
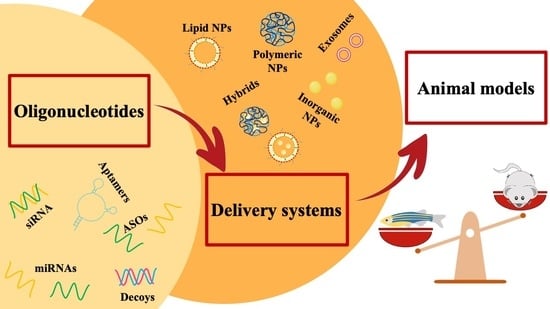
Nanoparticles-Based Oligonucleotides Delivery in Cancer: Role of Zebrafish as Animal Model


Abstract
:
1. Introduction
2. ON Therapeutics
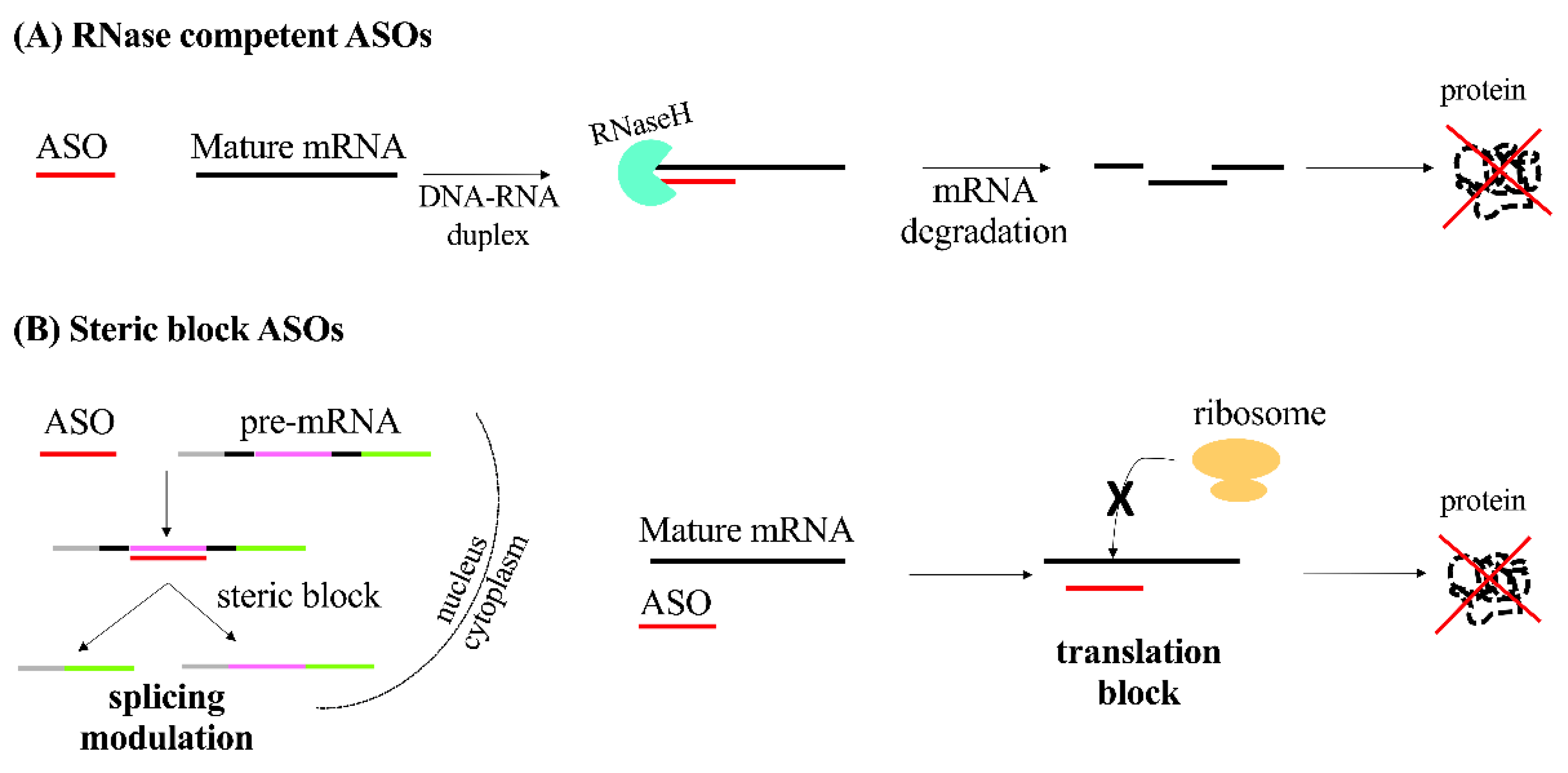
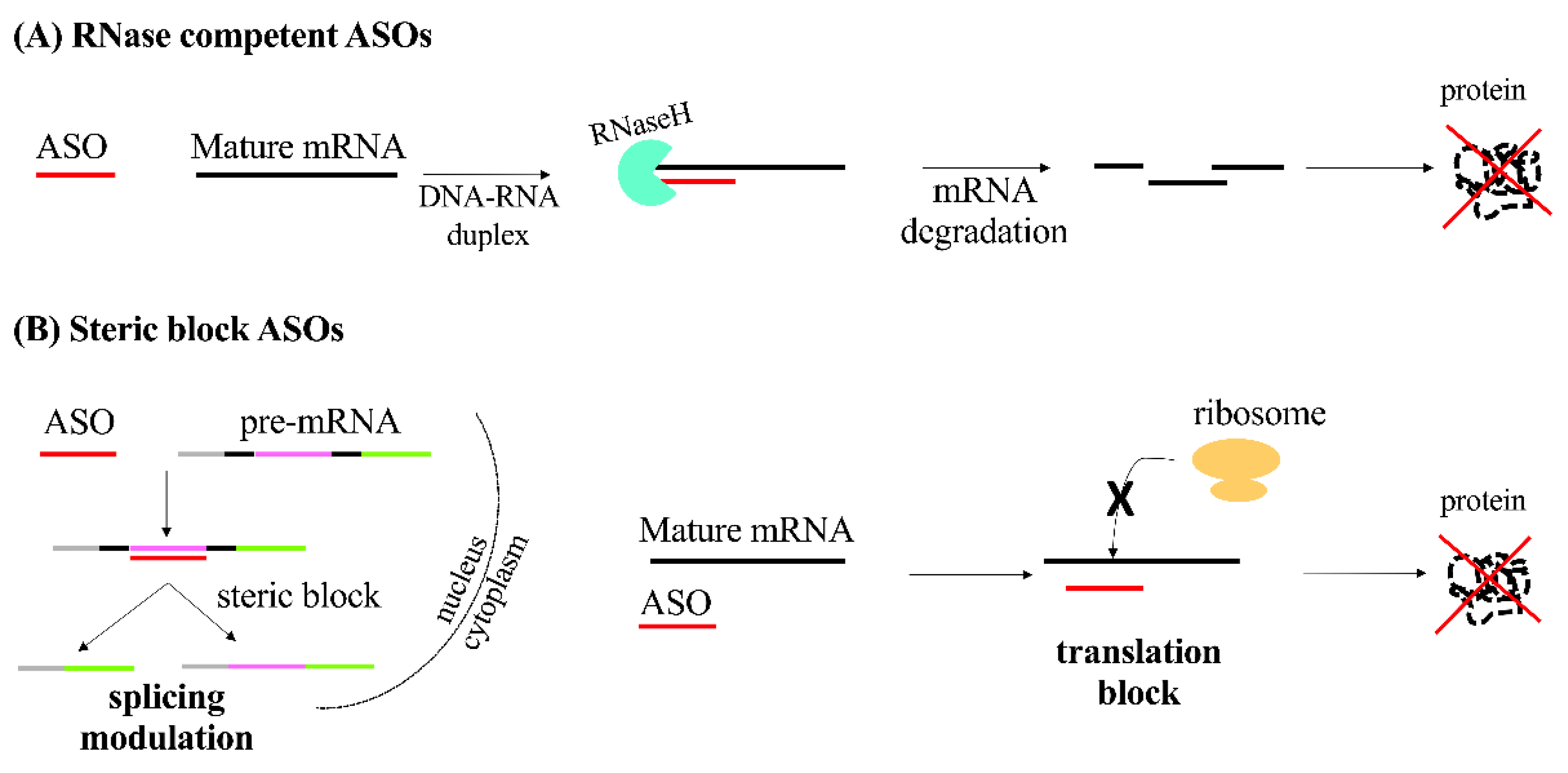
2.1. Antisense Oligonucleotides (ASOs)
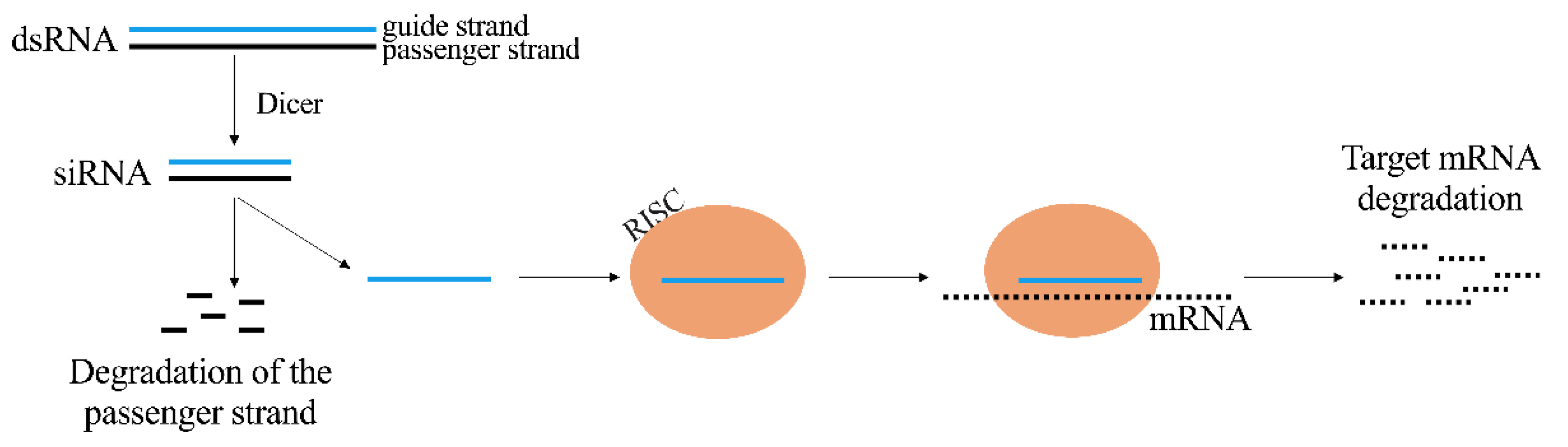
2.2. siRNA
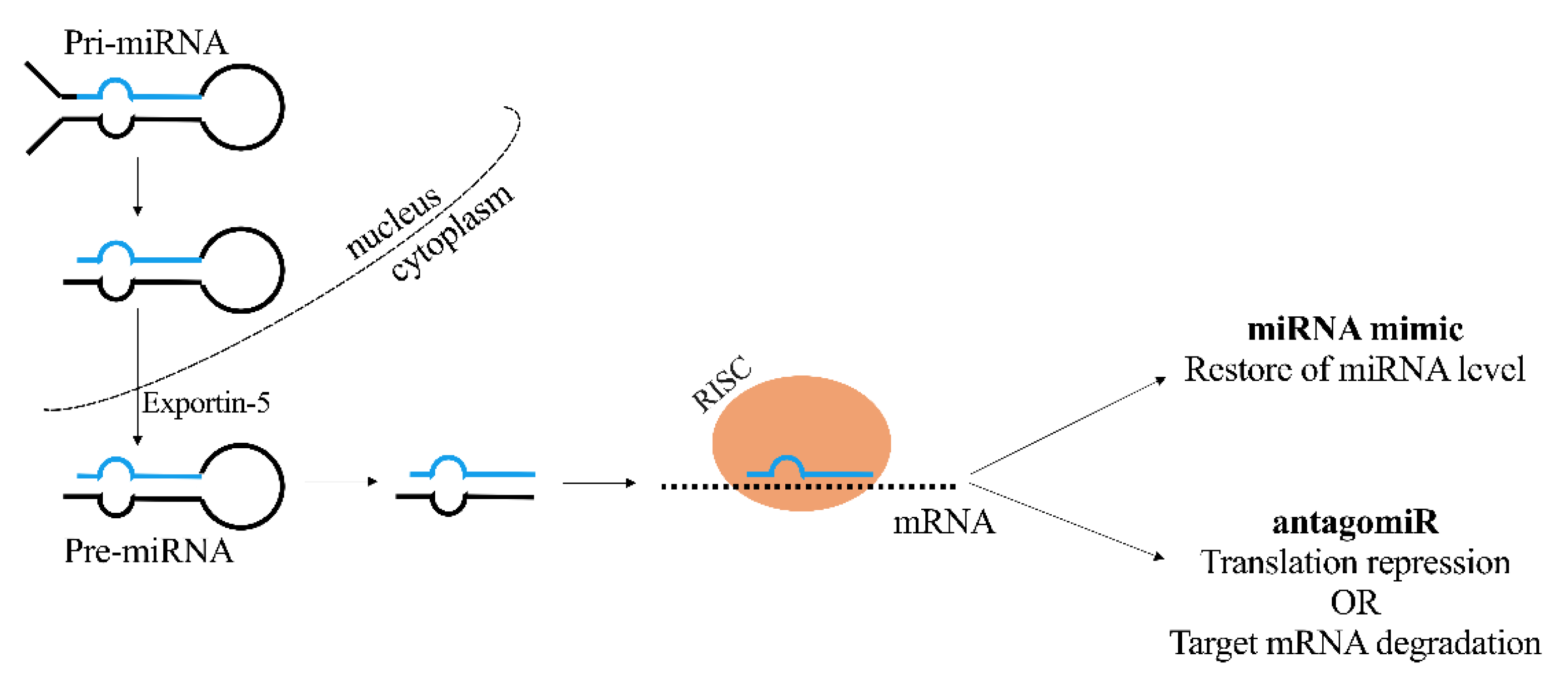
2.3. miRNAs
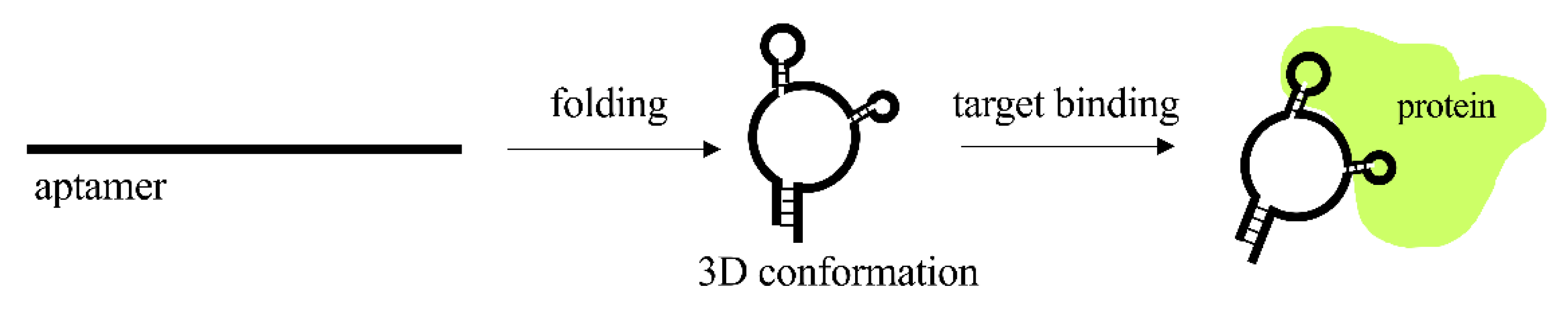
2.4. Aptamers
2.5. Decoys
3. Platforms for the Delivery of Oligonucleotides (ONs)
3.1. Lipid Nanoparticles
3.2. Polymeric Nanoparticles
3.3. Inorganic Nanoparticles
3.4. Exosomes
3.5. Hybrids
4. Zebrafish (ZF) as an Animal Model to Mimic Human Cancer
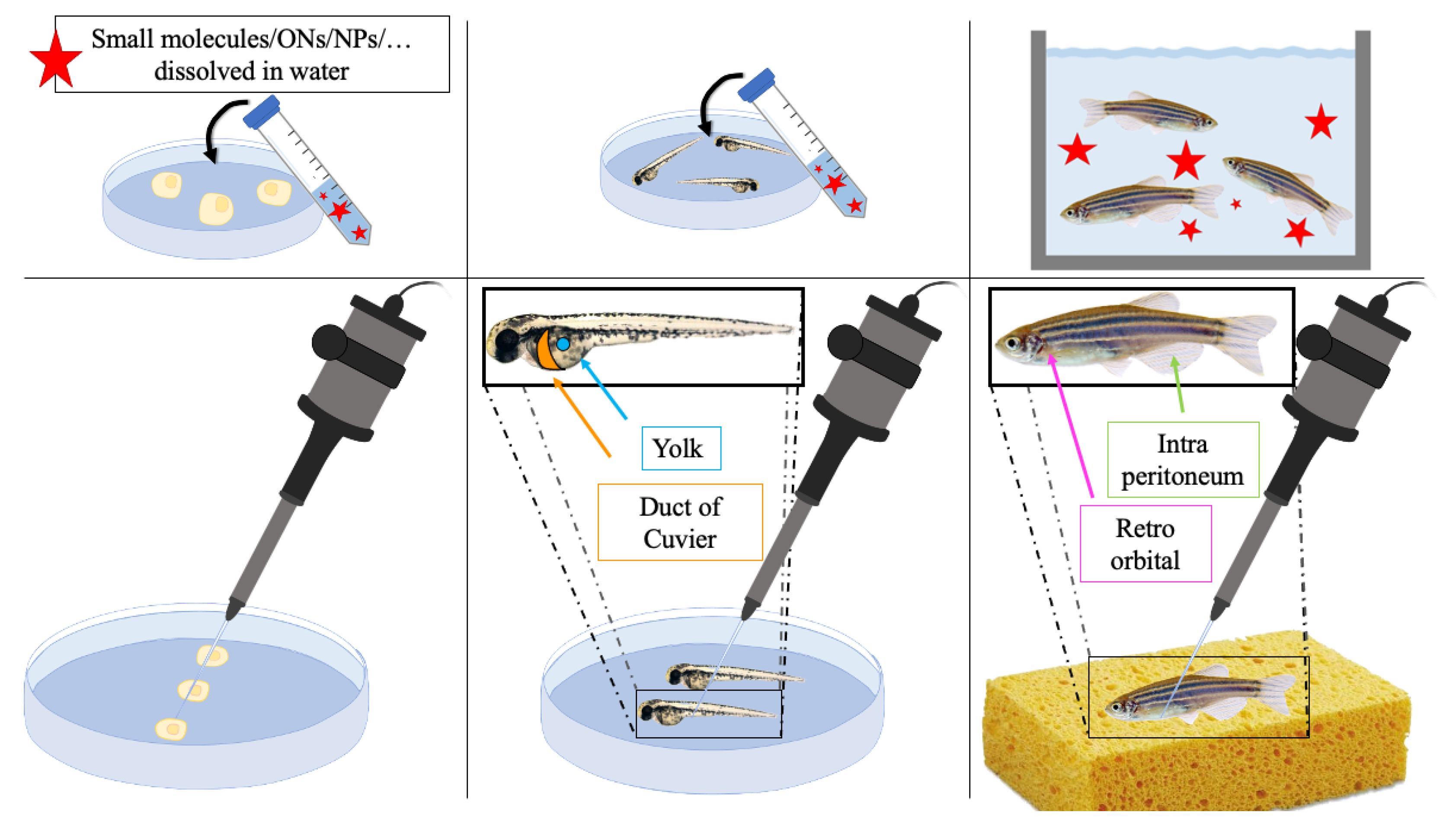
ZF for In Vivo Characterization of New Drugs
5. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Scherman, D.; Rousseau, A.; Bigey, P.; Escriou, V. Genetic Pharmacology: Progresses in SiRNA Delivery and Therapeutic Applications. Gene Ther. 2017, 24, 151–156. [Google Scholar] [CrossRef]
- Xiong, H.; Veedu, R.N.; Diermeier, S.D. Recent Advances in Oligonucleotide Therapeutics in Oncology. Int. J. Mol. Sci. 2021, 22, 3295. [Google Scholar] [CrossRef]
- Smith, C.; Zain, R. Therapeutic Oligonucleotides: State of the Art. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 605–630. [Google Scholar] [CrossRef]
- Wittrup, A.; Lieberman, J. Knocking down Disease: A Progress Report on SiRNA Therapeutics. Nat. Rev. Genet. 2015, 16, 543–552. [Google Scholar] [CrossRef]
- Lorenzer, C.; Dirin, M.; Winkler, A.-M.; Baumann, V.; Winkler, J. Going beyond the Liver: Progress and Challenges of Targeted Delivery of SiRNA Therapeutics. J. Controlled Release 2015, 203, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Giudice, V.; Mensitieri, F.; Izzo, V.; Filippelli, A.; Selleri, C. Aptamers and Antisense Oligonucleotides for Diagnosis and Treatment of Hematological Diseases. Int. J. Mol. Sci. 2020, 21, 3252. [Google Scholar] [CrossRef]
- Statello, L.; Ali, M.; Kanduri, C. In Vivo Administration of Therapeutic Antisense Oligonucleotides. Methods Mol. Biol. 2021, 2254, 273–282. [Google Scholar] [CrossRef]
- Wu, H.; Lima, W.F.; Zhang, H.; Fan, A.; Sun, H.; Crooke, S.T. Determination of the Role of the Human RNase H1 in the Pharmacology of DNA-like Antisense Drugs. J. Biol. Chem. 2004, 279, 17181–17189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Vickers, T.A.; Sun, H.; Liang, X.; Crooke, S.T. Binding of Phosphorothioate Oligonucleotides with RNase H1 Can Cause Conformational Changes in the Protein and Alter the Interactions of RNase H1 with Other Proteins. Nucleic Acids Res. 2021, 49, 2721–2739. [Google Scholar] [CrossRef] [PubMed]
- Monia, B.P.; Lesnik, E.A.; Gonzalez, C.; Lima, W.F.; McGee, D.; Guinosso, C.J.; Kawasaki, A.M.; Cook, P.D.; Freier, S.M. Evaluation of 2′-Modified Oligonucleotides Containing 2′-Deoxy Gaps as Antisense Inhibitors of Gene Expression. J. Biol. Chem. 1993, 268, 14514–14522. [Google Scholar] [CrossRef]
- Larrouy, B.; Bolziau, C.; Sproat, B.; Toulmé, J.-J. RNase H Is Responsible for the Non-Specific Inhibition of in Vitro Translation by 2′-O-Alkyl Chimeric Oligonucleotides: High Affinity or Selectivity, a Dilemma to Design Antisense Oligomers. Nucleic Acids Res. 1995, 23, 3434–3440. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.-H.; Sun, H.; Nichols, J.G.; Crooke, S.T. RNase H1-Dependent Antisense Oligonucleotides Are Robustly Active in Directing RNA Cleavage in Both the Cytoplasm and the Nucleus. Mol. Ther. 2017, 25, 2075–2092. [Google Scholar] [CrossRef] [Green Version]
- Dominski, Z.; Kole, R. Restoration of Correct Splicing in Thalassemic Pre-MRNA by Antisense Oligonucleotides. Proc. Natl. Acad. Sci. USA 1993, 90, 8673–8677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.N.; Singh, N.N. Mechanism of splicing regulation of spinal muscular atrophy genes. In RNA Metabolism in Neurodegenerative Diseases; Sattler, R., Donnelly, C.J., Eds.; Advances in Neurobiology; Springer International Publishing: Cham, Switzerland, 2018; Volume 20, pp. 31–61. [Google Scholar] [CrossRef]
- Wan, L.; Dreyfuss, G. Splicing-Correcting Therapy for SMA. Cell 2017, 170, 5. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Straub, V.; Hemmings, R.; Haas, M.; Schlosser-Weber, G.; Stoyanova-Beninska, V.; Mercuri, E.; Muntoni, F.; Sepodes, B.; Vroom, E.; et al. Development of Exon Skipping Therapies for Duchenne Muscular Dystrophy: A Critical Review and a Perspective on the Outstanding Issues. Nucleic Acid Ther. 2017, 27, 251–259. [Google Scholar] [CrossRef]
- Baker, B.F.; Lot, S.S.; Condon, T.P.; Cheng-Flournoy, S.; Lesnik, E.A.; Sasmor, H.M.; Bennett, C.F. 2′-O-(2-Methoxy)Ethyl-Modified Anti-Intercellular Adhesion Molecule 1 (ICAM-1) Oligonucleotides Selectively Increase the ICAM-1 MRNA Level and Inhibit Formation of the ICAM-1 Translation Initiation Complex in Human Umbilical Vein Endothelial Cells. J. Biol. Chem. 1997, 272, 11994–12000. [Google Scholar] [CrossRef] [Green Version]
- Boiziau, C.; Kurfurst, R.; Cazenave, C.; Roig, V.; Thuong, N.T.; Toulmé, J.-J. Inhibition of Translation Initiation by Antisense Oligonucleotides via an RNase-H Independent Mechanism. Nucleic Acids Res. 1991, 19, 1113–1119. [Google Scholar] [CrossRef] [Green Version]
- Calvo, S.E.; Pagliarini, D.J.; Mootha, V.K. Upstream Open Reading Frames Cause Widespread Reduction of Protein Expression and Are Polymorphic among Humans. Proc. Natl. Acad. Sci. USA 2009, 106, 7507–7512. [Google Scholar] [CrossRef] [Green Version]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-Nucleotide RNAs Mediate RNA Interference in Cultured Mammalian Cells. Nature 2001, 411, 494–498. [Google Scholar] [CrossRef]
- Liu, J.; Carmell, M.A.; Rivas, F.V.; Marsden, C.G.; Thomson, J.M.; Song, J.-J.; Hammond, S.M.; Joshua-Tor, L.; Hannon, G.J. Argonaute2 Is the Catalytic Engine of Mammalian RNAi. Science 2004, 305, 1437–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, T.C. The microRNA machinery. In microRNA: Basic Science; Santulli, G., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2015; Volume 887, pp. 15–30. ISBN 978-3-319-22379-7. [Google Scholar] [CrossRef]
- Schürmann, N.; Trabuco, L.G.; Bender, C.; Russell, R.B.; Grimm, D. Molecular Dissection of Human Argonaute Proteins by DNA Shuffling. Nat. Struct. Mol. Biol. 2013, 20, 818–826. [Google Scholar] [CrossRef]
- Kim, D.-H.; Behlke, M.A.; Rose, S.D.; Chang, M.-S.; Choi, S.; Rossi, J.J. Synthetic DsRNA Dicer Substrates Enhance RNAi Potency and Efficacy. Nat. Biotechnol. 2005, 23, 222–226. [Google Scholar] [CrossRef] [Green Version]
- Bramsen, J.B.; Laursen, M.B.; Damgaard, C.K.; Lena, S.W.; Ravindra Babu, B.; Wengel, J.; Kjems, J. Improved Silencing Properties Using Small Internally Segmented Interfering RNAs. Nucleic Acids Res. 2007, 35, 5886–5897. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Pendergraff, H.; Liu, J.; Kordasiewicz, H.B.; Cleveland, D.W.; Swayze, E.E.; Lima, W.F.; Crooke, S.T.; Prakash, T.P.; Corey, D.R. Single-Stranded RNAs Use RNAi to Potently and Allele-Selectively Inhibit Mutant Huntingtin Expression. Cell 2012, 150, 895–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrne, M.; Tzekov, R.; Wang, Y.; Rodgers, A.; Cardia, J.; Ford, G.; Holton, K.; Pandarinathan, L.; Lapierre, J.; Stanney, W.; et al. Novel Hydrophobically Modified Asymmetric RNAi Compounds (Sd-RxRNA) Demonstrate Robust Efficacy in the Eye. J. Ocul. Pharmacol. Ther. 2013, 29, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Liang, D.; Chen, C.; Tang, X. Caged SiRNAs with Single CRGD Modification for Photoregulation of Exogenous and Endogenous Gene Expression in Cells and Mice. Biomacromolecules 2018, 19, 2526–2534. [Google Scholar] [CrossRef]
- Alterman, J.F.; Godinho, B.M.D.C.; Hassler, M.R.; Ferguson, C.M.; Echeverria, D.; Sapp, E.; Haraszti, R.A.; Coles, A.H.; Conroy, F.; Miller, R.; et al. A Divalent SiRNA Chemical Scaffold for Potent and Sustained Modulation of Gene Expression throughout the Central Nervous System. Nat. Biotechnol. 2019, 37, 884–894. [Google Scholar] [CrossRef] [Green Version]
- Krützfeldt, J.; Rajewsky, N.; Braich, R.; Rajeev, K.G.; Tuschl, T.; Manoharan, M.; Stoffel, M. Silencing of MicroRNAs In Vivo with ‘Antagomirs’. Nature 2005, 438, 685–689. [Google Scholar] [CrossRef]
- Krützfeldt, J.; Kuwajima, S.; Braich, R.; Rajeev, K.G.; Pena, J.; Tuschl, T.; Manoharan, M.; Stoffel, M. Specificity, Duplex Degradation and Subcellular Localization of Antagomirs. Nucleic Acids Res. 2007, 35, 2885–2892. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.F.; Stoval, G.M.; Ellington, A.D. Aptamer Therapeutics Advance. Curr. Opin. Chem. Biol. 2006, 10, 282–289. [Google Scholar] [CrossRef]
- Shigdar, S.; Schrand, B.; Giangrande, P.H.; de Franciscis, V. Aptamers: Cutting Edge of Cancer Therapies. Mol. Ther. 2021. [Google Scholar] [CrossRef]
- Byun, J. Recent Progress and Opportunities for Nucleic Acid Aptamers. Life 2021, 11, 193. [Google Scholar] [CrossRef]
- Tuerk, C.; Gold, L. Systematic Evolution of Ligands by Exponential Enrichment: RNA Ligands to Bacteriophage T4 DNA Polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef]
- Robertson, D.; Joyce, G. Selection In Vitro of an RNA Enzyme That Specifically Cleaves Single-Stranded DNA. Nature 1990, 344, 467–468. [Google Scholar] [CrossRef] [PubMed]
- Ellington, A.D.; Szostak, J.W. In Vitro Selection of RNA Molecules That Bind Specific Ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Oberthür, D.; Achenbach, J.; Gabdulkhakov, A.; Buchner, K.; Maasch, C.; Falke, S.; Rehders, D.; Klussmann, S.; Betzel, C. Crystal Structure of a Mirror-Image L-RNA Aptamer (Spiegelmer) in Complex with the Natural L-Protein Target CCL2. Nat. Commun. 2015, 6, 6923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mann, M.J. Transcription Factor Decoys: A New Model for Disease Intervention. Ann. N. Y. Acad. Sci. 2005, 1058, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Hecker, M.; Wagner, A.H. Transcription Factor Decoy Technology: A Therapeutic Update. Biochem. Pharmacol. 2017, 144, 29–34. [Google Scholar] [CrossRef]
- Juliano, R.L. Intracellular Trafficking and Endosomal Release of Oligonucleotides: What We Know and What We Don’t. Nucleic Acid Ther. 2018, 28, 166–177. [Google Scholar] [CrossRef]
- Crooke, S.T.; Wang, S.; Vickers, T.A.; Shen, W.; Liang, X. Cellular Uptake and Trafficking of Antisense Oligonucleotides. Nat. Biotechnol. 2017, 35, 230–237. [Google Scholar] [CrossRef]
- Liang, X.; Sun, H.; Shen, W.; Stanley, T.C. Identification and Characterization of Intracellular Proteins That Bind Oligonucleotides with Phosphorothioate Linkages. Nucleic Acids Res. 2015, 43, 2927–2945. [Google Scholar] [CrossRef] [Green Version]
- Doherty, G.J.; McMahon, H.T. Mechanisms of Endocytosis. Annu. Rev. Biochem. 2009, 78, 857–902. [Google Scholar] [CrossRef] [Green Version]
- Marchese, A.; Paing, M.M.; Temple, B.R.S.; Trejo, J. G Protein–Coupled Receptor Sorting to Endosomes and Lysosomes. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 601–629. [Google Scholar] [CrossRef] [Green Version]
- Lajoie, P.; Nabi, I.R. Lipid rafts, caveolae, and their endocytosis. In International Review of Cell and Molecular Biology; Elsevier: Amsterdam, The Netherlands, 2010; Volume 282, pp. 135–163. [Google Scholar] [CrossRef]
- Alam, M.R.; Ming, X.; Dixit, V.; Fisher, M.; Chen, X.; Juliano, R.L. The Biological Effect of an Antisense Oligonucleotide Depends on Its Route of Endocytosis and Trafficking. Oligonucleotides 2010, 20, 103–109. [Google Scholar] [CrossRef] [Green Version]
- Koller, E.; Vincent, T.M.; Chappell, A.; De, S.; Manoharan, M.; Bennett, C.F. Mechanisms of Single-Stranded Phosphorothioate Modified Antisense Oligonucleotide Accumulation in Hepatocytes. Nucleic Acids Res. 2011, 39, 4795–4807. [Google Scholar] [CrossRef] [Green Version]
- Huotari, J.; Helenius, A. Endosome Maturation: Endosome Maturation. EMBO J. 2011, 30, 3481–3500. [Google Scholar] [CrossRef] [PubMed]
- Goldenring, J.R. Recycling Endosomes. Curr. Opin. Cell Biol. 2015, 35, 117–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luzio, J.P.; Pryor, P.R.; Bright, N.A. Lysosomes: Fusion and Function. Nat. Rev. Mol. Cell Biol. 2007, 8, 622–632. [Google Scholar] [CrossRef] [PubMed]
- Mariño, G.; Madeo, F.; Kroemer, G. Autophagy for Tissue Homeostasis and Neuroprotection. Curr. Opin. Cell Biol. 2011, 23, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Johannes, L.; Wunder, C. Retrograde Transport: Two (or More) Roads Diverged in an Endosomal Tree? Traffic 2011, 12, 956–962. [Google Scholar] [CrossRef] [PubMed]
- Sandvig, K.; Skotland, T.; van Deurs, B.; Klokk, T.I. Retrograde Transport of Protein Toxins through the Golgi Apparatus. Histochem. Cell Biol. 2013, 140, 317–326. [Google Scholar] [CrossRef]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in Oligonucleotide Drug Delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef]
- Stein, C.A.; Hansen, J.B.; Lai, J.; Wu, S.; Voskresenskiy, A.; H⊘g, A.; Worm, J.; Hedtjärn, M.; Souleimanian, N.; Miller, P.; et al. Efficient Gene Silencing by Delivery of Locked Nucleic Acid Antisense Oligonucleotides, Unassisted by Transfection Reagents. Nucleic Acids Res. 2010, 38, e3. [Google Scholar] [CrossRef] [Green Version]
- Biscans, A.; Caiazzi, J.; Davis, S.; McHugh, N.; Sousa, J.; Khvorova, A. The Chemical Structure and Phosphorothioate Content of Hydrophobically Modified SiRNAs Impact Extrahepatic Distribution and Efficacy. Nucleic Acids Res. 2020, 48, 7665–7680. [Google Scholar] [CrossRef] [PubMed]
- Hammond, S.M.; Aartsma-Rus, A.; Alves, S.; Borgos, S.E.; Buijsen, R.A.M.; Collin, R.W.J.; Covello, G.; Denti, M.A.; Desviat, L.R.; Echevarría, L.; et al. Delivery of Oligonucleotide-based Therapeutics: Challenges and Opportunities. EMBO Mol. Med. 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Palmerston Mendes, L.; Pan, J.; Torchilin, V. Dendrimers as Nanocarriers for Nucleic Acid and Drug Delivery in Cancer Therapy. Molecules 2017, 22, 1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarach, P.; Janaszewska, A. Recent Advances in Preclinical Research Using PAMAM Dendrimers for Cancer Gene Therapy. Int. J. Mol. Sci. 2021, 22, 2912. [Google Scholar] [CrossRef] [PubMed]
- Moss, K.H.; Popova, P.; Hadrup, S.R.; Astakhova, K.; Taskova, M. Lipid Nanoparticles for Delivery of Therapeutic RNA Oligonucleotides. Mol. Pharm. 2019, 16, 2265–2277. [Google Scholar] [CrossRef]
- de la Fuente, I.F.; Sawant, S.S.; Tolentino, M.Q.; Corrigan, P.M.; Rouge, J.L. Viral Mimicry as a Design Template for Nucleic Acid Nanocarriers. Front. Chem. 2021, 9, 613209. [Google Scholar] [CrossRef]
- Thi, T.T.H.; Suys, E.J.A.; Lee, J.S.; Nguyen, D.H.; Park, K.D.; Truong, N.P. Lipid-Based Nanoparticles in the Clinic and Clinical Trials: From Cancer Nanomedicine to COVID-19 Vaccines. Vaccines 2021, 9, 359. [Google Scholar] [CrossRef]
- Tam, Y.; Chen, S.; Cullis, P. Advances in Lipid Nanoparticles for SiRNA Delivery. Pharmaceutics 2013, 5, 498–507. [Google Scholar] [CrossRef] [Green Version]
- Inglut, C.T.; Sorrin, A.J.; Kuruppu, T.; Vig, S.; Cicalo, J.; Ahmad, H.; Huang, H.-C. Immunological and Toxicological Considerations for the Design of Liposomes. Nanomaterials 2020, 10, 190. [Google Scholar] [CrossRef] [Green Version]
- Pilkington, E.H.; Suys, E.J.A.; Trevaskis, N.L.; Wheatley, A.K.; Zukancic, D.; Algarni, A.; Al-Wassiti, H.; Davis, T.P.; Pouton, C.W.; Kent, S.J.; et al. From Influenza to COVID-19: Lipid Nanoparticle MRNA Vaccines at the Frontiers of Infectious Diseases. Acta Biomater. 2021. [Google Scholar] [CrossRef]
- Jyotsana, N.; Sharma, A.; Chaturvedi, A.; Budida, R.; Scherr, M.; Kuchenbauer, F.; Lindner, R.; Noyan, F.; Sühs, K.-W.; Stangel, M.; et al. Lipid Nanoparticle-Mediated SiRNA Delivery for Safe Targeting of Human CML in Vivo. Ann. Hematol. 2019, 98, 1905–1918. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.; Yu, B.; Wang, X.; Lu, Y.; Schmidt, C.R.; Lee, R.J.; Lee, L.J.; Jacob, S.T.; Ghoshal, K. Cationic Lipid Nanoparticles for Therapeutic Delivery of SiRNA and MiRNA to Murine Liver Tumor. Nanomed. Nanotechnol. Biol. Med. 2013, 9, 1169–1180. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.; Liao, J.-Z.; Xiang, G.-Y.; Zhao, P.-X.; Ye, F.; Zhao, Q.; He, X.-X. MiR-101 and Doxorubicin Codelivered by Liposomes Suppressing Malignant Properties of Hepatocellular Carcinoma. Cancer Med. 2017, 6, 651–661. [Google Scholar] [CrossRef]
- Wang, L.; Liang, T.-T. CD59 Receptor Targeted Delivery of MiRNA-1284 and Cisplatin-Loaded Liposomes for Effective Therapeutic Efficacy against Cervical Cancer Cells. AMB Express 2020, 10, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Pei, J.; Kumar, D.; Sakabe, I.; Boudreau, H.; Gokhale, P.; Kasid, U. Antisense Oligonucleotides: Target Validation and Development of Systemically Delivered Therapeutic Nanoparticles. Methods Mol. Biol 2007, 361, 163–185. [Google Scholar] [CrossRef] [PubMed]
- Ling, H.; Fabbri, M.; Calin, G.A. MicroRNAs and Other Non-Coding RNAs as Targets for Anticancer Drug Development. Nat. Rev. Drug Discov. 2013, 12, 847–865. [Google Scholar] [CrossRef] [Green Version]
- Beg, M.S.; Brenner, A.J.; Sachdev, J.; Borad, M.; Kang, Y.-K.; Stoudemire, J.; Smith, S.; Bader, A.G.; Kim, S.; Hong, D.S. Phase I Study of MRX34, a Liposomal MiR-34a Mimic, Administered Twice Weekly in Patients with Advanced Solid Tumors. Investig. New Drugs 2017, 35, 180–188. [Google Scholar] [CrossRef]
- Hong, D.S.; Kang, Y.-K.; Borad, M.; Sachdev, J.; Ejadi, S.; Lim, H.Y.; Brenner, A.J.; Park, K.; Lee, J.-L.; Kim, T.-Y.; et al. Phase 1 Study of MRX34, a Liposomal MiR-34a Mimic, in Patients with Advanced Solid Tumours. Br. J. Cancer 2020, 122, 1630–1637. [Google Scholar] [CrossRef]
- Wang, Y.; Miao, L.; Satterlee, A.; Huang, L. Delivery of Oligonucleotides with Lipid Nanoparticles. Adv. Drug Deliv. Rev. 2015, 87, 68–80. [Google Scholar] [CrossRef] [Green Version]
- Thomas, T.J.; Tajmir-Riahi, H.-A.; Pillai, C.K.S. Biodegradable Polymers for Gene Delivery. Molecules 2019, 24, 3744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gómez-Aguado, I.; Rodríguez-Castejón, J.; Vicente-Pascual, M.; Rodríguez-Gascón, A.; Solinís, M.Á.; del Pozo-Rodríguez, A. Nanomedicines to Deliver MRNA: State of the Art and Future Perspectives. Nanomaterials 2020, 10, 364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conte, R.; Valentino, A.; Di Cristo, F.; Peluso, G.; Cerruti, P.; Di Salle, A.; Calarco, A. Cationic Polymer Nanoparticles-Mediated Delivery of MiR-124 Impairs Tumorigenicity of Prostate Cancer Cells. Int. J. Mol. Sci. 2020, 21, 869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, G.; Zhu, Y.; Jing, A.; Wang, J.; Hu, F.; Feng, W.; Xiao, Z.; Chen, B. Cationic MicroRNA-Delivering Nanocarriers for Efficient Treatment of Colon Carcinoma in Xenograft Model. Gene Ther. 2016, 23, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Truong, N.P.; Jia, Z.; Burgess, M.; Payne, L.; McMillan, N.A.J.; Monteiro, M.J. Self-Catalyzed Degradable Cationic Polymer for Release of DNA. Biomacromolecules 2011, 12, 3540–3548. [Google Scholar] [CrossRef]
- Truong, N.P.; Gu, W.; Prasadam, I.; Jia, Z.; Crawford, R.; Xiao, Y.; Monteiro, M.J. An Influenza Virus-Inspired Polymer System for the Timed Release of SiRNA. Nat. Commun. 2013, 4, 1902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, Y.; Kang, C.-S.; Yuan, X.-B.; Zhou, X.; Xu, P.; Han, L.; Wang, G.X.; Jia, Z.; Zhong, Y.; Yu, S.; et al. Co-Delivery of as-MiR-21 and 5-FU by Poly(Amidoamine) Dendrimer Attenuates Human Glioma Cell Growth in Vitro. J. Biomater. Sci. Polym. Ed. 2010, 21, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Rajasekaran, D.; Srivastava, J.; Ebeid, K.; Gredler, R.; Akiel, M.; Jariwala, N.; Robertson, C.L.; Shen, X.-N.; Siddiq, A.; Fisher, P.B.; et al. Combination of Nanoparticle-Delivered SiRNA for Astrocyte Elevated Gene-1 (AEG-1) and All-Trans. Retinoic Acid (ATRA): An Effective Therapeutic Strategy for Hepatocellular Carcinoma (HCC). Bioconjug. Chem. 2015, 26, 1651–1661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizeq, B.R.; Younes, N.N.; Rasool, K.; Nasrallah, G.K. Synthesis, Bioapplications, and Toxicity Evaluation of Chitosan-Based Nanoparticles. Int. J. Mol. Sci. 2019, 20, 5776. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Tan, Y.F.; Wong, Y.S.; Liew, M.W.J.; Venkatraman, S. Recent Advances in Chitosan-Based Carriers for Gene Delivery. Mar. Drugs 2019, 17, 381. [Google Scholar] [CrossRef] [Green Version]
- Tezgel, Ö.; Szarpak-Jankowska, A.; Arnould, A.; Auzély-Velty, R.; Texier, I. Chitosan-Lipid Nanoparticles (CS-LNPs): Application to SiRNA Delivery. J. Colloid Interface Sci. 2018, 510, 45–56. [Google Scholar] [CrossRef]
- Kaban, K.; Salva, E.; Akbuga, J. In Vitro Dose Studies on Chitosan Nanoplexes for MicroRNA Delivery in Breast Cancer Cells. Nucleic Acid Ther. 2017, 27, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Cosco, D.; Cilurzo, F.; Maiuolo, J.; Federico, C.; Di Martino, M.T.; Cristiano, M.C.; Tassone, P.; Fresta, M.; Paolino, D. Delivery of MiR-34a by Chitosan/PLGA Nanoplexes for the Anticancer Treatment of Multiple Myeloma. Sci. Rep. 2015, 5, 17579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Y.; Jiang, Z.; Saha, K.; Kim, C.S.; Kim, S.T.; Landis, R.F.; Rotello, V.M. Gold Nanoparticles for Nucleic Acid Delivery. Mol. Ther. 2014, 22, 1075–1083. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Gupta, S.; Fitzgerald, T.J.; Bogdanov, A.A. Dual Radiosensitization and Anti-STAT3 Anti-Proliferative Strategy Based on Delivery of Gold Nanoparticle-Oligonucleotide Nanoconstructs to Head and Neck Cancer Cells. Nanotheranostics 2018, 2, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crew, E.; Rahman, S.; Razzak-Jaffar, A.; Mott, D.; Kamundi, M.; Yu, G.; Tchah, N.; Lee, J.; Bellavia, M.; Zhong, C.-J. MicroRNA Conjugated Gold Nanoparticles and Cell Transfection. Anal. Chem. 2012, 84, 26–29. [Google Scholar] [CrossRef]
- Gigante, A.; Li, M.; Junghänel, S.; Hirschhäuser, C.; Knauer, S.; Schmuck, C. Non-Viral Transfection Vectors: Are Hybrid Materials the Way Forward? MedChemComm 2019, 10, 1692–1718. [Google Scholar] [CrossRef] [PubMed]
- Shahabipour, F.; Barati, N.; Johnston, T.P.; Derosa, G.; Maffioli, P.; Sahebkar, A. Exosomes: Nanoparticulate Tools for RNA Interference and Drug Delivery. J. Cell. Physiol. 2017, 232, 1660–1668. [Google Scholar] [CrossRef] [Green Version]
- Syn, N.L.; Wang, L.; Chow, E.K.-H.; Lim, C.T.; Goh, B.-C. Exosomes in Cancer Nanomedicine and Immunotherapy: Prospects and Challenges. Trends Biotechnol. 2017, 35, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Kase, Y.; Uzawa, K.; Wagai, S.; Yoshimura, S.; Yamamoto, J.-I.; Toeda, Y.; Okubo, M.; Eizuka, K.; Ando, T.; Nobuchi, T.; et al. Engineered Exosomes Delivering Specific Tumor-Suppressive RNAi Attenuate Oral Cancer Progression. Sci. Rep. 2021, 11, 5897. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Zhou, Y.; Chen, X.; Ning, T.; Chen, H.; Guo, Q.; Zhang, Y.; Liu, P.; Zhang, Y.; Li, C.; et al. Pancreatic Cancer-Targeting Exosomes for Enhancing Immunotherapy and Reprogramming Tumor Microenvironment. Biomaterials 2021, 268, 120546. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Gu, C.; Gan, Y.; Shao, L.; Chen, H.; Zhu, H. Exosome-Mediated SiRNA Delivery to Suppress Postoperative Breast Cancer Metastasis. J. Control. Release 2020, 318, 1–15. [Google Scholar] [CrossRef]
- Tao, H.; Xu, H.; Zuo, L.; Li, C.; Qiao, G.; Guo, M.; Zheng, L.; Leitgeb, M.; Lin, X. Exosomes-Coated Bcl-2 SiRNA Inhibits the Growth of Digestive System Tumors Both In Vitro and In Vivo. Int. J. Biol. Macromol. 2020, 161, 470–480. [Google Scholar] [CrossRef]
- Bai, J.; Duan, J.; Liu, R.; Du, Y.; Luo, Q.; Cui, Y.; Su, Z.; Xu, J.; Xie, Y.; Lu, W. Engineered Targeting TLyp-1 Exosomes as Gene Therapy Vectors for Efficient Delivery of SiRNA into Lung Cancer Cells. Asian J. Pharm. Sci. 2020, 15, 461–471. [Google Scholar] [CrossRef]
- Ohno, S.; Takanashi, M.; Sudo, K.; Ueda, S.; Ishikawa, A.; Matsuyama, N.; Fujita, K.; Mizutani, T.; Ohgi, T.; Ochiya, T.; et al. Systemically Injected Exosomes Targeted to EGFR Deliver Antitumor MicroRNA to Breast Cancer Cells. Mol. Ther. 2013, 21, 185–191. [Google Scholar] [CrossRef] [Green Version]
- Naseri, Z.; Kazemi Oskuee, R.; Jaafari, M.R.; Forouzandeh, M. Exosome-Mediated Delivery of Functionally Active MiRNA-142-3p Inhibitor Reduces Tumorigenicity of Breast Cancer in Vitro and in Vivo. Int. J. Nanomed. 2018, 13, 7727–7747. [Google Scholar] [CrossRef] [Green Version]
- Liang, G.; Zhu, Y.; Ali, D.J.; Tian, T.; Xu, H.; Si, K.; Sun, B.; Chen, B.; Xiao, Z. Engineered Exosomes for Targeted Co-Delivery of MiR-21 Inhibitor and Chemotherapeutics to Reverse Drug Resistance in Colon Cancer. J. Nanobiotechnol. 2020, 18, 10. [Google Scholar] [CrossRef]
- Shaabani, E.; Sharifiaghdam, M.; De Keersmaecker, H.; De Rycke, R.; De Smedt, S.; Faridi-Majidi, R.; Braeckmans, K.; Fraire, J.C. Layer by Layer Assembled Chitosan-Coated Gold Nanoparticles for Enhanced SiRNA Delivery and Silencing. Int. J. Mol. Sci. 2021, 22, 831. [Google Scholar] [CrossRef]
- Hossen, M.N.; Wang, L.; Chinthalapally, H.R.; Robertson, J.D.; Fung, K.-M.; Wilhelm, S.; Bieniasz, M.; Bhattacharya, R.; Mukherjee, P. Switching the Intracellular Pathway and Enhancing the Therapeutic Efficacy of Small Interfering RNA by Auroliposome. Sci. Adv. 2020, 6, eaba5379. [Google Scholar] [CrossRef]
- Raby, L.; Völkel, P.; Le Bourhis, X.; Angrand, P.-O. Genetic Engineering of Zebrafish in Cancer Research. Cancers 2020, 12, 2168. [Google Scholar] [CrossRef] [PubMed]
- Hason, M.; Bartůněk, P. Zebrafish Models of Cancer—New Insights on Modeling Human Cancer in a Non-Mammalian Vertebrate. Genes 2019, 10, 935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letrado, P.; de Miguel, I.; Lamberto, I.; Díez-Martínez, R.; Oyarzabal, J. Zebrafish: Speeding Up the Cancer Drug Discovery Process. Cancer Res. 2018, 78, 6048–6058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evensen, L.; Johansen, P.L.; Koster, G.; Zhu, K.; Herfindal, L.; Speth, M.; Fenaroli, F.; Hildahl, J.; Bagherifam, S.; Tulotta, C.; et al. Zebrafish as a Model System for Characterization of Nanoparticles against Cancer. Nanoscale 2016, 8, 862–877. [Google Scholar] [CrossRef] [Green Version]
- Franco, G.D.; Usai, A.; Funel, N.; Palmeri, M.; Montesanti, I.E.R.; Bianchini, M.; Gianardi, D.; Furbetta, N.; Guadagni, S.; Vasile, E.; et al. Use of Zebrafish Embryos as Avatar of Patients with Pancreatic Cancer: A New Xenotransplantation Model towards Personalized Medicine. World J. Gastroenterol. 2020, 26, 2792–2809. [Google Scholar] [CrossRef] [PubMed]
- Jing, L.; Zon, L.I. Zebrafish as a Model for Normal and Malignant Hematopoiesis. Dis. Model. Mech. 2011, 4, 433–438. [Google Scholar] [CrossRef] [Green Version]
- Lee, L.M.J.; Seftor, E.A.; Bonde, G.; Cornell, R.A.; Hendrix, M.J.C. The Fate of Human Malignant Melanoma Cells Transplanted into Zebrafish Embryos: Assessment of Migration and Cell Division in the Absence of Tumor Formation. Dev. Dyn. 2005, 233, 1560–1570. [Google Scholar] [CrossRef]
- Lam, S.H.; Chua, H.L.; Gong, Z.; Lam, T.J.; Sin, Y.M. Development and Maturation of the Immune System in Zebrafish, Danio Rerio: A Gene Expression Profiling, in Situ Hybridization and Immunological Study. Dev. Comp. Immunol. 2004, 28, 9–28. [Google Scholar] [CrossRef]
- Pontes, K.C.d.S.; Groenewoud, A.; Cao, J.; Ataide, L.M.S.; Snaar-Jagalska, E.; Jager, M.J. Evaluation of (Fli:GFP) Casper Zebrafish Embryos as a Model for Human Conjunctival Melanoma. Investig. Opthalmol. Vis. Sci. 2017, 58, 6065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, A.M.; Zon, L.I. Zebrafish Tumor Assays: The State of Transplantation. Zebrafish 2009, 6, 339–346. [Google Scholar] [CrossRef]
- Haldi, M.; Ton, C.; Seng, W.L.; McGrath, P. Human Melanoma Cells Transplanted into Zebrafish Proliferate, Migrate, Produce Melanin, Form Masses and Stimulate Angiogenesis in Zebrafish. Angiogenesis 2006, 9, 139–151. [Google Scholar] [CrossRef]
- He, S.; Lamers, G.E.; Beenakker, J.M.; Cui, C.; Ghotra, V.P.; Danen, E.H.; Meijer, A.H.; Spaink, H.P.; Snaar-Jagalska, B.E. Neutrophil-mediated Experimental Metastasis Is Enhanced by VEGFR Inhibition in a Zebrafish Xenograft Model. J. Pathol. 2012, 227, 431–445. [Google Scholar] [CrossRef] [Green Version]
- Pruvot, B.; Jacquel, A.; Droin, N.; Auberger, P.; Bouscary, D.; Tamburini, J.; Muller, M.; Fontenay, M.; Chluba, J.; Solary, E. Leukemic Cell Xenograft in Zebrafish Embryo for Investigating Drug Efficacy. Haematologica 2011, 96, 612–616. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Tang, C.; Cui, K.; Ang, B.-T.; Wong, S.T.C. A Screening Platform for Glioma Growth and Invasion Using Bioluminescence Imaging: Laboratory Investigation. J. Neurosurg. 2009, 111, 238–246. [Google Scholar] [CrossRef]
- Harfouche, R.; Basu, S.; Soni, S.; Hentschel, D.M.; Mashelkar, R.A.; Sengupta, S. Nanoparticle-Mediated Targeting of Phosphatidylinositol-3-Kinase Signaling Inhibits Angiogenesis. Angiogenesis 2009, 12, 325–338. [Google Scholar] [CrossRef]
- Veinotte, C.J.; Dellaire, G.; Berman, J.N. Hooking the Big One: The Potential of Zebrafish Xenotransplantation to Reform Cancer Drug Screening in the Genomic Era. Dis. Model. Mech. 2014, 7, 745–754. [Google Scholar] [CrossRef] [Green Version]
- Cabezas-Sainz, P.; Guerra-Varela, J.; Carreira, M.J.; Mariscal, J.; Roel, M.; Rubiolo, J.A.; Sciara, A.A.; Abal, M.; Botana, L.M.; López, R.; et al. Improving Zebrafish Embryo Xenotransplantation Conditions by Increasing Incubation Temperature and Establishing a Proliferation Index with ZFtool. BMC Cancer 2018, 18, 3. [Google Scholar] [CrossRef] [PubMed]
- Vittori, M.; Motaln, H.; Turnšek, T.L. The Study of Glioma by Xenotransplantation in Zebrafish Early Life Stages. J. Histochem. Cytochem. 2015, 63, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.J.; Teraoka, H.; Heideman, W.; Peterson, R.E. Zebrafish as a Model Vertebrate for Investigating Chemical Toxicity. Toxicol. Sci. 2005, 86, 6–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vu, M.N.; Kelly, H.G.; Wheatley, A.K.; Peng, S.; Pilkington, E.H.; Veldhuis, N.A.; Davis, T.P.; Kent, S.J.; Truong, N.P. Cellular Interactions of Liposomes and PISA Nanoparticles during Human Blood Flow in a Microvascular Network. Small 2020, 16, 2002861. [Google Scholar] [CrossRef]
- Brown, H.K.; Schiavone, K.; Tazzyman, S.; Heymann, D.; Chico, T.J. Zebrafish Xenograft Models of Cancer and Metastasis for Drug Discovery. Expert Opin. Drug Discov. 2017, 12, 379–389. [Google Scholar] [CrossRef]
- Long, Y.; Li, L.; Li, Q.; He, X.; Cui, Z. Transcriptomic Characterization of Temperature Stress Responses in Larval Zebrafish. PLoS ONE 2012, 7, e37209. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.-L.; Qi, W.; Han, F.; Shao, J.Z.; Gao, J.Q. Toxicity Evaluation of Biodegradable Chitosan Nanoparticles Using a Zebrafish Embryo Model. Int. J. Nanomed. 2011, 6, 3351. [Google Scholar] [CrossRef] [Green Version]
- Ghotra, V.P.S.; He, S.; de Bont, H.; van der Ent, W.; Spaink, H.P.; van de Water, B.; Snaar-Jagalska, B.E.; Danen, E.H.J. Automated Whole Animal Bio-Imaging Assay for Human Cancer Dissemination. PLoS ONE 2012, 7, e31281. [Google Scholar] [CrossRef] [PubMed]
- van Pomeren, M.; Brun, N.R.; Peijnenburg, W.J.G.M.; Vijver, M.G. Exploring Uptake and Biodistribution of Polystyrene (Nano)Particles in Zebrafish Embryos at Different Developmental Stages. Aquat. Toxicol. 2017, 190, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Teijeiro-Valiño, C.; Yebra-Pimentel, E.; Guerra-Varela, J.; Csaba, N.; Alonso, M.J.; Sánchez, L. Assessment of the Permeability and Toxicity of Polymeric Nanocapsules Using the Zebrafish Model. Nanomedicine 2017, 12, 2069–2082. [Google Scholar] [CrossRef]
- Dal, N.K.; Kocere, A.; Wohlmann, J.; Van Herck, S.; Bauer, T.A.; Resseguier, J.; Bagherifam, S.; Hyldmo, H.; Barz, M.; De Geest, B.G.; et al. Zebrafish Embryos Allow Prediction of Nanoparticle Circulation Times in Mice and Facilitate Quantification of Nanoparticle–Cell Interactions. Small 2020, 16, 1906719. [Google Scholar] [CrossRef]
- Baboci, L.; Capolla, S.; Di Cintio, F.; Colombo, F.; Mauro, P.; Dal Bo, M.; Argenziano, M.; Cavalli, R.; Toffoli, G.; Macor, P. The Dual Role of the Liver in Nanomedicine as an Actor in the Elimination of Nanostructures or a Therapeutic Target. J. Oncol. 2020, 2020, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, Y.; Takamiya, M.; Jensen, P.B.; Ojea-Jiménez, I.; Claude, H.; Antony, C.; Kjaer-Sorensen, K.; Grabher, C.; Boesen, T.; Gilliland, D.; et al. Differential Nanoparticle Sequestration by Macrophages and Scavenger Endothelial Cells Visualized in Vivo in Real-Time and at Ultrastructural Resolution. ACS Nano 2020, 14, 1665–1681. [Google Scholar] [CrossRef]
- Lieschke, G.J.; Currie, P.D. Animal Models of Human Disease: Zebrafish Swim into View. Nat. Rev. Genet. 2007, 8, 353–367. [Google Scholar] [CrossRef]
- Renshaw, S.A.; Loynes, C.A.; Trushell, D.M.I.; Elworthy, S.; Ingham, P.W.; Whyte, M.K.B. A Transgenic Zebrafish Model of Neutrophilic Inflammation. Blood 2006, 108, 3976–3978. [Google Scholar] [CrossRef] [PubMed]
- Reitman, M.L. Of Mice and Men-Environmental Temperature, Body Temperature, and Treatment of Obesity. FEBS Lett. 2018, 592, 2098–2107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, A.; Hurlstone, A.F. The Use of RNAi Technologies for Gene Knockdown in Zebrafish. Brief. Funct. Genom. 2011, 10, 189–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slijkerman, R.; van Diepen, H.; Albert, S.; Dona, M.; Venselaar, H.; Zang, J.; Neuhauss, S.; Peters, T.; Broekman, S.; Pennings, R.; et al. Antisense Oligonucleotide-Based Treatment of Retinitis Pigmentosa Caused by Mutations in USH2A Exon 13. Genetics 2020, 28. [Google Scholar] [CrossRef]
- Yang, T.; Fogarty, B.; LaForge, B.; Aziz, S.; Pham, T.; Lai, L.; Bai, S. Delivery of Small Interfering RNA to Inhibit Vascular Endothelial Growth Factor in Zebrafish Using Natural Brain Endothelia Cell-Secreted Exosome Nanovesicles for the Treatment of Brain Cancer. AAPS J. 2017, 19, 475–486. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ONs | Nucleic Acid Composition | Lenght (Nucleotides) | Mechanisms of Action | Effects |
|---|---|---|---|---|
| ASO | ssDNA or ssRNA | 18–30 | Formation of RNA-DNA duplex recognized by RNASEH1 | Silencing of gene expression after RNA degradation |
| Steric block | Modulation of alternative splicing or translation block | |||
| siRNA | dsRNA | 21 | RISC is guided to the complementary target transcripts | Degradation of the target mRNA, gene silencing |
| miRNA mimics | dsRNA | ~20 | Increased level of a specific miRNA | Restore of miRNAs level |
| antagomiR | ssRNA | ~20 | Direct binding to RISC | Translation repression or miRNA degradation |
| Aptamers | ssDNA or ssRNA | 20–100 | Mimic of the promoter sequence | Block of protein translation |
| Decoys | dsDNA or dsRNA | 15–20 | Mimic binding site of transcription factors | Transcription silencing |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bozzer, S.; Bo, M.D.; Toffoli, G.; Macor, P.; Capolla, S. Nanoparticles-Based Oligonucleotides Delivery in Cancer: Role of Zebrafish as Animal Model. Pharmaceutics 2021, 13, 1106. https://doi.org/10.3390/pharmaceutics13081106
Bozzer S, Bo MD, Toffoli G, Macor P, Capolla S. Nanoparticles-Based Oligonucleotides Delivery in Cancer: Role of Zebrafish as Animal Model. Pharmaceutics. 2021; 13(8):1106. https://doi.org/10.3390/pharmaceutics13081106
Chicago/Turabian StyleBozzer, Sara, Michele Dal Bo, Giuseppe Toffoli, Paolo Macor, and Sara Capolla. 2021. "Nanoparticles-Based Oligonucleotides Delivery in Cancer: Role of Zebrafish as Animal Model" Pharmaceutics 13, no. 8: 1106. https://doi.org/10.3390/pharmaceutics13081106
APA StyleBozzer, S., Bo, M. D., Toffoli, G., Macor, P., & Capolla, S. (2021). Nanoparticles-Based Oligonucleotides Delivery in Cancer: Role of Zebrafish as Animal Model. Pharmaceutics, 13(8), 1106. https://doi.org/10.3390/pharmaceutics13081106