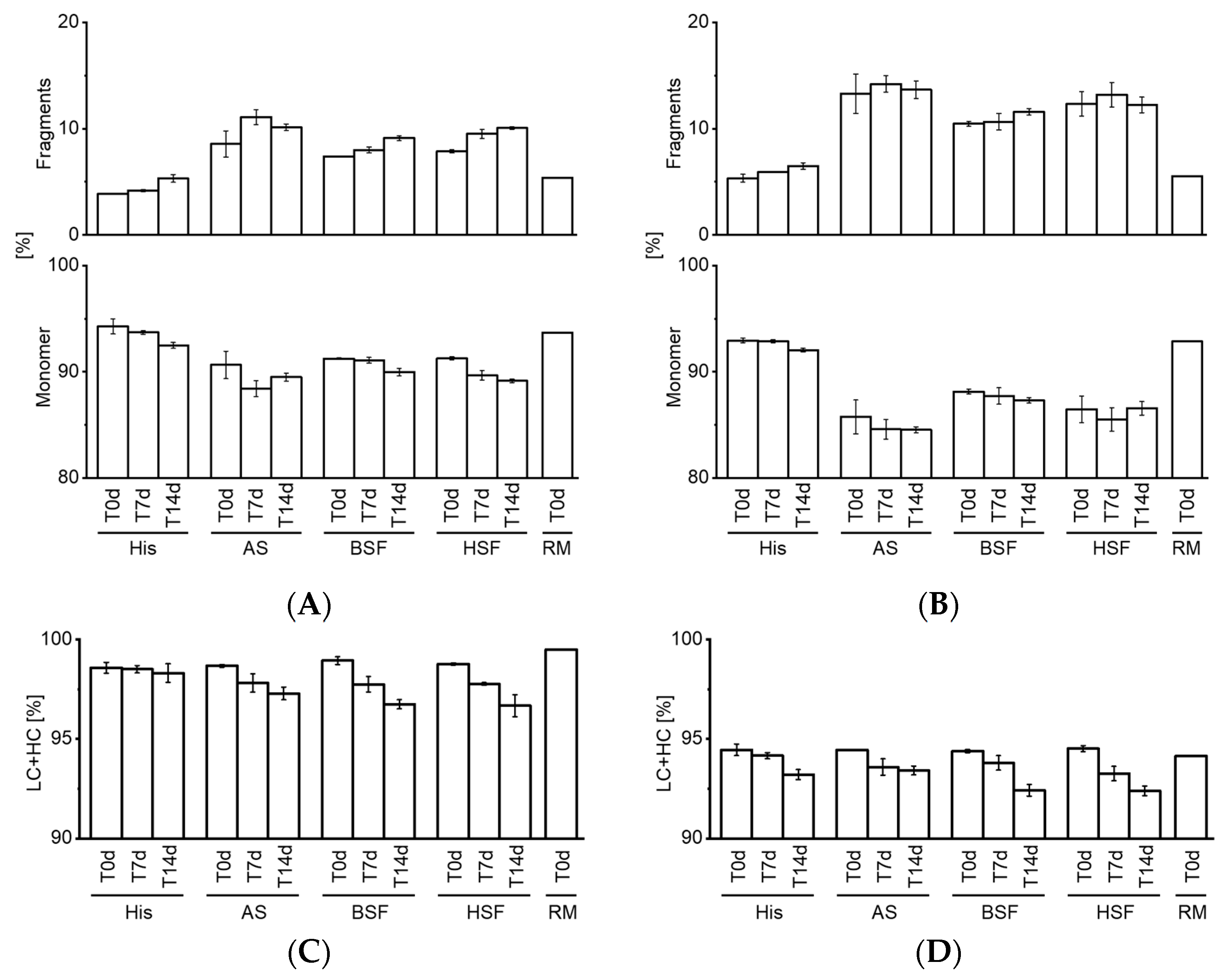
7]. Our CE-SDS results confirmed an increase in fragments in all fluids over time. Interestingly, comparing the RM to T0d values showed that protein fragments increased strikingly upon exposure to AS, BSF, and, HSF. This increase at T0d was not observed under accelerated stability conditions. Fragmentation was also more pronounced in AS, BSF, and, HSF compared to accelerated stability conditions.
Both mAbs degraded to a similar extent under physiological conditions, whereas they remained fairly stable under accelerated stability conditions. Particularly, SbVP formation and protein fragmentation were dissimilar under accelerated stability conditions and simulated physiological conditions. This reaffirms the importance of assessing the in vivo protein stability under a simulated physiological environment. Despite the differences in SbVP formation of mAbs in HSF, no relevant differences were observed between AS, BSF, and, HSF. Although all fluids remained stable, the cloudy appearance of BSF could hamper certain analyses (e.g., visible particle inspection) and appeared less suitable as a physiological in vitro model compared to AS and HSF. Artificial fluids such as AS may form SbVPs due to salt precipitation and therefore would be less applicable for particle analysis [
12,
15]. Fluids such as AS and the prepared filtrates, which are devoid of macromolecules, enable direct analysis in fluids; however, the matrix composition and osmolality of the prepared filtrates are more reflective of the physiological environment compared to AS. By stripping molecules based on molecular weight, theoretically, certain endogenous molecules such as peptides, nutrients (amino acids, lipids, carbohydrates), electrolytes, as well as small organic molecules and thiols remain in the filtrate [
14]. Compared to neat serum, fluids devoid of cells and macromolecules such as enzymes may underestimate the impact on a protein’s stability.
Selecting a higher MW cut-off (e.g., 50 kDa) may retain certain serum proteases which can impact protein degradation. While this improves the physiological relevance of the model, the presence of enzymes could accelerate protein degradation. On the other hand, the presence of macromolecules can limit the stability of the fluid itself and cause analytical interference [
5,
13]. Thus, depending on the analytical methods used, future studies may select a higher MW cut-off to increase the physiological relevance while still allowing analysis in the fluids directly. Our developed protein-free serum remained stable over two weeks and may enable even longer incubation periods to detect protein liabilities among candidates. Moreover, avoiding sample work-up procedures such as purification or labeling is advantageous as they can be laborious and may alter the stability of a therapeutic protein. Overall, AS and protein-stripped serum appeared suitable to screen protein liabilities in vitro.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




