Appendix A
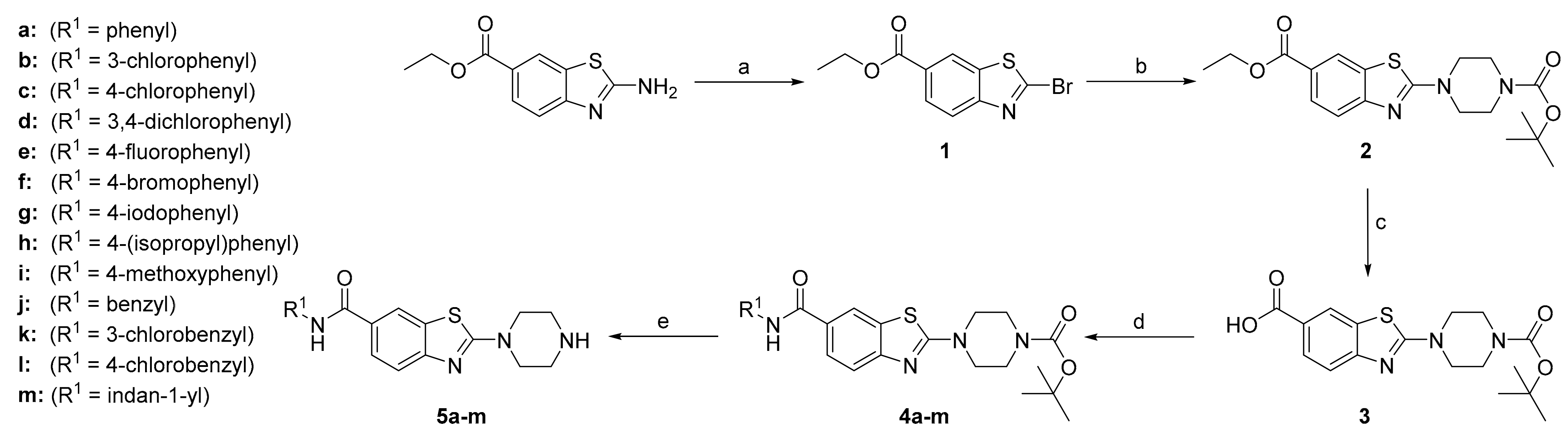
Ethyl 2-bromobenzo[d]thiazole-6-carboxylate (1): Ethyl 2-aminobenzo[d]thiazole-6-carboxylate (4.00 g, 18.00 mmol) and CuBr2 (8.04 g, 35.99 mmol) were dissolved in acetonitrile (90 mL). tert-Butyl nitrite (4.28 mL, 35.99 mmol, 867 mg mL−1) was added in an ice bath, and the reaction mixture was stirred for 1 h at room temperature. The solvent was then removed under reduced pressure, and the residue was taken up in ethyl acetate (150 mL) and saturated NH4Cl (50 mL). The organic phase was additionally washed with saturated NH4Cl (2 × 50 mL) and brine (50 mL). The organic phase was dried over Na2SO4, filtered, and the solvent was evaporated under reduced pressure. Yield: 3.95 g (77%); brown amorphous powder; Rf (DCM:MeOH = 30:1) = 0.74; 1H NMR (400 MHz, CDCl3, 25 °C, TMS): δ = 8.55 (d, J = 1.5 Hz, 1H, Ar-H7), 8.16 (dd, J1 = 8.5 Hz, J2 = 1.5 Hz, 1H, Ar-H5), 8.02 (d, J = 8.5 Hz, 1H, Ar-H4), 4.43 (q, J = 7.1 Hz, 2H, COO-CH2-CH3), 1.43 ppm (t, J = 7.1 Hz, 3H, COO-CH2-CH3); 13C NMR (101 MHz, CDCl3, 25 °C, TMS): δ = 165.8, 155.1, 142.4, 137.3, 127.9, 127.8, 122.9, 122.5, 61.5, 14.4 ppm; LC–MS (ESI+): m/z 285.9 [M+H]+ (calcd. m/z = 284.9 for C10H8BrNO2S).
Ethyl 2-(4-Boc-piperazin-1-yl)benzo[d]thiazole-6-carboxylate (2): Ethyl 2-bromobenzo[d]thiazole-6-carboxylate (1.98 g, 6.91 mmol) and 1-Boc-piperazine (3.22 g, 17.3 mmol) were dissolved in THF (100 mL). The reaction mixture was then stirred overnight at room temperature. The precipitate formed was pressure filtered off, and the solvent was evaporated under reduced pressure. The residue was taken up in ethyl acetate (100 mL) and washed with 1% (w/v) citric acid (3 × 50 mL) and saturated NaCl (50 mL). The organic phase was dried over Na2SO4, filtered, and the solvent was evaporated under reduced pressure. Yield: 2.63 g (97%); yellow amorphous powder; Rf(EtOAc:Hex = 1:3) = 0.29; 1H NMR (400 MHz, CDCl3, 25 °C, TMS): δ = 8.32 (d, J = 1.5 Hz, 1H, Ar-H7), 8.01 (dd, J1 = 8.5 Hz, J2 = 1.8 Hz, 1H, Ar-H5), 7.53 (d, J = 8.5 Hz, 1H, Ar-H4), 4.38 (q, J = 7.1 Hz, 2H, COO-CH2-CH3), 3.69–3.63 (m, 4H, 2 × piperazine-CH2), 3.63–3.56 (m, 4H, 2 × piperazine-CH2), 1.49 (s, 9H, COC(CH3)3), 1.40 ppm (t, J = 7.1 Hz, 3H, COO-CH2-CH3); 13C NMR (101 MHz, CDCl3, 25 °C, TMS): δ = 170.7, 166.4, 156.3, 154.5, 130.6, 128.0, 123.7, 122.8, 118.5, 80.6 (2C), 60.9, 48.2 (2C), 28.4 (3C), 14.4 ppm; LC–MS (ESI+): m/z 392.1 [M+H]+ (calcd. m/z = 391.2 for C19H25N3O4S).
2-(4-Boc-piperazin-1-yl)benzo[d]thiazole-6-carboxylic acid (3): Compound 2 (2.60 g, 6.64 mmol) was suspended in EtOH (96%, 50 mL) and NaOH(aq) (33.2 mL, 2 M, 66.4 mmol) was added. The mixture was heated to 100 °C and left to stir for 1 h. Afterwards, the reaction mixture was cooled and acidified to pH 3, using 2 M HCl(aq). The precipitate that formed was then filtered off under pressure and left to dry. Yield: 2.25 g (93%); yellow amorphous powder; Rf(EtOAc:Hex = 1:1) = 0,16; 1H NMR (400 MHz, CDCl3, 25 °C, TMS): δ = 8.39 (d, J = 1.6 Hz, 1H, Ar-H7), 8.08 (dd, J1 = 8.5 Hz, J2 = 1.6 Hz, 1H, Ar-H5), 7.58 (d, J = 8.5 Hz, 1H, Ar-H4), 3.73–3.66 (m, 4H, 2 × piperazine-CH2), 3.65–3.58 (m, 4H, 2 × piperazine-CH2), 1.50 ppm (s, 9H, COC(CH3)3); 13C NMR (101 MHz, [D6]DMSO, 25 °C, TMS): δ = 170.9, 167.6, 156.5, 154.2, 130.9, 128.1, 123.8, 123.6, 118.4, 79.8 (2C), 48.2 (2C), 28.5 ppm (3C); LC–MS (ESI+): m/z 364.1 [M+H]+ (calcd. m/z = 363.1 for C17H21N3O4S).
General procedure for amide coupling (4a–m)
The amide coupling reactions were performed under an argon atmosphere. 2-Substituted benzo[d]thiazole-6-carboxylic acid (1 equiv) was dissolved in DMF. EDC (1.2 equiv), HOBT (1.3 equiv) and DIPEA (2.5 equiv) were added in an ice bath. After 20 min, the respective amine (1.2 equiv) was added, and the reaction mixture was stirred for 1–3 days at room temperature. When the activated ester that formed with HOBt was too stable to further react with the respective aniline at r.t. (according to liquid chromatography–mass spectrometry analysis), the temperature of the reaction mixture was increased to 70 °C (for 4b–d, 4g) and stirred overnight. The solvent was then removed under reduced pressure, and the residue was taken up in ethyl acetate (~100 mL) and washed with 1 M NaOH (3 × 50 mL), 1% (w/v) citric acid (3 × 50 mL) and with saturated NaCl (50 mL). The organic phase was dried over Na2SO4, filtered, and the solvent was evaporated under reduced pressure. The crude product was purified with precipitation from a mixture of ethyl acetate and hexane.
4-(6-(Phenylcarbamoyl)benzo[d]thiazol-2-yl)-1-Boc-piperazine (4a): Yield: 216 mg (49%); yellow amorphous powder; Rf(EtOAc:Hex = 2:1) = 0.54; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 10.18 (s, 1H, Ar-NH-COR), 8.41 (d, J = 1.8 Hz, 1H, Ar-H7), 7.91 (dd, J1 = 8.5 Hz, J2 = 1.8 Hz, 1H, Ar-H5), 7.78 (d, J = 7.6 Hz, 2H, 2 × Ar-H), 7.55 (d, J = 8.5 Hz, 1H, Ar-H4), 7.35 (t, J = 7.9 Hz, 2H, 2 × Ar-H), 7.09 (t, J = 7.4 Hz, 1H, Ar-H), 3.69–3.58 (m, 4H, 2 × piperazine-CH2), 3.55–3.45 (m, 4H, 2 × piperazine-CH2), 1.44 ppm (s, 9H, COC(CH3)3); 13C (101 MHz, [D6]DMSO, 25 °C, TMS): δ = 170.4, 165.5, 155.5, 154.3, 139.9, 130.8, 129.0 (2C), 128.2, 126.6, 123.9, 121.7, 120.7 (2C), 118.3, 79.8 (2C), 48.2 (2C), 28.5 ppm (3C); LC–MS (ESI+): m/z 439.1 [M+H]+ (calcd. m/z = 438.2 for C23H26N4O3S).
4-(6-((3-Chlorophenyl)carbamoyl)benzo[d]thiazol-2-yl)-1-Boc-piperazine (4b): Yield: 67 mg (15%); light pink amorphous powder; Rf(EtOAc:Hex = 2:1) = 0.61; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 10.34 (s, 1H, Ar-NH-COR), 8.41 (d, J = 1.9 Hz, 1H, Ar-H7), 7.98 (t, J = 2.0 Hz, 1H, Ar-H), 7.91 (dd, J1 = 8.5 Hz, J2 = 1.9 Hz, 1H, Btz-H5), 7.71 (ddd, J1 = 8.2 Hz, J2 = 2.0 Hz, J3 = 0.9 Hz, 1H, Ar-H), 7.56 (d, J = 8.5 Hz, 1H, Ar-H4), 7.38 (t, J = 8.2 Hz, 1H, Ar-H), 7.15 (ddd, J1 = 8.2 Hz, J2 = 2.0 Hz, J3 = 0.9 Hz, 1H, Ar-H), 3.65–3.60 (m, 4H, 2 × piperazine-CH2), 3.53–3.47 (m, 4H, 2 × piperazine-CH2), 1.44 ppm (s, 9H, COC(CH3)3); LC–MS (ESI+): m/z 472.8 [M+H]+ (calcd. m/z = 471.1 for C24H26Cl2N4O3S).
4-(6-((4-Chlorophenyl)carbamoyl)benzo[d]thiazol-2-yl)-1-Boc-piperazine (4c): Yield: 258 mg (48%); off-white amorphous powder; Rf(EtOAc:Hex = 1:2) = 0.39; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 10.31 (s, 1H, Ar-NH-COR), 8.40 (d, J = 1.8 Hz, 1H, Ar-H7), 7.91 (dd, J1 = 8.5, J2 = 1.8 Hz, 1H, Ar-H5), 7.82 (d, J = 8.9 Hz, 2H, 2 × Ar-H), 7.55 (d, J = 8.4 Hz, 1H, Ar-H4), 7.41 (d, J = 8.9 Hz, 2H, 2 × Ar-H), 3.69–3.58 (m, 4H, 2 × piperazine-CH2), 3.54–3.46 (m, 4H, 2 × piperazine-CH2), 1.43 ppm (s, 9H, COC(CH3)3); LC–MS (ESI+): m/z 473.1 [M+H]+ (calcd. m/z = 472.1 for C23H25ClN4O3S).
4-(6-((3,4-Dichlorophenyl)carbamoyl)benzo[d]thiazol-2-yl)-1-Boc-piperazine (4d): Yield: 274 mg (20%); off-white amorphous powder; Rf(EtOAc:Hex = 1:2) = 0,15; 1H NMR (400 MHz, CDCl3, 25 °C, TMS): δ = 8.21 (d, J = 1.8 Hz, 1H, Ar-H7), 7.91 (d, J = 2.5 Hz, 1H, Ar-H20), 7.84 (s, 1H, Ar-NH-COR), 7.74 (dd, J1 = 8.5 Hz, J2 = 1.8 Hz, 1H, Ar-H5), 7.58 (d, J = 8.5 Hz, 1H, Ar-H4), 7.48 (dd, J1 = 8.7, J2 = 2.5 Hz, 1H, Ar-H24), 7.42 (d, J = 8.7 Hz, 1H, Ar-H23), 3.70–3.64 (m, 6.5, 3.6 Hz, 4H, 2 × piperazine-CH2), 3.63–3.58 (m, 4H, 2 × piperazine-CH2), 1.50 ppm (s, 9H, COC(CH3)3); LC–MS (ESI+): m/z 506.9 [M+H]+ (calcd. m/z = 506,1 for C23H24Cl2N4O3S).
4-(6-((4-Fluorophenyl)carbamoyl)benzo[d]thiazol-2-yl)-1-Boc-piperazine (4e): Yield: 151 mg (59%); off-white amorphous powder; Rf(EtOAc:Hex = 2:1) = 0.43; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 10.24 (s, 1H, Ar-NH-COR), 8.41 (d, J = 1.6 Hz, 1H, Ar-H7), 7.91 (dd, J1 = 8.5 Hz, J2 = 1.6 Hz, 1H, Ar-H5), 7.83–7.76 (m, 2H, 2 × Ar-H), 7.55 (d, J = 8.5 Hz, 1H, Ar-H4), 7.20 (t, J = 8.9 Hz, 2H, 2 × Ar-H), 3.70–3.58 (m, 4H, 2 × piperazine-CH2), 3.56–3.46 (m, 4H, 2 × piperazine-CH2), 1.44 ppm (s, 9H, COC(CH3)3); 19F NMR (376 MHz, [D6]DMSO, 25 °C, TMS): δ = -119.20 ppm; 13C (101 MHz, [D6]DMSO, 25 °C, TMS): δ = 170.4, 165.4, 158.6 (d, J = 239.9 Hz), 155.5, 154.2, 136.2, 136.2, 130.8, 128.0, 126.5, 122.5, 122.5, 121.7, 118.3, 115.7, 115.5, 79.8 (2C), 48.2 (2C), 28.5 ppm (3C); LC–MS (ESI+): m/z 457.1 [M+H]+ (calcd. m/z = 456.2 for C23H25FN4O3S).
4-(6-((4-Bromophenyl)carbamoyl)benzo[d]thiazol-2-yl)-1-Boc-piperazine (4f): Yield: 203 mg (58%); yellow amorphous powder; Rf(EtOAc:Hex = 2:1) = 0.46; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 10.30 (s, 1H, Ar-NH-COR), 8.40 (d, J = 1.9 Hz, 1H, Ar-H7), 7.91 (dd, J1 = 8.5 Hz, J2 = 1.9 Hz, 1H, Ar-H5), 7.81–7.73 (m, 2H, 2 × Ar-H), 7.58–7.49 (m, 3H, 3 × Ar-H), 3.68–3.57 (m, 4H, 2 × piperazine-CH2), 3.56–3.45 (m, 4H, 2 × piperazine-CH2), 1.43 ppm (s, 9H, COC(CH3)3); LC–MS (ESI+): m/z 571.4 [M+H]+ (calcd. m/z = 516.1 for C23H25BrN4O3S).
4-(6-((4-Iodophenyl)carbamoyl)benzo[d]thiazol-2-yl)-1-Boc-piperazine (4g): Yield: 77 mg (20%); yellow amorphous powder; Rf(EtOAc:Hex = 2:1) = 0.58; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 10.27 (s, 1H, Ar-NH-COR), 8.40 (d, J = 1.9 Hz, 1H, Ar-H7), 7.90 (dd, J1 = 8.5 Hz, J2 = 1.9 Hz, 1H, Ar-H5), 7.72–7.66 (m, 2H, 2 × Ar-H), 7.66–7.60 (m, 2H, 2 × Ar-H), 7.54 (d, J = 8.5 Hz, 1H, Ar-H4), 3.66–3.58 (m, 4H, 2 × piperazine-CH2), 3.54–3.45 (m, 4H, 2 × piperazine-CH2), 1.43 ppm (s, 9H, COC(CH3)3); LC–MS (ESI+): m/z 565.0 [M+H]+ (calcd. m/z = 564.0 for C23H25IN4O3S).
4-(6-((4-Isopropylphenyl)carbamoyl)benzo[d]thiazol-2-yl)-1-Boc-piperazine (4h): Yield: 151 mg (50%); off-white amorphous powder; Rf(EtOAc:Hex = 2:1) = 0.63; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 10.10 (s, 1H, Ar-NH-COR), 8.40 (d, J = 1.8 Hz, 1H, Ar-H7), 7.91 (dd, J1 = 8.5 Hz, J2 = 1.8 Hz, 1H, Ar-H5), 7.68 (d, J = 8.5 Hz, 2H, 2 × Ar-H), 7.54 (d, J = 8.5 Hz, 1H, Ar-H4), 7.21 (d, J = 8.5 Hz, 2H, 2 × Ar-H), 3.68–3.59 (m, 4H, 2 × piperazine-CH2), 3.54–3.47 (m, 4H, 2 × piperazine-CH2), 2.86 (dt, J1 = 13.7 Hz, J2 = 6.9 Hz, 1H, Ar-CH-(CH3)2), 1.44 (s, 9H, COC(CH3)3), 1.20 ppm (d, J = 6.9 Hz, 6H, Ar-CH-(CH3)2); LC–MS (ESI+): m/z 481.1 [M+H]+ (calcd. m/z = 480.2 for C26H32N4O3S).
4-(6-((4-(Methylhydroxy)phenyl)carbamoyl)benzo[d]thiazol-2-yl)-1-Boc-piperazine (4i): Yield: 333 mg (79%); yellow amorphous powder; Rf(EtOAc:Hex = 2:1) = 0.58; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 10.08 (s, 1H, Ar-NH-COR), 8.40 (d, J = 1.8 Hz, 1H, Ar-H7), 7.91 (dd, J1 = 8.5 Hz, J2 = 1.8 Hz, 1H, Ar-H5), 7.73–7.64 (m, 2H, 2 × Ar-H), 7.53 (d, J = 8.5 Hz, 1H¸, Ar-H4), 6.98–6.87 (m, 2H, 2 × Ar-H), 3.74 (s, 3H, Ar-O-CH3), 3.66–3.58 (m, 4H, 2 × piperazine-CH2), 3.54–3.46 (m, 4H, 2 × piperazine-CH2), 1.44 ppm (s, 9H, COC(CH3)3); LC–MS (ESI+): m/z 469.2 [M+H]+ (calcd. m/z = 468.2 for C24H28N4O4S).
4-(6-(Benzylcarbamoyl)benzo[d]thiazol-2-yl)-1-Boc-piperazine (4j): Yield: 265 mg (71%); yellow amorphous powder; Rf(EtOAc:Hex = 2:1) = 0.33; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 8.96 (t, J = 6.0 Hz, 1H, Ar-CH2-NH-COR), 8.32 (d, J = 1.8 Hz, 1H, Ar-H7), 7.84 (dd, J1 = 8.5, J2 = 1.8 Hz, 1H, Ar-H5), 7.49 (d, J = 8.5 Hz, 1H, Ar-H4), 7.35–7.29 (m, 4H, 4 × Ar-H), 7.27–7.21 (m, 1H, Ar-H), 4.48 (d, J = 6.0 Hz, 2H, Ar-CH2-NHCO), 3.61 (m, 4H, 2 × piperazine-CH2), 3.54–3.44 (m, 4H, 2 × piperazine-CH2), 1.43 ppm (s, 9H, COC(CH3)3); LC–MS (ESI+): m/z 452.7 [M+H]+ (calcd. m/z = 452.2 for C24H26Cl2N4O3S).
4-(6-((3-Chlorobenzyl)carbamoyl)benzo[d]thiazol-2-yl)1-Boc-piperazine (4k): Yield: 196 mg (64%); white amorphous powder; Rf(EtOAc:Hex = 1:1) = 0.19; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 9.01 (t, J = 6.0 Hz, 1H, CH2-NH-COR), 8.33 (d, J = 1.8 Hz, 1H, Ar-H7), 7.84 (dd, J1 = 8.5 Hz, J2 = 1.8 Hz, 1H, Ar-H5), 7.50 (d, J = 8.5 Hz, 1H, Ar-H4), 7.39–7.34 (m, 2H, 2 × Ar-H), 7.33–7.27 (m, 2H, 2 × Ar-H), 4.48 (d, J = 6.0 Hz, 2H, RCO-NH-CH2-Ph), 3.64–3.58 (m, 4H, 2 × piperazine-CH2), 3.55–3.44 (m, 4H, 2 × piperazine-CH2), 1,43 ppm (s, 9H, COC(CH3)3); LC–MS (ESI+): m/z 487.1 [M+H]+ (calcd. m/z = 486.1 for C24H27ClN4O3S).
4-(6-((4-Chlorobenzyl)carbamoyl)benzo[d]thiazol-2-yl)-1-Boc-piperazine (4l): Yield: 200 mg (65%); yellow amorphous powder; Rf(EtOAc:Hex = 1:1) = 0.19; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 8.99 (t, J = 5.9 Hz, 1H, CH2-NH-COR), 8.32 (d, J = 1.7 Hz, 1H, Ar-H7), 7.83 (dd, J1 = 8.5 Hz, J2 = 1.7 Hz, 1H, Ar-H5), 7.49 (d, J = 8.5 Hz, 1H, Ar-H4), 7.42–7.31 (m, 4H, 4 × Ar-H), 4.46 (d, J = 5.9 Hz, 2H, RCO-NH-CH2-Ph), 3.66–3.56 (m, 4H, 2 × piperazine-CH2), 3.53–3.43 (m, 4H, 2 × piperazine-CH2), 1.43 ppm (s, 9H, COC(CH3)3); 13C (101 MHz, [D6]DMSO, 25 °C, TMS): δ = 170.2, 166.3, 155.2, 154.2, 139.4, 131.7, 130.8, 129.6 (2C), 128.7 (2C), 127.6, 126.0, 121.3, 118.3, 79.8, 48.2 (2C), 42.5 (2C), 28.5 ppm (3C); LC–MS (ESI+): m/z 487.4 [M+H]+ (calcd. m/z = 486.1 for C24H27ClN4O3S).
4-(6-((Indane-1-yl)carbamoyl)benzo[d]thiazol-2-yl)-1-Boc-piperazine (4m): Yield: 165 mg (63%); white amorphous powder; Rf(EtOAc:Hex = 2:1) = 0.39; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 8.68 (d, J = 8.3 Hz, 1H, CH-NH-COR), 8.35 (d, J = 1.6 Hz, 1H, Ar-H), 7.87 (dd, J1 = 8.5 Hz, J2 = 1.6 Hz, 1H, Ar-H), 7.48 (d, J = 8.5 Hz, 1H, Ar-H), 7.32–7.13 (m, 4H, 4 × Ar-H), 5.56 (dd, J1 = 16.1 Hz, J2 = 8.3 Hz, 1H, indane-H), 3.66–3.55 (m, 1H, indane-H), 3.53–3.46 (m, 4H, 2 × piperazine-CH2), 3.05–2.95 (m, 4H, 2 × piperazine-CH2), 2.91–2.79 (m, 1H, indane-H), 2.05–1.92 ppm (m, 1H, indane-H), not visible (1H, indane-H); LC–MS (ESI+): m/z 479.1 [M+H]+ (calcd. m/z = 478.2 for C26H30N4O3S).
General procedure for acidolysis used for the synthesis of compounds 5a-m, 9a-j and 14
4-Boc-protected benzothiazole (1 equiv) was dissolved in dichloromethane. Trifluoroacetic acid (25 equiv) was added, and the reaction mixture was stirred overnight at room temperature. Afterwards, the reaction mixture was diluted with dichloromethane to 25 mL and washed with 1 M NaOH (3 × 25 mL) and saturated NaCl (25 mL). The organic phase was dried over Na2SO4, filtered, and the solvent was evaporated under reduced pressure.
4-(6-(Phenylcarbamoyl)benzo[d]thiazol-2-yl)piperazine (5a): Yield: 91 mg (74%); off-white amorphous powder; Rf(DCM:MeOH = 9:1) = 0.0; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 10.16 (s, 1H, Ar-NH-COR), 8.38 (d, J = 1.8 Hz, 1H, Ar-H7), 7.90 (dd, J1 = 8.5 Hz, J2 = 1.8 Hz, 1H, Ar-H5), 7.82–7.75 (m, 2H, 2 × Ar-H), 7.51 (d, J = 8.5 Hz, 1H, Ar-H4), 7.39–7.30 (m, 2H, 2 × Ar-H), 7.13–7.04 (m, 1H, Ar-H), 3.58–3.48 (m, 4H, 2 × piperazine-CH2), 2.87–2.76 ppm (m, 4H, 2 × piperazine-CH2); 13C (101 MHz, [D6]DMSO, 25 °C, TMS): δ = 170.6, 165.5, 155.7, 139.9, 130.6, 129.0 (2C), 127.8, 126.5, 123.9, 121.5, 120.7 (2C), 118.1, 49.9 (2C), 45.5 ppm (2C); HRMS (ESI+) m/z calcd. calcd. for C18H18N4OS+H+: 339.1274 [M+H]+: found 339.1269; HPLC: tr = 2.27 min (97.4% at 254 nm).
4-(6-((3-Chlorophenyl)carbamoyl)benzo[d]thiazol-2-yl)piperazine (5b): Yield: 43 mg (91%); off-white amorphous powder; Rf(DCM:MeOH = 9:1) = 0.07; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 10.32 (s, 1H, Ar-NH-COR), 8.38 (d, J = 1.9 Hz, 1H, Ar-H7), 7.98 (t, J = 2.0 Hz, 1H, Ar-H), 7.90 (dd, J1 = 8.5 Hz, J2 = 1.9 Hz, 1H, Ar-H5), 7.71 (ddd, J1 = 8.1 Hz, J2 = 2.0 Hz, J3 = 0.9 Hz, 1H, Ar-H), 7.52 (d, J = 8.5 Hz, 1H, Ar-H4), 7.38 (t, J = 8.1 Hz, 1H, Ar-H), 7.14 (ddd, J1 = 8.1 Hz, J2 = 2.0 Hz, J3 = 0.9 Hz, 1H, Ar-H), 3.58–3.51 (m, 4H, 2 × piperazine-CH2), 2.85–2.79 ppm (m, 4H, 2 × piperazine-CH2), not visible (NH); 13C (101 MHz, [D6]DMSO, 25 °C, TMS): δ = 170.7, 165.8, 155.9, 141.4, 133.4, 130.8, 130.6, 127.3, 126.6, 123.5, 121.7, 120.0, 119.0, 118.1, 49.9 (2C), 45.5 (2C) ppm; HRMS (ESI+) m/z calcd. calcd. for C18H17ClN4OS+H+: 373.0884 [M+H]+: found 373.0880; HPLC: tr = 5.31 min (95.5% at 254 nm).
4-(6-((4-Chlorophenyl)carbamoyl)benzo[d]thiazol-2-yl)piperazine (5c): Yield: 61 mg (31%); white powder; Rf(DCM:MeOH = 9:1) = 0.09; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 10.29 (s, 1H, Ar-NH-COR), 8.38 (d, J = 1.8 Hz, 1H, Ar-H7), 7.89 (dd, J1 = 8.5, J2 = 1.8 Hz, 1H, Ar-H5), 7.85–7.78 (m, 2H, 2 × Ar-H), 7.51 (d, J = 8.5 Hz, 1H, Ar-H4), 7.44–7.37 (m, 2H, 2 × Ar-H), 3.57–3.50 (m, 4H, 2 × piperazine-CH2), 2.84–2.78 ppm (m, 4H, 2 × piperazine-CH2), not visible (NH); 13C (101 MHz, [D6]DMSO, 25 °C, TMS): δ = 170.7, 165.6, 155.8, 138.9, 130.6, 129.0 (2C), 127.5, 127.4, 126.6, 122.2 (2C), 121.6, 118.1, 49.9 (2C), 45.5 ppm (2C); HRMS (ESI+) m/z calcd. calcd. for C18H17ClN4OS+H+: 373.0884 [M+H]+: found 373.0878; HPLC: tr = 5.27 min (97.2% at 254 nm).
4-(6-((3,4-Dichlorophenyl)carbamoyl)benzo[d]thiazol-2-yl)piperazine (5d): Yield: 118 mg (68%); off-white amorphous powder; Rf(EtOAc:Hex = 1:1) = 0; 1H NMR (400 MHz, CDCl3, 25 °C, TMS): δ = 8.20 (d, J = 1.9 Hz, 1H, Ar-H7), 7.90 (d, J = 2.4 Hz, 1H, Ar-H20), 7.79 (s, 1H, Ar-NH-COR), 7.73 (dd, J1 = 8.5 Hz, J2 = 1.9 Hz, 1H, Ar-H5), 7.57 (d, J = 8.5 Hz, 1H, Ar-H4), 7.48 (dd, J1 = 8.7 Hz, J2 = 2.4 Hz, 1H, Ar-H24), 7.42 (d, J = 8.7 Hz, 1H, Ar-H23), 3.70–3.65 (m, 4H, 2 × piperazine-CH2), 3.06–2.99 ppm (m, 4H, 2 × piperazine-CH2), not visible (NH); 13C NMR (101 MHz, [D6]DMSO, 25 °C, TMS): δ = 170.8, 165.8, 156.0, 140.1, 131.3, 131.0, 130.7, 127.1, 126.7, 125.2, 121.7, 121.7, 120.6, 118.1, 49.9 (2C), 45.5 ppm (2C); HRMS (ESI+) m/z calcd. calcd. for C18H16Cl2N4OS+H+: 407.0495 [M+H]+: found 407.0487; HPLC: tr = 6.57 min (95.3% at 254 nm).
4-(6-((4-Fluorophenyl)carbamoyl)benzo[d]thiazol-2-yl)piperazine (5e): Yield: 74 mg (95%); white amorphous powder; Rf(DCM:MeOH = 9:1) = 0.05; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 10.22 (s, 1H, Ar-NH-COR), 8.38 (d, J = 1.8 Hz, 1H, Ar-H7), 7.90 (dd, J1 = 8.5 Hz, J2 = 1.8 Hz, 1H, Ar-H5), 7.84–7.74 (m, 2H, 2 × Ar-H), 7.52 (d, J = 8.5 Hz, 1H, Ar-H4), 7.24–7.14 (m, 2H, 2 × Ar-H), 3.59–3.50 (m, 4H, 2 × piperazine-CH2), 2.83 ppm (m, 4.3 Hz, 4H, 2 × piperazine-CH2), not visible (NH); 19F NMR (376 MHz, [D6]DMSO, 25 °C, TMS): δ = -119.26 ppm; 13C (101 MHz, [D6]DMSO, 25 °C, TMS): δ = 170.6, 165.4, 158.60 (d, J = 239.8 Hz), 155.7, 136.2 (d, J = 2.6 Hz), 130.6, 127.6, 126.5, 122.5, 122.4, 121.5, 118.1, 115.7, 115.5, 49.9 (2C), 45.5 ppm (2C); HRMS (ESI+) m/z calcd. calcd. for C18H17FN4OS+H+: 357.1180 [M+H]+: found 357.1173; HPLC: tr = 3.27 min (99.0% at 254 nm).
4-(6-((4-Bromophenyl)carbamoyl)benzo[d]thiazol-2-yl)piperazine (5f): Yield: 70 mg (72%); off-white amorphous powder; Rf(DCM:MeOH = 9:1) = 0.05; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 10.28 (s, 1H, Ar-NH-COR), 8.38 (d, J = 1.8 Hz, 1H, Ar-H7), 7.89 (dd, J1 = 8.5 Hz, J2 = 1.8 Hz, 1H, Ar-H5), 7.79–7.73 (m, 2H, 2 × Ar-H), 7.56–7.47 (m, 3H, 3 × Ar-H), 3.59–3.48 (m, 4H, 2 × piperazine-CH2), 2.86–2.77 ppm (m, 4H, 2 × piperazine-CH2), not visible (NH); 13C (101 MHz, [D6]DMSO, 25 °C, TMS): δ = 170.7, 165.6, 155.8, 139.3, 131.9 (2C), 130.6, 127.5, 126.6, 122.6 (2C), 121.6, 118.1, 115.5, 49.9 (2C), 45.5 ppm (2C); HRMS (ESI+) m/z calcd. calcd. for C18H17BrN4OS+H+: 417.0379 [M+H]+: found 417.0372; HPLC: tr = 5.45 min (96.5% at 254 nm).
4-(6-((4-Iodophenyl)carbamoyl)benzo[d]thiazol-2-yl)piperazine (5g): Yield: 44 mg (83%); light yellow amorphous powder; Rf(DCM:MeOH = 9:1) = 0.09; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 10.25 (s, 1H, Ar-NH-COR), 8.37 (s, 1H, Ar-H), 7.89 (d, J = 8.5 Hz, 1H, Ar-H), 7.71–7.60 (m, 4H, 4 × Ar-H), 7.50 (d, J = 8.1 Hz, 1H, Ar-H), 3.57–3.51 (m, 4H, 2 × piperazine-CH2), 2.84–2.79 ppm (m, 4H, 2 × piperazine-CH2), not visible (NH); 13C (101 MHz, [D6]DMSO, 25 °C, TMS): δ = 170.6, 165.6, 155.8, 139.8, 137.7 (2C), 130.6, 127.5, 126.6, 122.8 (2C), 121.6, 118.1, 87.4, 49.8 (2C), 45.5 ppm (2C); HRMS (ESI+) m/z calcd. for C18H17IN4OS+H+: 465.0234 [M+H]+: found 465.0241; HPLC: tr = 5.03 min (98.7% at 254 nm).
4-(6-((4-Isopropylphenyl)carbamoyl)benzo[d]thiazol-2-yl)piperazine (5h): Yield: 44 mg (80%); off-white amorphous powder; Rf(DCM:MeOH = 9:1) = 0.06; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 10.08 (s, 1H, Ar-NH-COR), 8.37 (d, J = 1.8 Hz, 1H, Ar-H7), 7.89 (dd, J1 = 8.5 Hz, J2 = 1.8 Hz, 1H, Ar-H5), 7.68 (d, J = 8.6 Hz, 2H, 2 × Ar-H), 7.50 (d, J = 8.5 Hz, 1H, Ar-H4), 7.21 (d, J = 8.6 Hz, 2H, 2 × Ar-H), 3.58–3.47 (m, 4H, 2 × piperazine-CH2), 2.90–2.78 (m, 5H, Ar-CH-(CH3)2 + 2 × piperazine-CH2), 1.20 ppm (d, J = 6.9 Hz, 6H, Ar-CH-(CH3)2); 13C (101 MHz, [D6]DMSO, 25 °C, TMS): δ = 170.5, 165.3, 155.6, 143.9, 137.6, 130.5, 127.9, 126.7 (2C), 126.5, 121.5, 120.8 (2C), 118.0, 49.9 (2C), 45.5 (2C), 33.4, 24.5 ppm (2C); HRMS (ESI+) m/z calcd. for C21H24N4OS+H+: 381.1744 [M+H]+: found 381.1737; HPLC: tr = 5.12 min (99.0% at 254 nm).
4-(6-((4-(Methylhydroxy)phenyl)carbamoyl)benzo[d]thiazol-2-yl)piperazine (5i): Yield: 43 mg (91%); off-white amorphous powder; Rf(DCM:MeOH = 9:1) = 0.08; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 10.04 (s, 1H, Ar-NH-COR), 8.36 (d, J = 1.8 Hz, 1H, Ar-H7), 7.89 (dd, J1 = 8.5 Hz, J2 = 1.8 Hz, 1H, Ar-H5), 7.71–7.63 (m, 2H, 2 × Ar-H), 7.50 (d, J = 8.5 Hz, 1H, Ar-H4), 6.95–6.89 (m, 2H, 2 × Ar-H), 3.74 (s, 3H, Ar-O-CH3), 3.58–3.48 (m, 4H, 2 × piperazine-CH2), 2.85–2.78 ppm (m, 4H, 2 × piperazine-CH2), not visible (NH); 13C (101 MHz, [D6]DMSO, 25 °C, TMS): δ = 170.5, 165.1, 155.9, 155.6, 132.9, 130.6, 128.0, 125.9, 122.3 (2C), 121.4, 118.0, 114.2 (2C), 55.6, 49.9 (2C), 45.5 ppm (2C); HRMS (ESI+) m/z calcd. for C19H20N4O2S+H+: 369.1380 [M+H]+: found 369.1374; HPLC: tr = 3.42 min (95.0% at 254 nm).
4-(6-((Benzyl)carbamoyl)benzo[d]thiazol-2-yl)piperazine (5j): Yield: 96 mg (81%); light pink amorphous powder; Rf(DCM:MeOH = 9:1) = 0.13; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 8.95 (t, J = 6.0 Hz, 1H, Ar-CH2-NH-COR), 8.30 (d, J = 1.8 Hz, 1H, Ar-H7), 7.83 (dd, J1 = 8.5 Hz, J2 = 1.8 Hz, 1H, Ar-H5), 7.45 (d, J = 8.5 Hz, 1H, Ar-H4), 7.35–7.29 (m, 4H, 4 × Ar-H), 7.26–7.20 (m, 1H, Ar-H), 4.48 (d, J = 6.0 Hz, 2H, Ar-CH2-NHCO), 3.56–3.48 (m, 4H, 2 × piperazine-CH2), 2.84–2.76 ppm (m, 4H, 2 × piperazine-CH2), not visible (NH); 13C (101 MHz, [D6]DMSO, 25 °C, TMS): δ = 170.3, 166.3, 155.3, 140.3, 130.6, 128.7 (2C), 127.7 (2C), 127.6, 127.2, 126.0, 121.1, 118.1, 49.3 (2C), 45.1 (2C), 43.1 ppm; HRMS (ESI+) m/z calcd. for C19H20N4OS+H+: 353.1430 [M+H]+: found 353.1424; HPLC: tr = 3.43 min (95.3% at 254 nm).
4-(6-((3-Chlorobenzyl)carbamoyl)benzo[d]thiazol-2-yl)piperazine (5k): Yield: 48 mg (74%); white amorphous powder; Rf(DCM:MeOH = 9:1) = 0.06; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 8.99 (t, J = 6.0 Hz, 1H, CH2-NH-COR), 8.30 (d, J = 1.9 Hz, 1H, Ar-H7), 7.82 (dd, J1 = 8.5 Hz, J2 = 1.9 Hz, 1H, Ar-H5), 7.46 (d, J = 8.5 Hz, 1H, Ar-H4), 7.39–7.33 (m, 2H, 2 × Ar-H), 7.33–7.27 (m, 2H, 2 × Ar-H), 4.47 (d, J = 6.0 Hz, 2H), 3.56–3.48 (m, 4H, 2 × piperazine-CH2), 2.85–2.76 ppm (m, 4H, 2 × piperazine-CH2), not visible (NH); 13C (101 MHz, [D6]DMSO, 25 °C, TMS): δ = 170.4, 166.4, 155.5, 142.9, 133.4, 130.6, 130.6, 127.5, 127.2, 127.1, 126.4, 126.0, 121.1, 118.1, 49.8 (2C), 45.5 (2C), 42.7 ppm; HRMS (ESI+) m/z calcd. for C19H19ClN4OS+H+: 387.1041 [M+H]+: found 387.1032; HPLC: tr = 4.26 min (98.0% at 254 nm).
4-(6-((4-Chlorobenzyl)carbamoyl)benzo[d]thiazol-2-yl)piperazine (5l): Yield: 59 mg (92%); off-white amorphous powder; Rf(DCM:MeOH = 9:1) = 0.07; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 8.98 (t, J = 6.0 Hz, 1H, t, J = 5.9 Hz, 1H, CH2-NH-COR), 8.29 (d, J = 1.8 Hz, 1H, Ar-H7), 7.82 (dd, J1 = 8.5 Hz, J2 = 1.8 Hz, 1H, Ar-H5), 7.46 (d, J = 8.5 Hz, 1H, Ar-H4), 7.41–7.30 (m, 4H, 4 × Ar-H), 4.46 (d, J = 6.0 Hz, 2H, RCO-NH-CH2-Ph), 3.57–3.46 (m, 4H, 2 × piperazine-CH2), 2.86–2.77 (m, 4H, 2 × piperazine-CH2), 2.77–2.69 ppm (m, 1H, NH); 13C (101 MHz, [D6]DMSO, 25 °C, TMS): δ = 170.4, 166.3, 155.4, 139.4, 131.7, 130.6, 129.6 (2C), 128.7 (2C), 127.3, 126.0, 121.1, 118.0, 49.7 (2C), 45.4 (2C), 42.5 ppm; HRMS (ESI+) m/z calcd. for C19H19ClN4OS+H+: 387.1041 [M+H]+: found 387.1034; HPLC: tr = 4.15 min (97.7% at 254 nm).
4-(6-((Indane-1-yl)carbamoyl)benzo[d]thiazol-2-yl)piperazine (5m): Yield: 52 mg (83%); white amorphous powder; Rf(DCM:MeOH = 9:1) = 0.07; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 8.66 (d, J = 8.3 Hz, 1H, CH-NH-CO), 8.32 (d, J = 1.7 Hz, 1H, Ar-H7), 7.86 (dd, J1 = 8.5 Hz, J2 = 1.7 Hz, 1H, Ar-H5), 7.44 (d, J = 8.5 Hz, 1H, Ar-H4), 7.29–7.14 (m, 4H, 4 × Ar-H), 5.56 (q, J = 8.3 Hz, 1H, indane-H), 3.55–3.48 (m, 4H, 2 × piperazine-CH2), 3.00 (ddd, J1 = 15.6 Hz, J2 = 8.8 Hz, J3 = 2.9 Hz, 1H, indane-H), 2.90–2.77 (m, 5H, 2 × piperazine-CH2, and indane-H), 2.47–2.40 (m, 1H), 2.05–1.92 ppm (m, 1H, indane-H), not visible (NH); 13C (101 MHz, [D6]DMSO, 25 °C, TMS): δ = 170.3, 166.2, 155.3, 144.8, 143.4, 130.4, 127.8, 127.5, 126.8, 126.2, 124.9, 124.5, 121.2, 118.0, 54.7, 49.8 (2C), 45.5 (2C), 33.2, 30.3 ppm; HRMS (ESI+) m/z calcd. for C21H22N4OS+H+: 379.1587 [M+H]+: found 379.1579; HPLC: tr = 4.22 min (96.6% at 254 nm).
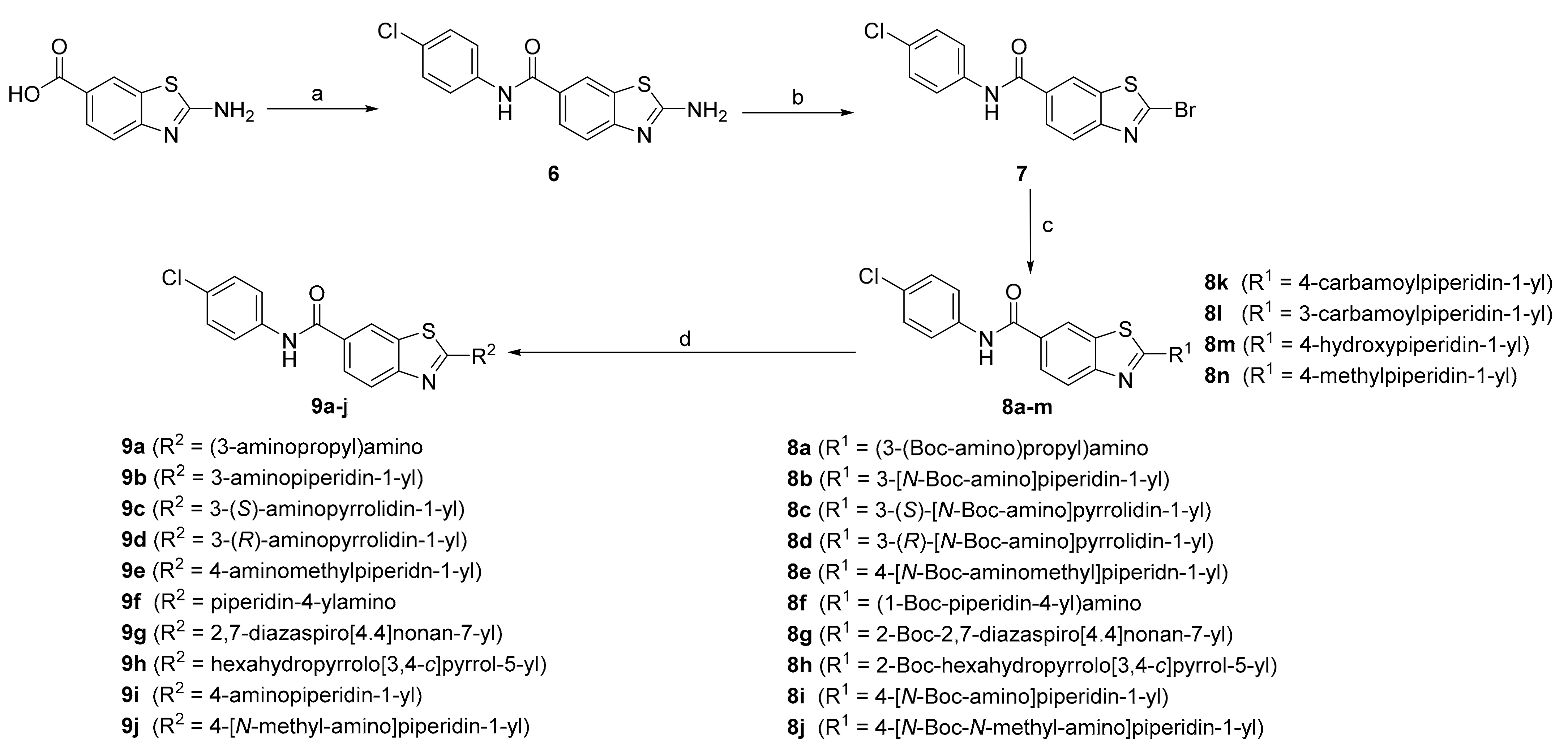
2-Amino-N-(4-chlorophenyl)benzo[d]thiazole-6-carboxamide (6): The reaction was performed under an argon atmosphere. 2-Aminobenzo[d]thiazole-6-carboxylic acid (2.13 g, 11.0 mmol) was dissolved in DMF (40 mL). EDC (2.04 g, 13.2 mmol), HOBt (1.93 g, 14.3 mmol) and N-methylmorpholine (2.86 mL, 27.4 mmol) were added in an ice bath. After 20 min, 4-chloroaniline (1.82 g, 14.3 mmol) was added to the reaction mixture, which was then left to stir for 2 days at room temperature. The solvent was removed under reduced pressure, and the residue was taken up in ethyl acetate (200 mL) and washed with 1 M NaOH (3 × 75 mL), 1% (w/v) citric acid (3 × 75 mL) and with saturated NaCl (100 mL). The organic phase was dried over Na2SO4, filtered, and the solvent was evaporated under reduced pressure. The product was purified with precipitation from a mixture of ethyl acetate and hexane. Yield: 1.82 g (55%); off-white amorphous powder; Rf(DCM:MeOH = 9:1) = 0.14; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 10.25 (s, 1H, Ar-NH-COR) 8.29 (d, J = 1.7 Hz, 1H, Ar-NH-COR), 7.87–7.80 (m, 5H, 5 × Ar-H), 7.45–7.37 ppm (m, 3H, Ar-H and NH2); 13C (101 MHz, [D6]DMSO, 25 °C, TMS): δ = 169.3, 165.7, 156.2, 138.9, 131.4, 129.0 (2C), 127.4; 127.4, 126.2, 122.1 (2C), 121.3, 117.4 ppm; LC–MS (ESI+): m/z 345.1 [M+H+CH3CN]+ (calcd. m/z = 303.0 for C14H10ClN3OS).
2-Bromo-N-(4-chlorophenyl)benzo[d]thiazole-6-carboxamide (7): Compound 6 (1.80 g, 5.93 mmol) and CuBr2 (2.65 g, 11.9 mmol) were dissolved in acetonitrile (90 mL). tert-Butyl nitrite (1.41 mL, 11.9 mmol, 867 mg/mL) was added in an ice bath, and the reaction mixture was stirred overnight at room temperature. The solvent was then removed under reduced pressure, and the residue was taken up in ethyl acetate (150 mL) and saturated NH4Cl (50 mL). The precipitate that formed was filtered off under pressure. The organic phase was additionally washed with saturated NH4Cl (3 × 75 mL) and saturated NaCl. The organic phase was dried over Na2SO4, filtered, and the solvent was evaporated under reduced pressure. Yield: 1.80 g (83%); orange amorphous powder; Rf(EtOAc:Hex = 1:2) = 0.34; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 10.58 (s, 1H, Ar-NH-COR), 8.73–8.70 (m, 1H, Ar-H), 8.16–8.12 (m, 1H, Ar-H), 8.09 (m, 1H, Ar-H), 7.87–7.80 (m, 2H, 2 × Ar-H), 7.48–7.40 ppm (m, 2H, 2 × Ar-H); 13C (101 MHz, [D6]DMSO, 25 °C, TMS): δ = 165.4, 154.2, 143.2, 138.5, 137.4, 132.6, 129.1 (2C), 127.9, 126.8, 122.7, 122.5, 122.3 ppm (2C); LC–MS (ESI-): m/z 365.0 [M-H]- (calcd. m/z = 365.9 for C14H8BrClN2OS).
General procedure for the synthesis of compounds 8a-n
The reaction was performed under an argon atmosphere. Compound 7 (1 equiv) was dissolved in THF, and triethylamine was added (2.5 equiv). The corresponding amine (1.25 equiv) was added to the reaction mixture, and the solution was stirred overnight at room temperature. The precipitate formed was filtered off. The solvent was then evaporated under reduced pressure, and the residue was dissolved in ethyl acetate (50–75 mL) and 1% (w/v) citric acid (30 mL). The organic phase was additionally washed with 1% (w/v) citric acid (2 × 30 mL) and saturated NaCl (30 mL). The organic phase was then dried over Na2SO4, filtered, and the solvent was evaporated under reduced pressure. The products were purified using column chromatography.
3-N-Boc-1-N-(6-((4-Chlorophenyl)carbamoyl)benzo[d]thiazol-2-yl)1,3-diaminopropane (8a): The product was purified using column chromatography (mobile phase: DCM:MeOH = 25:1). Yield: 159 mg (42%); white amorphous powder; Rf(DCM:MeOH = 20:1) = 0.19; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 10.26 (s, 1H, Ar-NH-COR), 8.34 (t, J = 5.3 Hz, 1H, Ar-NH-CH2), 8.29 (d, J = 1.7 Hz, 1H, Ar-H), 7.89–7.79 (m, 3H, 3 × Ar-H), 7.46 (d, J = 8.4 Hz, 1H, Ar-H), 7.43–7.38 (m, 2H, 2 × Ar-H), 6.91 (t, J = 5.3 Hz, 1H, CO-NH-CH2), 3.40 (dd, J1 = 12.6 Hz, J2 = 6.7 Hz, 2H, CH2), 3.02 (dd, J1 = 12.6 Hz, J2 = 6.7 Hz, 2H, CH2), 1.72 (p, J = 6.7 Hz, 2H, CH2), 1.38 ppm (s, 9H, COC(CH3)3); LC–MS (ESI+): m/z 461.2 [M+H]+ (calcd. m/z = 460.1 for C22H25N4O3S).
3-(N-Boc-Amino)-1-(6-((4-chlorophenyl)carbamoyl)benzo[d]thiazol-2-yl)piperidine (8b): The product was purified using column chromatography (mobile phase: DCM:MeOH = 40:1). Yield: 108 mg (33%); light orange amorphous powder; Rf(DCM:MeOH = 30:1) = 0.08; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 10.28 (s, 1H, Ar-NH-COR), 8.36 (d, J = 1.8 Hz, 1H, Ar-H7), 7.89 (dd, J1 = 8.5 Hz, J2 = 1.8 Hz, 1H, Ar-H5), 7.86–7.78 (m, 2H, 2 × Ar-H), 7.51 (d, J = 8.5 Hz, 1H, Ar-H4), 7.43–7.37 (m, 2H, 2 × Ar-H), 7.08 (d, J = 7.1 Hz, 1H, CH-NH-Boc), 4.05–3.97 (m, 1H, piperidine-H), 3.84–3.74 (m, 1H, piperidine-H), 3.53–3.42 (m, 1H, piperidine-H), 3.11 (dd, J1 = 12.6 Hz, J2 = 9.1 Hz, 1H, piperidine-H), 1.92–1.78 (m, 2H, 2 × piperidine-H), 1.63–1.32 ppm (m, 11H, 2 × piperidine-H and COC(CH3)3); LC–MS (ESI+): m/z 487.4 [M+H]+ (calcd. m/z = 486.1 for C24H27ClN4O3S).
3-(S)-(N-Boc-Amino)-1-(6-((4-chlorophenyl)carbamoyl)benzo[d]thiazol-2-yl)pyrrolidine (8c): The product was purified using column chromatography (mobile phase: DCM:MeOH = 40:1). Yield: 179 mg (56%); white amorphous powder; Rf(DCM:MeOH = 9:1) = 0.32; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 10.28 (s, 1H, Ar-NH-COR), 8.38 (d, J = 1.9 Hz, 1H, Ar-H7), 7.89 (dd, J1 = 8.5 Hz, J2 = 1.9 Hz, 1H, Ar-H5), 7.85–7.79 (m, 2H, 2 × Ar-H), 7.54 (d, J = 8.5 Hz, 1H, Ar-H4), 7.44–7.38 (m, 2H, 2 × Ar-H), 7.37–7.33 (m, 1H, CH-NH-Boc), 4.24–4.14 (m, 1H, pyrrolidine-H), 3.79–3.51 (m, 3H, 3 × pyrrolidine-H), 3.43–3.36 (m, 1H, pyrrolidine-H), 2.28–2.17 (m, 1H, pyrrolidine-H), 2.02–1.91 (m, 1H, pyrrolidine-H), 1.40 ppm (s, 9H, COC(CH3)3); [α]D25 = 9.5 (c = 0.86 in DMF); LC–MS (ESI+): m/z 473.3 [M+H]+ (calcd. m/z = 472.1 for C23H25ClN4O3S).
3-(R)-(N-Boc-Amino)-1-(6-((4-chlorophenyl)carbamoyl)benzo[d]thiazol-2-yl)pyrrolidine (8d): The product was purified using column chromatography (mobile phase: DCM:MeOH = 30:1). Yield: 130 mg (67%); white amorphous powder; Rf(DCM:MeOH = 9:1) = 0.50; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 10.28 (s, 1H, Ar-NH-COR), 8.38 (d, J = 1.9 Hz, 1H, Ar-H7), 7.89 (dd, J1 = 8.5 Hz, J2 = 1.9 Hz, 1H, Ar-H5), 7.85–7.79 (m, 2H, 2 × Ar-H), 7.54 (d, J = 8.5 Hz, 1H, Ar-H4), 7.45–7.38 (m, 2H, 2 × Ar-H), 7.35 (d, J = 6.3 Hz, 1H, CH-NH-Boc), 4.22–4.15 (m, 1H, pyrrolidine-H), 3.78–3.51 (m, 3H, 3 × pyrrolidine-H), 3.45–3.36 (m, 1H, pyrrolidine-H), 2.28–2.17 (m, 1H, pyrrolidine-H), 2.01–1.92 (m, 1H, pyrrolidine-H), 1.40 ppm (s, 9H, COC(CH3)3); [α]D25 = − 9.5 (c = 0,69 in DMF); LC–MS (ESI+): m/z 473.0 [M+H]+ (calcd. m/z = 472.1 for C23H25ClN4O3S).
4-(N-Boc-Aminomethyl)-1-(6-((4-chlorophenyl)carbamoyl)benzo[d]thiazol-2-yl)piperidine (8e): The product was purified using column chromatography (mobile phase: EtOAc:Hex = 1:1). Yield: 67 mg (20%); white amorphous powder; Rf(EtOAc:Hex = 2:1) = 0.34; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 10.28 (s, 1H, Ar-NH-COR), 8.37 (d, J = 1.9 Hz, 1H, Ar-H7), 7.89 (dd, J1 = 8.5 Hz, J2 = 1.9 Hz, 1H, Ar-H5), 7.84–7.79 (m, 2H, 2 × Ar-H), 7.50 (d, J = 8.5 Hz, 1H, Ar-H4), 7.43–7.37 (m, 2H, 2 × Ar-H), 6.96 (t, J = 5.8 Hz, 1H, CH2-NH-Boc), 4.15–4.00 (m, 2H, 2 × methylpiperdine-H), 3.24–3.13 (m, 2H, 2 × methylpiperdine-H), 2.86 (t, J = 6.2 Hz, 2H, 2 × methylpiperdine-H), 1.80–1.64 (m, 3H, 3 × methylpiperdine-H), 1.38 (s, 9H, COC(CH3)3), 1.27–1.11 ppm (m, 2H, 2 × methylpiperdine-H); LC–MS (ESI): m/z 501.4 [M+H]+ (calcd. m/z = 500.2 for C25H29ClN4O3S).
1-Boc-4-((6-((4-Chlorophenyl)carbamoyl)benzo[d]thiazol-2-yl)amino)piperidine (8f): The product was purified using column chromatography (mobile phase: DCM:MeOH = 20:1). Yield: 49 mg (15%); white amorphous powder; Rf(DCM:MeOH = 20:1) = 0.21; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 10.26 (s, 1H, Ar-NH-COR), 8.38 (d, J = 7.4 Hz, 1H, Ar-NH-CH), 8.29 (d, J = 1.8 Hz, 1H, Ar-H), 7.88–7.78 (m, 3H, 3 × Ar-H), 7.47 (d, J = 8.5 Hz, 1H, Ar-H), 7.44–7.36 (m, 2H, 2 × Ar-H), 4.02–3.81 (m, 3H, 3 × piperidine-H), 3.05–2.88 (m, 2H, 2 × piperidine-H), 2.01–1.93 (m, 2H, 2 × piperidine-H), 1.45–1.30 ppm (m, 11H, COC(CH3)3+ 2 × piperidine-H); LC–MS (ESI+): m/z 487.1 [M+H]+ (calcd. m/z = 486.1 for C24H27ClN4O3S).
2-Boc-7-(6-((4-Chlorophenyl)carbamoyl)benzo[d]thiazol-2-yl)-2,7-diazaspiro[4.4]nonane (8g): The product was purified using column chromatography (mobile phase: DCM:MeOH = 20:1). Yield: 141 mg (51%); off-white amorphous powder; Rf(DCM:MeOH = 9:1) = 0.44; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 10.28 (s, 1H, Ar-NH-COR), 8.39 (d, J = 1.9 Hz, 1H, Ar-H7), 7.90 (dd, J1 = 8.5 Hz, J2 = 1.9 Hz, 1H, Ar-H5), 7.86–7.79 (m, 2H, 2 × Ar-H), 7.53 (d, J = 8.5 Hz, 1H, Ar-H4), 7.44–7.36 (m, 2H, 2 × Ar-H), 3.65 (s, 2H, CH2), 3.52 (s, 2H, CH2), 2.08–2.01 (m, 2H, CH2), 1.97–1.84 (m, 2H, CH2), 1,40 ppm (s, 9H, COC(CH3)3). covered with solvent (2 × CH2); LC–MS (ESI+): m/z 513.2 [M+H]+ (calcd. m/z = 512.2 for C26H29ClN4O3S).
2-Boc-5-(6-((4-Chlorophenyl)carbamoyl)benzo[d]thiazol-2-yl)hexahydropyrrolo[3,4-c]pyrrole (8h): The product was purified using column chromatography (mobile phase: DCM:MeOH = 20:1). Yield: 66 mg (24%); white amorphous powder; Rf(DCM:MeOH = 20:1) = 0.14; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 10.28 (s, 1H, Ar-NH-COR), 8.39 (d, J = 1.9 Hz, 1H, Ar-H7), 7.90 (dd, J1 = 8.5 Hz, J2 = 1.9 Hz, 1H, Ar-H5), 7.85–7.78 (m, 2H, 2 × Ar-H), 7.54 (d, J = 8.5 Hz, 1H, Ar-H4), 7.43–7.37 (m, 2H, 2 × Ar-H), 3.83–3.73 (m, 2H, 2 × CH), 3.61–3.50 (m, 2H, 2 × CH), 3.49–3.39 (m, 2H, 2 × CH), 3.26–3.18 (m, 2H, 2 × CH), 3.12–3.01 (m, 2H, 2 × CH), 1.39 ppm (s, 9H, COC(CH3)3); LC–MS (ESI+): m/z 499.0 [M+H]+ (calcd. m/z = 498.1 for C25H27ClN4O3S).
4-(N-Boc-Amino)-1-(6-((4-chlorophenyl)carbamoyl)benzo[d]thiazol-2-yl)piperidine (8i): The product was purified using column chromatography (mobile phase: DCM:MeOH = 30:1). Yield: 75 mg (28%); white amorphous powder; Rf(DCM:MeOH = 30:1) = 0.28; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 10.28 (s, 1H, Ar-NH-COR), 8.37 (d, J = 1.9 Hz, 1H, Ar-H7), 7.89 (dd, J1 = 8.5 Hz, J2 = 1.9 Hz, 1H, Ar-H5), 7.86–7.78 (m, 2H, 2 × Ar-H), 7.51 (d, J = 8.5 Hz, 1H, Ar-H4), 7.43–7.37 (m, 2H, 2 × Ar-H), 6.95 (d, J = 7.9 Hz, 1H, CH-NH-Boc), 4.02 (d, J = 13.5 Hz, 2H, 2 × piperidine-H), 3.64–3.52 (m, 1H, piperidine-H), 3.31–3.26 (m, 2H, 2 × piperidine-H), 1.93–1.79 (m, 2H, 2 × piperidine-H), 1.50–1.41 (m, 2H, 2 × piperidine-H), 1.39 ppm (s, 9H, COC(CH3)3); 13C (101 MHz, [D6]DMSO, 25 °C, TMS): δ = 168.9, 164.5, 154.9, 154.2, 137.8, 129.9, 127.9 (2C), 126.4, 126.4, 125.5, 121.1 (2C), 120.5, 116.9, 77.1, 46.6 (2C), 46.2, 30.5 (2C), 27.6 ppm (3C); LC–MS (ESI+): m/z 487.1 [M+H]+ (calcd. m/z = 486.1 for C24H27ClN4O3S).
4-(N-Boc-N-Methylamino)-1-(6-((4-chlorophenyl)carbamoyl)benzo[d]thiazol-2-yl)piperidine (8j): The product was purified using column chromatography (mobile phase: DCM:MeOH = 20:1). Yield: 155 mg (50%); off-white amorphous powder; Rf(DCM:MeOH = 20:1) = 0.12; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 10.29 (s, 1H, Ar-NH-COR), 8.38 (d, J = 1.9 Hz, 1H, Ar-H7), 7.90 (dd, J1 = 8.5 Hz, J2 = 1.9 Hz, 1H, Ar-H5), 7.85–7.79 (m, 2H, 2 × Ar-H), 7.52 (d, J = 8.5 Hz, 1H, Ar-H4), 7.44–7.36 (m, 2H, 2 × Ar-H), 4.24–3.96 (m, 3H, 3 × piperidine-H), 3.31–3.23 (m, 2H, 2 × piperidine-H), 2.67 (s, 3H, N-CH3) 1.83–1.65 (m, 4H, 4 × piperidine-H), 1.41 ppm (s, 9H, COC(CH3)3); LC–MS (ESI+): m/z 501.2 [M+H]+ (calcd. m/z = 500.2 for C25H29ClN4O3S).
2-(4-Carbamoylpiperidin-1-yl)-N-(4-chlorophenyl)benzo[d]thiazole-6-carboxamide (8k): The product was purified using column chromatography (mobile phase: DCM:MeOH = 17.5:1). Yield: 31 mg (23%); white amorphous powder; Rf(DCM:MeOH = 17:1) = 0.08; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 10.29 (s, 1H, Ar-NH-COR), 8.38 (d, J = 1.9 Hz, 1H, Ar-H7), 7.90 (dd, J1 = 8.5 Hz, J2 = 1.9 Hz, 1H, Ar-H5), 7.85–7.79 (m, 2H, 2 × Ar-H), 7.52 (d, J = 8.5 Hz, 1H, Ar-H4), 7.43–7.38 (m, 2H, 2 × Ar-H), 7.36 (s, 1H, CH-CONHa), 6.87 (s, 1H, CH-CONHb), 4.08 (d, J = 13.0 Hz, 2H, 2 × piperidine-H), 3.29–3.21 (m, 2H, 2 × piperidine-H), 2.45–2.39 (m, 1H, piperidine-H), 1.90–1.79 (m, 2H, 2 × piperidine-H), 1.68–1.56 ppm (m, 2H, 2 × piperidine-H); 13C (101 MHz, [D6]DMSO, 25 °C, TMS): δ = 176.1, 170.2, 165.6, 155.9, 138.9, 130.8, 129.0 (2C), 127.5, 127.4, 126.6, 122.2 (2C), 121.6, 118.0, 48.4, 41.5 (2C), 28.2 ppm (2C); HRMS (ESI+) m/z calcd. for C20H19ClN4O2S+H+: 415.0990 [M+H]+: found 415.0966; HPLC: tr = 5.41 min (99.0% at 254 nm).
2-(3-Carbamoylpiperidin-1-yl)-N-(4-chlorophenyl)benzo[d]thiazole-6-carboxamide (8l): The product was purified using column chromatography (mobile phase: DCM:MeOH = 15:1). Yield: 73 mg (65%); white amorphous powder; Rf(DCM:MeOH = 9:1) = 0.27; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 10.29 (s, 1H, Ar-NH-COR), 8.38 (s, 1H, Ar-H), 7.90 (d, J = 8.5 Hz, 1H, Ar-H), 7.82 (d, J = 8.4 Hz, 2H, 2 × Ar-H), 7.52 (d, J = 8.5 Hz, 1H, Ar-H), 7.46 (s, 1H, CO-NHa), 7.41 (d, J = 8.4 Hz, 2H, 2 × Ar-H), 6.98 (s, 1H, CO-NHb), 4.15–4.07 (m, 1H, piperidine-H), 4.00–3.93 (m, 1H, piperidine- H), 3.30–3.19 (m, 2H, 2 × piperidine-H), 2.00–1.92 (m, 1H, piperidine- H), 1.85–1.77 (m, 1H, piperidine- H), 1.71–1.50 ppm (m, 3H, 3 × piperidine-H); 13C (101 MHz, [D6]DMSO, 25 °C, TMS): δ = 174.8, 170.2, 165.6, 155.9, 138.9, 130.8, 129.0 (2C), 127.5, 127.4, 126.6, 122.2 (2C), 121.6, 118.0, 51.0, 49.2, 41.8, 27.8, 24.3 ppm; HRMS (ESI+) m/z calcd. for C20H19ClN4O2S+H+: 415.0990 [M+H]+: found 415.0985; HPLC: tr = 5.54 min (99.9% at 254 nm).
2-(4-Hydroxypiperidin-1-yl)-N-(4-chlorophenyl)benzo[d]thiazole-6-carboxamide (8m): The product was purified using column chromatography (mobile phase: EtOAc:Hex = 2:1). Yield: 49 mg (46%); white amorphous powder; Rf(DCM:MeOH = 20:1) = 0.07; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 10.28 (s, 1H, Ar-NH-COR), 8.37 (d, J = 1.8 Hz, 1H, Ar-H7), 7.89 (dd, J1 = 8.5 Hz, J2 = 1.8 Hz, 1H, Ar-H5), 7.86–7.79 (m, 2H, 2 × Ar-H), 7.50 (d, J = 8.5 Hz, 1H, Ar-H4), 7.44–7.37 (m, 2H, 2 × Ar-H), 4.88 (d, J = 4.1 Hz, 1H, CH-OH), 3.94–3.83 (m, 2H, 2 × piperidine-H), 3.84–3.76 (m, 1H, piperidine-H), 3.46–3.37 (m, 2H, 2 × piperidine-H), 1.93–1.80 (m, 2H, 2 × piperidine-H), 1.56–1.41 ppm (m, 2H, 2 × piperidine-H); 13C (101 MHz, [D6]DMSO, 25 °C, TMS): δ = 170.1, 165.6, 156.0, 138.9, 130.9, 129.0 (2C), 127.4, 126.6, 122.2 (2C), 121.6, 118.0, 65.5, 46.41 (2C), 33.81 (2C) ppm; HRMS (ESI+) m/z calcd. for C19H18ClN3O2S+H+: 388.0881 [M+H]+: found 388.0885; HPLC: tr = 6.00 min (96.2% at 254 nm).
2-(4-Methylpiperidin-1-yl)-N-(4-chlorophenyl)benzo[d]thiazole-6-carboxamide (8n): The product was purified using column chromatography (mobile phase: DCM:MeOH = 67:1). Yield: 27 mg (29%); white amorphous powder; Rf(DCM:MeOH = 66:1) = 0.12; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 10.28 (s, 1H, Ar-NH-COR), 8.37 (d, J = 1.9 Hz, 1H, Ar-H7), 7.89 (dd, J1 = 8.5 Hz, J2 = 1.9 Hz, 1H, Ar-H5), 7.85–7.78 (m, 2H, 2 × Ar-H), 7.50 (d, J = 8.5 Hz, 1H, Ar-H4), 7.44–7.38 (m, 2H, 2 × Ar-H), 4.11–4.02 (m, 2H, 2 × piperidine-H), 3.25–3.17 (m, 2H, 2 × piperidine-H), 1.80–1.62 (m, 3H, 3 × piperidine-H), 1.28–1.14 (m, 2H, 2 × piperidine-H), 0.96 ppm (d, J = 6.4 Hz, 3H, CH3); 13C (101 MHz, [D6]DMSO, 25 °C, TMS): δ = 170.1, 165.6, 156.0, 138.9, 130.8, 128.9 (2C), 127.4, 127.4, 126.6, 122.2 (2C), 121.5, 117.9, 49.0 (2C), 33.5 (2C), 30.5, 22.0 ppm; HRMS (ESI+) m/z calcd. for C20H20N3OS+H+: 386.1088 [M+H]+: found 386.1083; HPLC: tr = 7.49 min (99.4% at 254 nm).
General procedure for the synthesis of compounds 9a-j
Acidolysis was performed in the same manner as described for the synthesis of compounds 5a–m.
1-N-(6-((4-Chlorophenyl)carbamoyl)benzo[d]thiazol-2-yl)1,3-diaminopropane (9a): Yield: 29 mg (62%); off-white amorphous powder; Rf(DCM:MeOH = 9:1) = 0.0; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 10.25 (s, 1H, Ar-NH-COR), 8.28 (d, J = 1.8 Hz, 1H, Ar-H), 7.88–7.78 (m, 3H, 3 × Ar-H), 7.48–7.36 (m, 3H, 3 × Ar-H), 3.44 (t, J = 6.8 Hz, 2H, N-CH2-CH2), 2.63 (t, J = 6.8 Hz, 2H, N-CH2-CH2), 1.66 ppm (p, J = 6.8 Hz, 2H), not visible (NH and NH2); 13C (101 MHz, [D6]DMSO, 25 °C, TMS): δ = 168.8, 165.7, 156.0, 138.9, 130.7, 128.9 (2C), 127.4, 127.2, 126.2, 122.1 (2C), 121.3, 117.5, 42.4 (2C), 33.0 ppm; HRMS (ESI+) m/z calcd. for C17H17ClN4OS+H+: 361.0884 [M+H]+: found 361.0877; HPLC: tr = 4.40 min (99.8% at 254 nm).
3-Amino-1-(6-((4-chlorophenyl)carbamoyl)benzo[d]thiazol-2-yl)piperidine (9b): Yield: 39 mg (89%); off-white amorphous powder; Rf(DCM:MeOH = 4:1) = 0.0; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 10.27 (s, 1H, Ar-NH-COR), 8.36 (d, J = 1.8 Hz, 1H, Ar-H7), 7.89 (dd, J1 = 8.5 Hz, J2 = 1.8 Hz, 1H, Ar-H5), 7.85–7.78 (m, 2H, 2 × Ar-H), 7.49 (d, J = 8.5 Hz, 1H, Ar-H4), 7.43–7.38 (m, 2H, 2 × Ar-H), 3.95 (dd, J1 = 29.1 Hz, J2 = 12.8 Hz, 2H, CH-NH2), 3.23–3.14 (m, 1H, piperidine-H), 2.93–2.86 (m, 1H, piperidine-H), 2.79–2.70 (m, 1H, piperidine-H), 1.93–1.84 (m, 1H, piperidine-H), 1.83–1.70 (m, 3H, 3 × piperidine-H), 1.60–1.46 (m, 1H, piperidine-H), 1.34–1.21 ppm (m, 1H, piperidine-H); 13C (101 MHz, [D6]DMSO, 25 °C, TMS): δ = 170.2, 165.6, 156.0, 138.9, 130.8, 129.0 (2C), 127.4, 127.3, 126.6, 122.2 (2C), 121.5, 117.9, 57.1, 48.9, 47.9, 33.7, 23.7 ppm; HRMS (ESI+) m/z calcd. for C19H19ClN4OS+H+: 387.1041 [M+H]+: found 387.1017; HPLC: tr = 5.43 min (97.9% at 254 nm).
3-(S)-Amino-1-(6-((4-chlorophenyl)carbamoyl)benzo[d]thiazol-2-yl)pyrrolidine (9c): Yield: 47 mg (80%); off-white amorphous powder; Rf(DCM:MeOH = 9:1) = 0.03; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 10.27 (s, 1H, Ar-NH-COR), 8.37 (d, J = 1.8 Hz, 1H, Ar-H7), 7.88 (dd, J1 = 8.5 Hz, J2 = 1.9 Hz, 1H, Ar-H5), 7.84–7.78 (m, 2H, 2 × Ar-H), 7.52 (d, J = 8.5 Hz, 1H, Ar-H4), 7.44–7.37 (m, 2H, 2 × Ar-H), 3.73–3.46 (m, 4H, CH-NH2 + 2 × pyrrolidine-H), 3.29–3.15 (m, 1H, pyrrolidine-H), 2.17–2.04 (m, 1H, pyrrolidine-H), 1.90–1.74 ppm (m, 3H, 3 × pyrrolidine-H); 13C (101 MHz, [D6]DMSO, 25 °C, TMS): δ = 167.0, 165.7, 156.4, 138.9, 130.9, 129.0 (2C), 127.4, 126.9, 126.5, 122.2 (2C), 121.7, 117.7, 58.3, 51.4, 48.5, 34.5 ppm; [α]D25 = 11.5 (c = 0.77 in DMF); HRMS (ESI+) m/z calcd. for C18H17ClN4OS+H+: 373.0884 [M+H]+: found 373.0878; HPLC: tr = 4.41 min (98.9% at 254 nm).
3-(R)-Amino-1-(6-((4-chlorophenyl)carbamoyl)benzo[d]thiazol-2-yl)pyrrolidine (9d): Yield: 41 mg (75%); off-white amorphous powder; Rf(DCM:MeOH = 9:1) = 0.0; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 10.27 (s, 1H, Ar-NH-COR), 8.37 (d, J = 1.8 Hz, 1H, Ar-H7), 7.88 (dd, J1 = 8.5 Hz, J2 = 1.9 Hz, 1H, Ar-H5), 7.85–7.79 (m, 2H, 2 × Ar-H), 7.52 (d, J = 8.5 Hz, 1H, Ar-H4), 7.44–7.37 (m, 2H, 2 × Ar-H), 3.73–3.48 (m, 4H, CH-NH2 and 2 × pyrrolidine-H), 3.27–3.18 (m, 1H, pyrrolidine-H), 2.16–2.07 (m, 1H, pyrrolidine-H), 1.96–1.74 ppm (m, 3H, 3 × pyrrolidine-H); 13C (101 MHz, [D6]DMSO, 25 °C, TMS): δ = 167.0, 165.7, 156.4, 138.9, 130.9, 129.0 (2C), 127.4, 126.9, 126.5, 122.2 (2C), 121.7, 117.7, 58.2, 51.4, 48.5, 34.4 ppm; [α]D25 = - 11.5 (c = 0.77 in DMF);HRMS (ESI+) m/z calcd. for C18H17ClN4OS+H+: 373.0884 [M+H]+: found 373.0879; HPLC: tr = 4.41 min (98.7% at 254 nm).
4-Aminomethyl-1-(6-((4-chlorophenyl)carbamoyl)benzo[d]thiazol-2-yl)piperidin (9e): Yield: 45 mg (94%); off-white amorphous powder; Rf(DCM:MeOH = 9:1) = 0.0; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 10.28 (s, 1H, Ar-NH-COR), 8.36 (d, J = 1.8 Hz, 1H, Ar-H7), 7.89 (dd, J1 = 8.5 Hz, J2 = 1.8 Hz, 1H, Ar-H5), 7.84–7.80 (m, 2H, 2 × Ar-H), 7.50 (d, J = 8.5 Hz, 1H, Ar-H4), 7.43–7.38 (m, 2H, 2 × Ar-H), 4.13–4.05 (m, 2H, CH2-NH2), 3.19 (td, J1 =12.7 Hz, J2 = 2.6 Hz, 2H, CH-CH2-NH2), 2.47–2.44 (m, 2H, 2 × piperidine-H), 1.87–1.80 (m, 2H, 2 × piperidine-H), 1.59–1.40 (m, 3H, 3 × piperidine-H), 1.26–1.12 ppm (m, 2H, 2 × piperidine-H); 13C (101 MHz, [D6]DMSO, 25 °C, TMS): δ = 170.2, 165.6, 156.1, 138.9, 130.8, 129.0 (2C), 127.4, 127.4, 126.6, 122.2 (2C), 121.5, 117.9, 49.0 (2C), 47.7, 29.5 ppm (3C); HRMS (ESI+) m/z calcd. for C20H21ClN4OS+H+: 401.1197 [M+H]+: found 401.1192; HPLC: tr = 5.49 min (97.7% at 254 nm).
4-((6-((4-Chlorophenyl)carbamoyl)benzo[d]thiazol-2-yl)amino)piperidine (9f): Yield: 39 mg (82%); off-white amorphous powder; Rf(DCM:MeOH = 9:1) = 0.0; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 10.24 (s, 1H, Ar-NH-COR), 8.34 (d, J = 7.3 Hz, 1H, Ar-NH-CH), 8.27 (d, J = 1.7 Hz, 1H, Ar-H), 7.87–7.79 (m, 3H, 3 × Ar-H), 7.45 (d, J = 8.4 Hz, 1H, Ar-H), 7.42–7.37 (m, 2H, 2 × Ar-H), 3.82–3.76 (m, 1H, piperidine-H), 2.99–2.90 (m, 2H, 2 × piperidine-H), 1.98–1.88 (m, 2H, 2 × piperidine-H), 1.34 ppm (ddd, J1 = 14.9 Hz, J2 = 11.7 Hz, J3 = 3.8 Hz, 2H, 2 × piperidine-H), not visible (2 × piperidine-H and NH); 13C (101 MHz, [D6]DMSO, 25 °C, TMS): δ = 167.6, 165.7, 156.1, 138.9, 130.7, 128.9 (2C), 127.4, 127.2, 126.2, 122.1 (2C), 121.3, 117.5, 52.5, 45.3 (2C), 33.4 ppm (2C); HRMS (ESI+) m/z calcd. for C19H19ClN4OS+H+: 387.1041 [M+H]+: found 387.1033; HPLC: tr = 4.45 min (95.1% at 254 nm).
7-(6-((4-Chlorophenyl)carbamoyl)benzo[d]thiazol-2-yl)-2,7-diazaspiro[4.4]nonane (9g): Yield: 68 mg (85%); off-white amorphous powder; Rf(DCM:MeOH = 9:1) = 0.0; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 10.27 (s, 1H, Ar-NH-COR), 8.39 (d, J = 1.8 Hz, 1H, Ar-H7), 7.89 (dd, J1 = 8.5 Hz, J2 = 1.8 Hz, 1H, Ar-H5), 7.84–7.79 (m, 2H, 2 × Ar-H), 7.52 (d, J = 8.5 Hz, 1H, Ar-H4), 7.43–7.37 (m, 2H, 2 × Ar-H), 3.69–3.40 (m, 4H, 2 × CH2), 2.94–2.86 (m, 2H, CH2), 2.81–2.70 (m, 2H, CH2), 2.07–1.97 (m, 2H, CH2), 1.81–1.68 ppm (m, 2H, CH2), not visible (NH); 13C (101 MHz, [D6]DMSO, 25 °C, TMS): δ = 166.9, 165.7, 156.3, 138.9, 130.9, 128.9 (2C), 127.4, 127.0, 126.6, 122.2 (2C), 121.7, 117.8, 59.3, 55.6, 49.9, 49.4, 45.8, 35.9, 35.5 ppm; HRMS (ESI+) m/z calcd. for C21H21ClN4OS+H+: 413.1197 [M+H]+: found 413.1190; HPLC: tr = 4.59 min (97.0% at 254 nm).
5-(6-((4-Chlorophenyl)carbamoyl)benzo[d]thiazol-2-yl)hexahydropyrrolo[3,4-c]pyrrole (9h): Yield: 30 mg (70%); off-white amorphous powder; Rf(DCM:MeOH = 9:1) = 0.0; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 10.28 (s, 1H, Ar-NH-COR), 8.38 (d, J = 1.9 Hz, 1H, Ar-H7), 7.89 (dd, J1 = 8.5 Hz, J2 = 1.9 Hz, 1H, Ar-H5), 7.85–7.79 (m, 2H, 2 × Ar-H), 7.53 (d, J = 8.5 Hz, 1H, Ar-H4), 7.43–7.38 (m, 2H, 2 × Ar-H), 3.82–3.75 (m, 2H, 2 × CH), 3.39–3.37 (m, 2H, 2 × CH ), 2.96–2.88 (m, 4H, 4 × CH), 2.74–2.69 ppm (m, 2H, 2 × CH), not visible (NH); 13C (101 MHz, [D6]DMSO, 25 °C, TMS): δ = 166.6, 165.7, 156.2, 138.9, 131.1, 128.9 (2C), 127.4, 127.1, 126.5, 122.2 (2C), 121.7, 117.9, 55.3 (2C), 53.2 (2C), 43.8 ppm (2C); HRMS (ESI+) m/z calcd. for C20H19ClN4OS+H+: 399.1040 [M+H]+: found 399.1034; HPLC: tr = 4.47 min (98.8% at 254 nm).
4-Amino-1-(6-((4-chlorophenyl)carbamoyl)benzo[d]thiazol-2-yl)piperidine (9i): Yield: 32 mg (73%); off-white amorphous powder; Rf(DCM:MeOH = 9:1) = 0.03; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 10.28 (s, 1H, Ar-NH-COR), 8.37 (d, J = 1.8 Hz, 1H, Ar-H7), 7.89 (dd, J1 = 8.5 Hz, J2 = 1.8 Hz, 1H, Ar-H5), 7.85–7.79 (m, 2H, 2 × Ar-H), 7.50 (d, J = 8.5 Hz, 1H, Ar-H4), 7.43–7.37 (m, 2H, 2 × Ar-H), 3.99 (d, J = 13.1 Hz, 2H, CH-NH2), 3.30–3.23 (m, 2H, 2 × piperidine-H), 2.91–2.83 (m, 1H, piperidine-H), 1.87–1.64 (m, 4H, 4 × piperidine-H), 1.37–1.23 ppm (m, 2H, 2 × piperidine-H); 13C (101 MHz, [D6]DMSO, 25 °C, TMS): δ = 170.1, 165.6, 156.1, 138.9, 130.9, 128.9 (2C), 127.4, 127.4, 126.6, 122.2 (2C), 121.5, 117.9, 48.0, 47.6 (2C), 34.8 ppm(2C); HRMS (ESI+) m/z calcd. for C19H18ClN3O2S+H+: 387.1041 [M+H]+: found 387.1033; HPLC: tr = 4.54 min (98.9% at 254 nm).
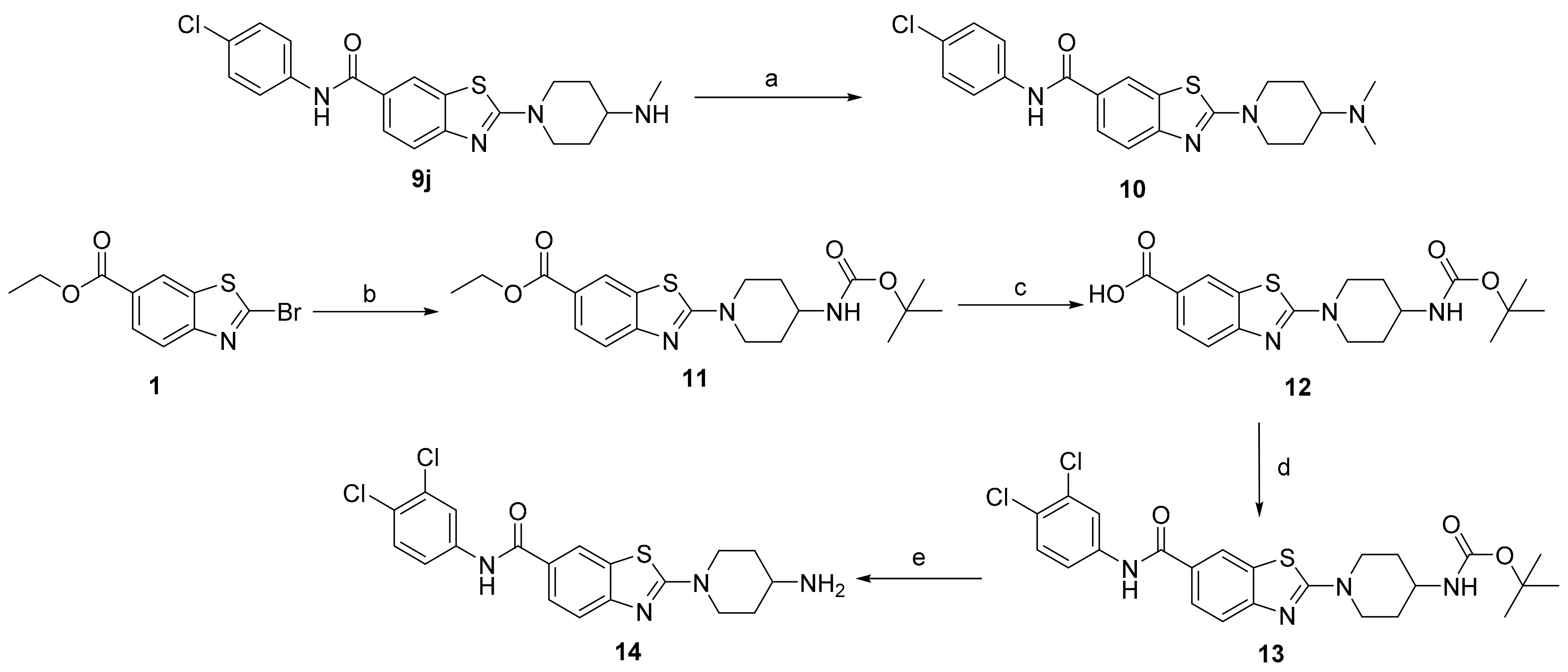
4-(N-Methylamino)-1-(6-((4-chlorophenyl)carbamoyl)benzo[d]thiazol-2-yl)piperidine (9j): Yield: 89 mg (74%); off-white amorphous powder; Rf(DCM:MeOH = 9:1) = 0.0; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 10.28 (s, 1H, Ar-NH-COR), 8.37 (d, J = 1.8 Hz, 1H, Ar-H7), 7.89 (dd, J1 = 8.5 Hz, J2 = 1.8 Hz, 1H, Ar-H5), 7.85–7.78 (m, 2H, 2 × Ar-H), 7.50 (d, J = 8.5 Hz, 1H, Ar-H4), 7.44–7.37 (m, 2H, 2 × Ar-H), 3.93–4.00 (m, 2H, 2 × piperidine-H), 3.32–3.26 (m, 2H, 2 × piperidine-H), 2.63–2.54 (m, 1H, piperidine-H), 2.30 (s, 3H, NH-CH3), 1.97–1.86 (m, 2H, 2 × piperidine-H), 1.83–1.67 (m, 1H, NH), 1.39–1.28 ppm (m, 2H, 2 × piperidine-H); 13C (101 MHz, [D6]DMSO, 25 °C, TMS): δ = 170.1, 165.6, 156.0, 138.9, 130.9, 128.9 (2C), 127.4, 127.4, 126.6, 122.2 (2C), 121.5, 118.0, 55.7, 47.3 (2C), 33.7, 31.3 ppm (2C); HRMS (ESI) m/z calcd. for C20H21ClN4OS+H+: 401.1197 [M+H]+: found 401.1192; HPLC: tr = 4.53 min (99.6% at 254 nm).
4-(N,N-Dimethylamino)-1-(6-((4-chlorophenyl)carbamoyl)benzo[d]thiazol-2-yl)piperidine (10): Compound 9j (60 mg, 0.15 mmol) was dissolved in a mixture of DCM (5 mL) and MeOH (5 mL). Formaldehyde(aq) (56 μL, 37% [w/w], 1.08 g/mL, 0.75 mmol) and acetic acid (8.6 μL, 100%, 1.05 g/mL, 0.15 mmol) were added. After 2 h, NaCNBH3 (15 mg, 0.24 mmol) was added. The reaction mixture was stirred overnight at room temperature. The solvent was evaporated under reduced pressure and the residue was taken up in DCM (30 mL). The organic phase was washed with 1 M NaOH (3 × 25 mL), saturated NaCl (25 mL) and dried over Na2SO4. The solvent was evaporated under reduced pressure. Yield: 56 mg (90%); white amorphous powder; Rf(DCM:MeOH = 9:1) = 0.03; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 10.29 (s, 1H, Ar-NH-COR), 8.38 (d, J = 1.9 Hz, 1H, Ar-H7), 7.90 (dd, J1 = 8.5 Hz, J2 = 1.9 Hz, 1H, Ar-H5), 7.88–7.78 (m, 2H, 2 × Ar-H), 7.51 (d, J = 8.5 Hz, 1H, Ar-H4), 7.44–7.37 (m, 2H, 2 × Ar-H), 4.08 (d, J = 13 Hz, 2H, 2 × piperidine-H), 3.29–3.16 (m, 2 × piperidine-H), 2.20 (s, 6H, N(CH3)2), 1.92–1.84 (m, 2H, 2 × piperidine-H), 1.55–1.42 ppm (m, 2H, 2 × piperidine-H), covered with solvent (1H, piperidine-H); 13C (101 MHz, [D6]DMSO, 25 °C, TMS): δ = 170.0, 165.6, 156.0, 138.9, 130.9, 128.9 (2C), 127.5, 127.4, 126.6, 122.2 (2C), 121.6, 118.0, 61.1, 48.0 (2C), 41.9 (2C), 27.9 ppm (2C); HRMS (ESI+) m/z calcd. for C21H23ClN4OS+H+: 415.1354 [M+H]+: found 415.1346; HPLC: tr = 5.02 min (96.9% at 254 nm).
Ethyl 2-(4-(N-Boc-amino)piperidin-1-yl)benzo[d]thiazole-6-carboxylate (11): Ethyl 2-bromobenzo[d]thiazole-6-carboxylate (750 mg, 2.62 mmol) and 4-(N-Boc-amino)piperidine (1.31 g, 6.55 mmol) were dissolved in THF (100 mL). The reaction mixture was then stirred at r.t. overnight. The precipitate was filtered off and the solvent was evaporated under reduced pressure. The residue was taken up in ethyl acetate (100 mL) and washed with 1% (w/v) citric acid (3 × 50 mL) and saturated NaCl (50 mL). The organic phase was dried over Na2SO4, filtered and the solvent was evaporated under reduced pressure. Yield: 1.05 g (99%); yellow amorphous powder; Rf(EtOAc:Hex = 1:2) = 0.16; 1H NMR (400 MHz, CDCl3, 25 °C, TMS): δ = 8.30 (d, J = 1.8 Hz, 1H, Ar-H7), 8.00 (dd, J1 = 8.5 Hz, J2 = 1.8 Hz, 1H, Ar-H5), 7.51 (d, J = 8.5 Hz, 1H, Ar-H4), 4.52–4.44 (m, 1H, CH-NH-CO), 4.37 (q, J = 7.1 Hz, 2H, COO-CH2-CH3), 4.19–4.10 (m, 2H, 2× piperidine-H), 3.82–3.70 (m, 1H, piperidine-H), 3.34–3.24 (m, 2H, 2× piperidine-H), 2.14–2.06 (m, 2H, 2× piperidine-H), 1.54–1.49 (m, 2H, 2× piperidine-H), 1.46 (s, 9H, COC(CH3)3), 1.40 ppm (t, J = 7.1 Hz, 3H, COO-CH2-CH3); LC–MS (ESI+): m/z 406.3 [M+H]+ (calcd. m/z = 405.2 for C20H27N3O4S).
2-(4-(N-Boc-Amino)piperidin-1-yl)benzo[d]thiazole-6-carboxylic acid (12): Compound 11 (1.00 g, 2.47 mmol) was suspended in ethanol (96%, 50 mL). Then, 2 M NaOH(aq) (12.3 mL, 24.6 mmol) was added. The mixture was heated to 100 °C and left to stir for 1 h. After that, the reaction mixture was cooled and acidified to pH 3 using 2 M HCl(aq). The precipitated product was then filtered off under pressure. Yield: 258 mg (93%); yellow amorphous powder; Rf(DCM:MeOH = 9:1) = 0.34; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 12.71 (s, 1H, COOH), 8.35 (d, J = 1.7 Hz, 1H, Ar-H7), 7.84 (dd, J1 = 8.5 Hz, J2 = 1.7 Hz, 1H, Ar-H5), 7.45 (d, J = 8.5 Hz, 1H, Ar-H4), 6.94 (d, J = 7.9 Hz, 1H, CH-NH-CO), 4.06–3.96 (m, J = 12.9 Hz, 2H, 2× piperidine-H), 3.68–3.51 (m, 1H, CH-NH), 1.86 (m, 2H, 2× piperidine-H), 1.50–1.33 ppm (m, 11H, 2× piperidine-H and COC(CH3)3), not visible (2× piperidine-H); LC–MS (ESI+): m/z 378.0 [M+H]+ (calcd. m/z = 377.1 for C18H23N3O4S).
The amide coupling procedure was used for the synthesis of 4-(N-Boc-amino)-1-(6-((3,4-dichlorophenyl)carbamoyl)benzo[d]thiazol-2-yl)piperidine (13): Yield: 258 mg (48%); off-white amorphous powder; Rf(EtOAc:Hex = 1:2) = 0.39; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 10.31 (s, 1H, Ar-NH-COR), 8.40 (d, J = 1.8 Hz, 1H, Ar-H7), 7.91 (dd, J1 = 8.5, J2 = 1.8 Hz, 1H, Ar-H5), 7.82 (d, J = 8.9 Hz, 2H, 2 × Ar-H), 7.55 (d, J = 8.5 Hz, 1H, Ar-H4), 7.41 (d, J = 8.9 Hz, 2H, 2 × Ar-H), 3.69–3.58 (m, 4H, 2 × piperazine-CH2), 3.54–3.46 (m, 4H, 2 × piperazine-CH2), 1.43 ppm (s, 9H, COC(CH3)3); LC–MS (ESI+): m/z 473.1 [M+H]+ (calcd. m/z = 472.1 for C23H25ClN4O3S).
The general procedure for the preparation of compounds 5a–m was used for the synthesis of 4-amino-1-(6-((3,4-dichlorophenyl)carbamoyl)benzo[d]thiazol-2-yl)piperidine (14): Yield: 220 mg (91%); off-white amorphous powder; Rf(DCM:MeOH = 9:1) = 0.02; 1H NMR (400 MHz, [D6]DMSO, 25 °C, TMS): δ = 10.47 (s, 1H, Ar-NH-COR), 8.40 (d, J = 1.8 Hz, 1H, Ar-H7), 8.19 (d, J = 2.4 Hz, 1H, Ar-H20), 7.91 (dd, J1 = 8.5, J2 = 1.8 Hz, 1H, Ar-H5), 7.79 (dd, J = 8.9, 2.4 Hz, 1H, Ar-H24), 7.61 (d, J = 8.9 Hz, 1H, Ar-H23), 7.50 (d, J = 8.5 Hz, 1H, Ar-H4), 3.99 (d, J = 13.2 Hz, 2H, CH-NH2), 3.33–3.20 (m, 2H, 2 × piperidin-H) 2.91–2.83 (m, 1H, CH-NH2), 1.88–1.78 (m, 2H, 2 × piperidin-H), 1.88–1.58 (m, 4H, 4 × piperidin-H), 1.37–1.25 ppm (m, 2H, 2 × piperidin-H); 13C (101 MHz, [D6]DMSO, 25 °C, TMS): δ = 170.2, 165.8, 156.3, 140.2, 131.2, 130.9 (2C), 126.9, 126.8, 125.2, 121.8 (2C), 120.7, 117.9, 48.0 (2C), 47.6 (2C), 34.8 ppm; HRMS (ESI+) m/z calcd. for C19H18Cl2N4OS+H+: 421.0651 [M+H]+: found 421.0645; HPLC: tr = 6.31 min (95.1% at 254 nm).


 ,
, 













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






























