
Identification of miRNA-Small Molecule Associations by Continuous Feature Representation Using Auto-Encoders




Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.1.1. Datasets for Training Auto-Encoders
2.1.2. Datasets for SM-miRNA Associations
2.2. Methods
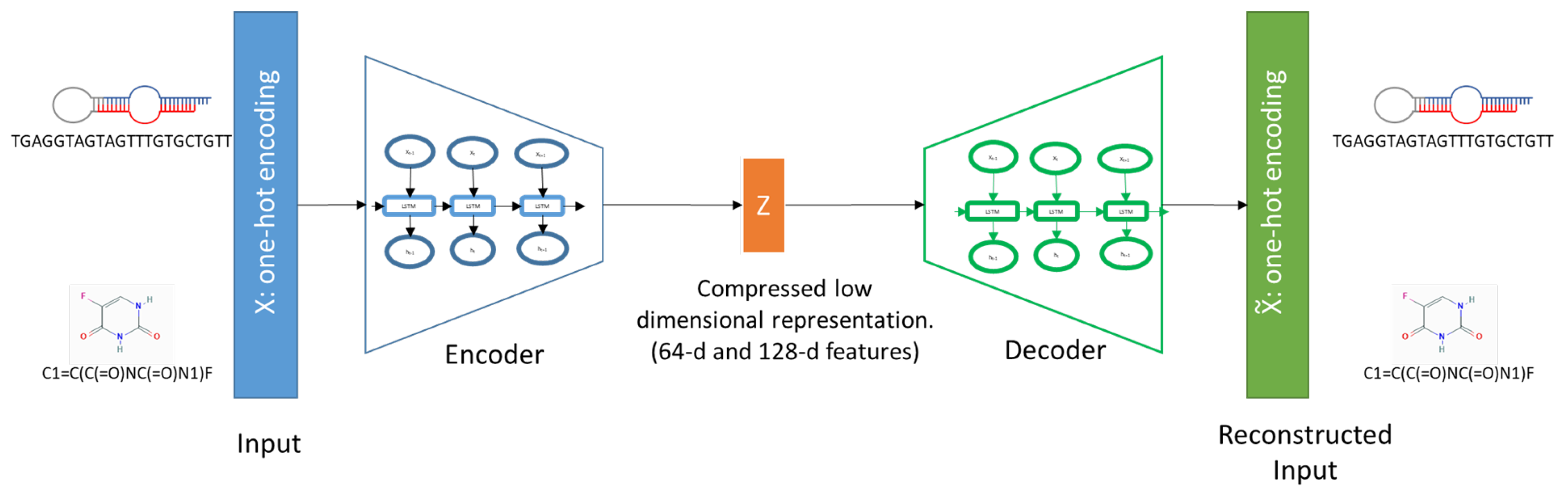
2.2.1. Long Short-Term Memory (LSTM) Sequence Auto-Encoders
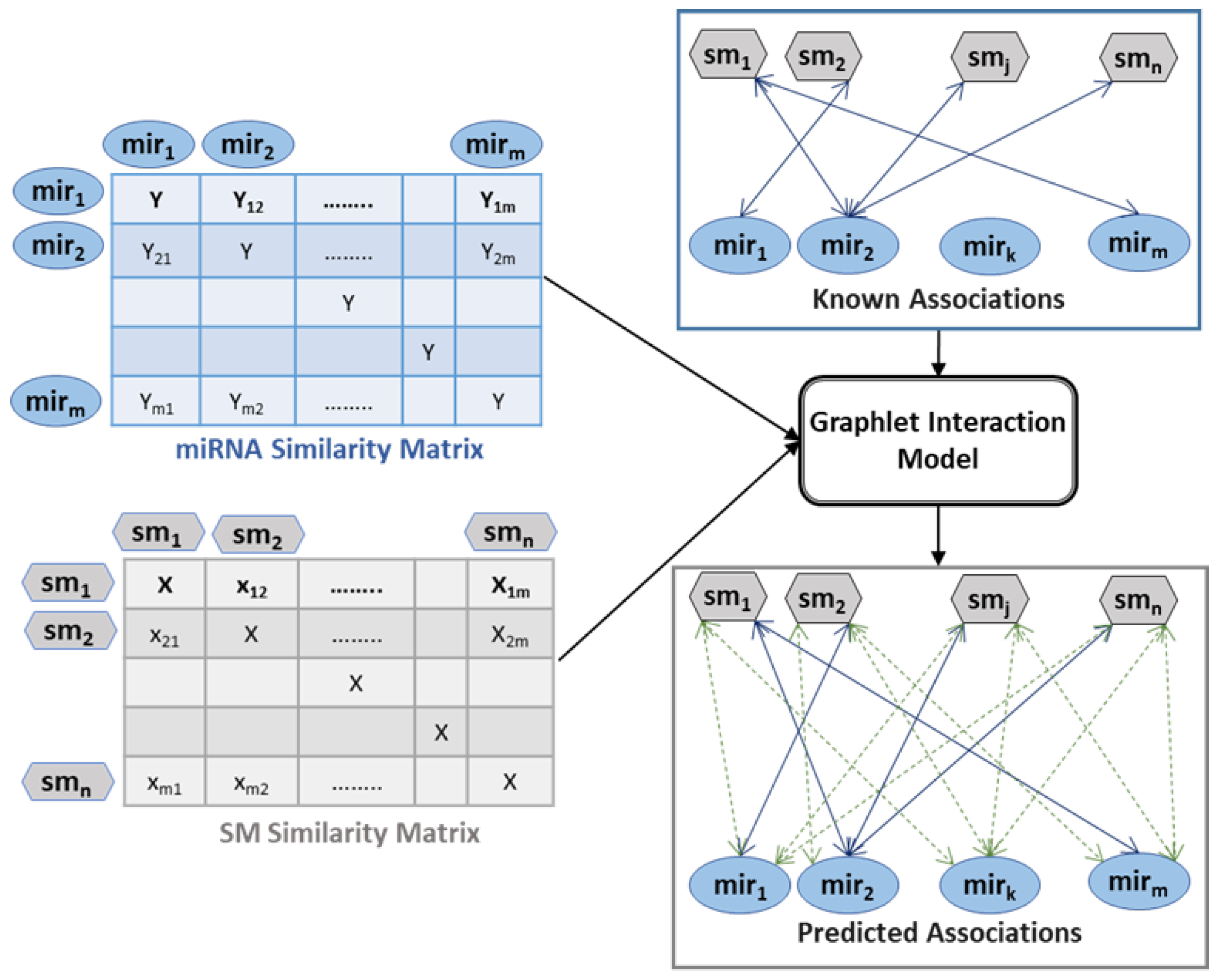
2.2.2. Similarity Calculation
2.2.3. Graphlet Interaction
2.2.4. Predicting Unknown Associations
2.2.5. Evaluation Methodology
- 139: Enoxacin (CID: 3229): An antibacterial drug that inhibits DNA synthesis. It is used to treat gonorrhea and urinary tract infections.
- 162: 5-Fluorouracil (CID: 3385): An antineoplastic agent that inhibits DNA synthesis and is also used for treating solid tumors that occur in different body parts, such as the breast, colon, and liver.
- 351: Vorinostat (CID:5311): An antineoplastic agent and a deacetylase inhibitor, which is used for treating cutaneous T cell lymphoma.
- 405: Estradiol (CID: 5757): A synthetic form of the steroid sex hormone, estradiol, which maintains fertility and female characteristics. Synthetic estradiol can be used as a hormone replacement therapy.
- 607: Gemcitabine (CID:60750): An antineoplastic agent used for treating advanced lung, breast, and pancreatic cancers.
- 736: Diethylstilbestrol (CID:448537): Used in the treatment of prostate and breast cancers.
3. Results
3.1. Evaluation
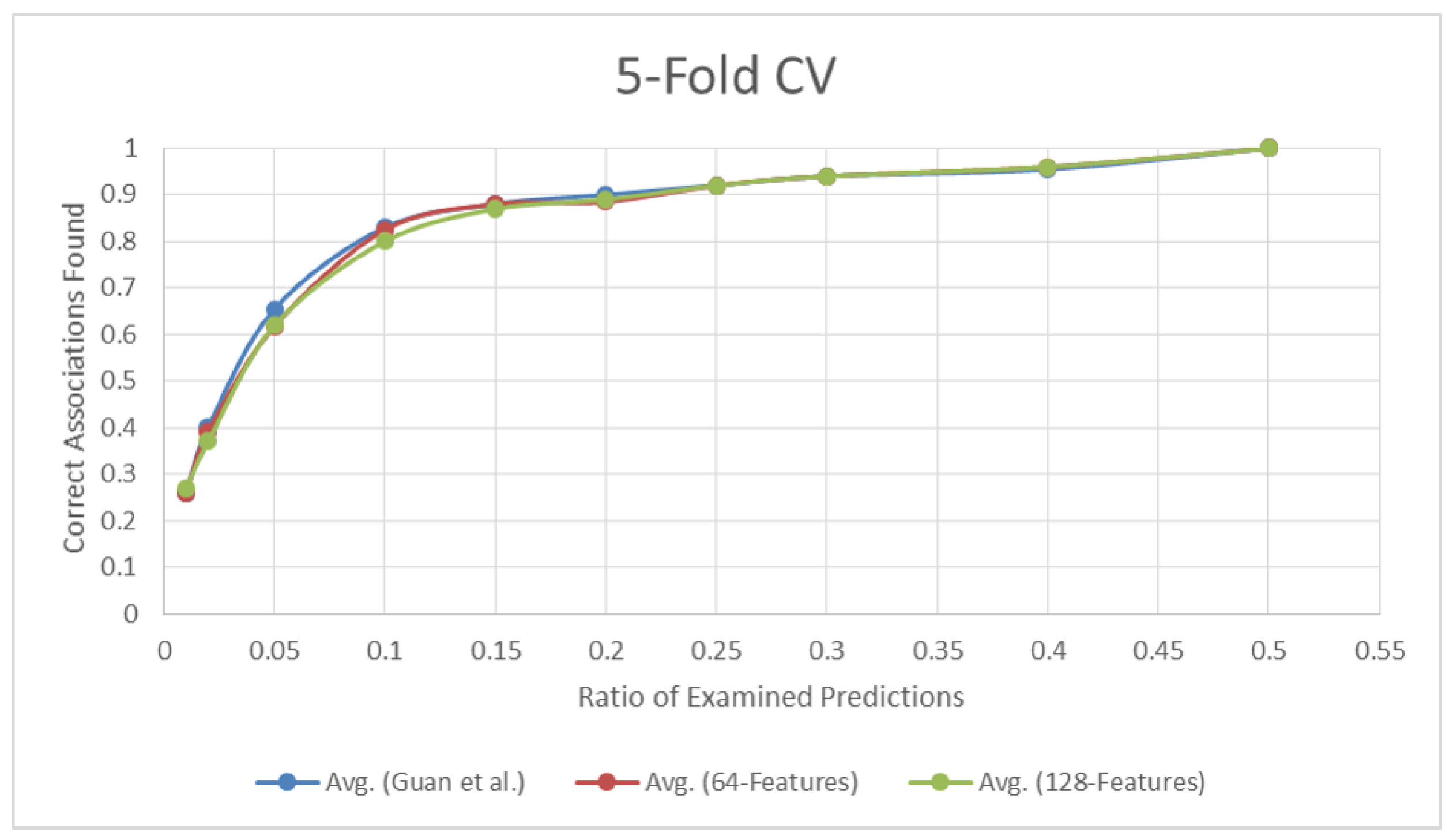
3.1.1. 5-Fold Cross Validation
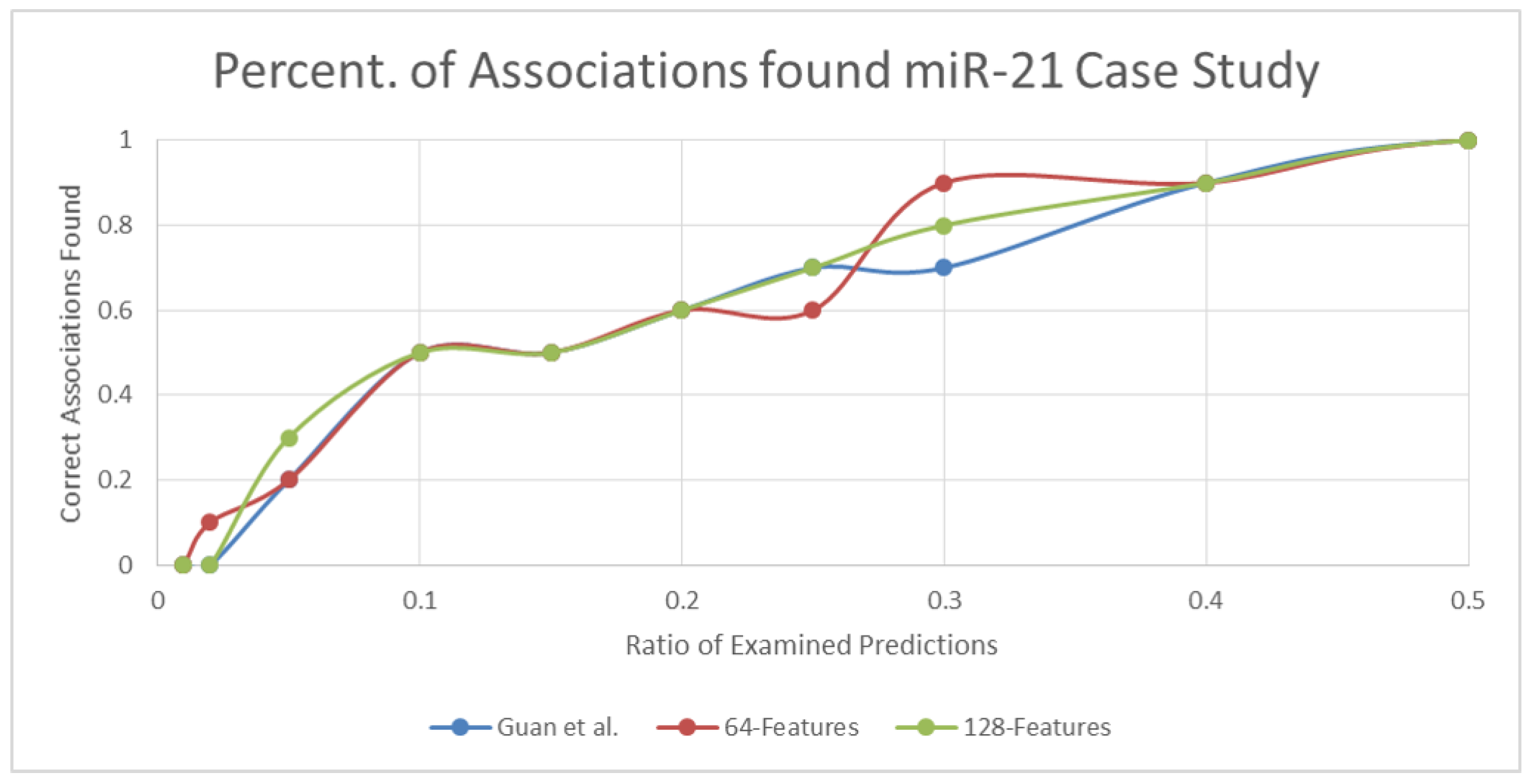
3.1.2. Case Study Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Olive, V.; Minella, A.C.; He, L. Outside the coding genome, mammalian microRNAs confer structural and functional complexity. Sci. Signal. 2015, 8, re2. [Google Scholar] [CrossRef] [Green Version]
- Ling, H.; Fabbri, M.; Calin, G.A. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat. Rev. Drug Discov. 2013, 12, 847–865. [Google Scholar] [CrossRef] [Green Version]
- Ambros, V. MicroRNA pathways in flies and worms: Growth, death, fat, stress, and timing. Cell 2003, 113, 673–676. [Google Scholar] [CrossRef] [Green Version]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [Green Version]
- Xu, P.; Wu, Q.; Yu, J.; Rao, Y.; Kou, Z.; Fang, G.; Shi, X.; Liu, W.; Han, H. A systematic way to infer the regulation relations of miRNAs on target genes and critical miRNAs in cancers. Front. Genet. 2020, 11, 278. [Google Scholar] [CrossRef] [PubMed]
- Peter, M. Targeting of mRNAs by multiple miRNAs: The next step. Oncogene 2010, 29, 2161–2164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, C.H.; Chang, N.W.; Shrestha, S.; Hsu, S.D.; Lin, Y.L.; Lee, W.H.; Yang, C.D.; Hong, H.C.; Wei, T.Y.; Tu, S.J.; et al. miRTarBase 2016: Updates to the experimentally validated miRNA-target interactions database. Nucleic Acids Res. 2016, 44, D239–D247. [Google Scholar] [CrossRef] [PubMed]
- Tong, Z.; Cui, Q.; Wang, J.; Zhou, Y. TransmiR v2.0: An updated transcription factor-microRNA regulation database. Nucleic Acids Res. 2019, 47, D253–D258. [Google Scholar] [CrossRef] [Green Version]
- Helwak, A.; Kudla, G.; Dudnakova, T.; Tollervey, D. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell 2013, 153, 654–665. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y.; Lee, Y.H.; Bae, Y.S. MiR-186, miR-216b, miR-337-3p, and miR-760 cooperatively induce cellular senescence by targeting α subunit of protein kinase CKII in human colorectal cancer cells. Biochem. Biophys. Res. Commun. 2012, 429, 173–179. [Google Scholar] [CrossRef]
- Pan, W.; Zhu, S.; Yuan, M.; Cui, H.; Wang, L.; Luo, X.; Li, J.; Zhou, H.; Tang, Y.; Shen, N. MicroRNA-21 and microRNA-148a contribute to DNA hypomethylation in lupus CD4+ T cells by directly and indirectly targeting DNA methyltransferase 1. J. Immunol. 2010, 184, 6773–6781. [Google Scholar] [CrossRef] [Green Version]
- Merkerova, M.; Vasikova, A.; Belickova, M.; Bruchova, H. MicroRNA expression profiles in umbilical cord blood cell lineages. Stem Cells Dev. 2010, 19, 17–26. [Google Scholar] [CrossRef]
- Fujita, Y.; Kojima, K.; Ohhashi, R.; Hamada, N.; Nozawa, Y.; Kitamoto, A.; Sato, A.; Kondo, S.; Kojima, T.; Deguchi, T.; et al. MiR-148a attenuates paclitaxel resistance of hormone-refractory, drug-resistant prostate cancer PC3 cells by regulating MSK1 expression. J. Biol. Chem. 2010, 285, 19076–19084. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Song, Y.; Wang, Z.; Yue, Z.; Xu, H.; Xing, C.; Liu, Z. Altered expression of MiR-148a and MiR-152 in gastrointestinal cancers and its clinical significance. J. Gastrointest. Surg. 2010, 14, 1170–1179. [Google Scholar] [CrossRef]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, P.; Guo, M.; Hay, B.A. MicroRNAs and the regulation of cell death. Trends Genet. 2004, 20, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.M.; Byrom, M.W.; Shelton, J.; Ford, L.P. Antisense inhibition of human miRNAs and indications for an involvement of miRNA in cell growth and apoptosis. Nucleic Acids Res. 2005, 33, 1290–1297. [Google Scholar] [CrossRef] [Green Version]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.R.; Yin, M.M.; Jiao, C.N.; Gao, Y.L.; Kong, X.Z.; Liu, J.X. MCCMF: Collaborative matrix factorization based on matrix completion for predicting miRNA-disease associations. BMC Bioinform. 2020, 21, 454. [Google Scholar] [CrossRef]
- Yin, J.; Chen, X.; Wang, C.C.; Zhao, Y.; Sun, Y.Z. Prediction of Small Molecule–MicroRNA Associations by Sparse Learning and Heterogeneous Graph Inference. Mol. Pharm. 2019, 16, 3157–3166. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Li, M.; Hu, C.; Duan, H. Clinical significance of serum miR-223, miR-25 and miR-375 in patients with esophageal squamous cell carcinoma. Mol. Biol. Rep. 2014, 41, 1257–1266. [Google Scholar] [CrossRef]
- Huang, Z.; Shi, J.; Gao, Y.; Cui, C.; Zhang, S.; Li, J.; Zhou, Y.; Cui, Q. HMDD v3.0: A database for experimentally supported human microRNA–disease associations. Nucleic Acids Res. 2019, 47, D1013–D1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Q.; Wang, Y.; Hao, Y.; Juan, L.; Teng, M.; Zhang, X.; Li, M.; Wang, G.; Liu, Y. miR2Disease: A manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 2009, 37, D98–D104. [Google Scholar] [CrossRef] [Green Version]
- Monroig, P.d.C.; Chen, L.; Zhang, S.; Calin, G.A. Small molecule compounds targeting miRNAs for cancer therapy. Adv. Drug Deliv. Rev. 2015, 81, 104–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, T.; Li, J.; Cheng, H.; Zhang, C.; Zhang, Y. Small-Molecule Regulators of MicroRNAs in Biomedicine. Drug Dev. Res. 2015, 76, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Guan, N.N.; Sun, Y.Z.; Ming, Z.; Li, J.Q.; Chen, X. Prediction of potential small molecule-associated microRNAs using graphlet interaction. Front. Pharmacol. 2018, 9, 1152. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [Google Scholar] [CrossRef]
- Bose, D.; Jayaraj, G.; Suryawanshi, H.; Agarwala, P.; Pore, S.K.; Banerjee, R.; Maiti, S. The tuberculosis drug streptomycin as a potential cancer therapeutic: Inhibition of miR-21 function by directly targeting its precursor. Angew. Chem. 2012, 124, 1043–1047. [Google Scholar] [CrossRef]
- Rossi, L.; Bonmassar, E.; Faraoni, I. Modification of miR gene expression pattern in human colon cancer cells following exposure to 5-fluorouracil in vitro. Pharmacol. Res. 2007, 56, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt-Eriksen, E.S.; Aarup, V.; Persson, R.; Hansen, H.F.; Munk, M.E.; Ørum, H. A locked nucleic acid oligonucleotide targeting microRNA 122 is well-tolerated in cynomolgus monkeys. Nucleic Acid Ther. 2012, 22, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Kutay, H.; Bai, S.; Datta, J.; Motiwala, T.; Pogribny, I.; Frankel, W.; Jacob, S.T.; Ghoshal, K. Downregulation of miR-122 in the rodent and human hepatocellular carcinomas. J. Cell. Biochem. 2006, 99, 671–678. [Google Scholar] [CrossRef] [Green Version]
- Chandrasekhar, S.; Pushpavalli, S.N.; Chatla, S.; Mukhopadhyay, D.; Ganganna, B.; Vijeender, K.; Srihari, P.; Reddy, C.R.; Ramaiah, M.J.; Bhadra, U. aza-Flavanones as potent cross-species microRNA inhibitors that arrest cell cycle. Bioorganic Med. Chem. Lett. 2012, 22, 645–648. [Google Scholar] [CrossRef]
- Chen, X.; Guan, N.N.; Sun, Y.Z.; Li, J.Q.; Qu, J. MicroRNA-small molecule association identification: From experimental results to computational models. Briefings Bioinform. 2020, 21, 47–61. [Google Scholar] [CrossRef] [PubMed]
- Bose, D.; Jayaraj, G.G.; Kumar, S.; Maiti, S. A molecular-beacon-based screen for small molecule inhibitors of miRNA maturation. ACS Chem. Biol. 2013, 8, 930–938. [Google Scholar] [CrossRef]
- Davies, B.P.; Arenz, C. A fluorescence probe for assaying micro RNA maturation. Bioorganic Med. Chem. 2008, 16, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Cha, W.; Fan, R.; Miao, Y.; Zhou, Y.; Qin, C.; Shan, X.; Wan, X.; Cui, T. MicroRNAs as novel endogenous targets for regulation and therapeutic treatments. Medchemcomm 2018, 9, 396–408. [Google Scholar] [CrossRef]
- Jiang, W.; Chen, X.; Liao, M.; Li, W.; Lian, B.; Wang, L.; Meng, F.; Liu, X.; Chen, X.; Jin, Y.; et al. Identification of links between small molecules and miRNAs in human cancers based on transcriptional responses. Sci. Rep. 2012, 2, 1–8. [Google Scholar] [CrossRef]
- Lv, Y.; Wang, S.; Meng, F.; Yang, L.; Wang, Z.; Wang, J.; Chen, X.; Jiang, W.; Li, Y.; Li, X. Identifying novel associations between small molecules and miRNAs based on integrated molecular networks. Bioinformatics 2015, 31, 3638–3644. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Meng, F.; Dai, E.; Yang, F.; Wang, S.; Chen, X.; Yang, L.; Wang, Y.; Jiang, W. Identification of associations between small molecule drugs and miRNAs based on functional similarity. Oncotarget 2016, 7, 38658. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Lei, K.; Wu, Z.; Li, W.; Liu, G.; Liu, J.; Cheng, F.; Tang, Y. Network-based identification of microRNAs as potential pharmacogenomic biomarkers for anticancer drugs. Oncotarget 2016, 7, 45584. [Google Scholar] [CrossRef] [Green Version]
- Qu, J.; Chen, X.; Sun, Y.Z.; Zhao, Y.; Cai, S.B.; Ming, Z.; You, Z.H.; Li, J.Q. In Silico prediction of small molecule-miRNA associations based on the HeteSim algorithm. Mol. Ther.-Nucleic Acids 2019, 14, 274–286. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Lin, K.; Wang, S.; Wang, L.; Cai, C.; Song, C.; Lai, L.; Pei, J. Deep learning for molecular generation. Future Med. Chem. 2019, 11, 567–597. [Google Scholar] [CrossRef]
- Elton, D.C.; Boukouvalas, Z.; Fuge, M.D.; Chung, P.W. Deep learning for molecular design—A review of the state of the art. Mol. Syst. Des. Eng. 2019, 4, 828–849. [Google Scholar] [CrossRef] [Green Version]
- Vamathevan, J.; Clark, D.; Czodrowski, P.; Dunham, I.; Ferran, E.; Lee, G.; Li, B.; Madabhushi, A.; Shah, P.; Spitzer, M.; et al. Applications of machine learning in drug discovery and development. Nat. Rev. Drug Discov. 2019, 18, 463–477. [Google Scholar] [CrossRef] [PubMed]
- Kotsias, P.C.; Arús-Pous, J.; Chen, H.; Engkvist, O.; Tyrchan, C.; Bjerrum, E.J. Direct steering of de novo molecular generation with descriptor conditional recurrent neural networks. Nat. Mach. Intell. 2020, 2, 254–265. [Google Scholar] [CrossRef]
- Zhang, H.; Li, J.; Ji, Y.; Yue, H. Understanding subtitles by character-level sequence-to-sequence learning. IEEE Trans. Ind. Inform. 2016, 13, 616–624. [Google Scholar] [CrossRef]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Félix, E.; Magariños, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M.; et al. ChEMBL: Towards direct deposition of bioassay data. Nucleic Acids Res. 2019, 47, D930–D940. [Google Scholar] [CrossRef]
- Knox, C.; Law, V.; Jewison, T.; Liu, P.; Ly, S.; Frolkis, A.; Pon, A.; Banco, K.; Mak, C.; Neveu, V.; et al. DrugBank 3.0: A comprehensive resource for ‘omics’ research on drugs. Nucleic Acids Res. 2010, 39, D1035–D1041. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Xiao, J.; Suzek, T.O.; Zhang, J.; Wang, J.; Bryant, S.H. PubChem: A public information system for analyzing bioactivities of small molecules. Nucleic Acids Res. 2009, 37, W623–W633. [Google Scholar] [CrossRef]
- Liu, X.; Wang, S.; Meng, F.; Wang, J.; Zhang, Y.; Dai, E.; Yu, X.; Li, X.; Jiang, W. SM2miR: A database of the experimentally validated small molecules’ effects on microRNA expression. Bioinformatics 2013, 29, 409–411. [Google Scholar] [CrossRef]
- Lu, M.; Zhang, Q.; Deng, M.; Miao, J.; Guo, Y.; Gao, W.; Cui, Q. An analysis of human microRNA and disease associations. PLoS ONE 2008, 3, e3420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruepp, A.; Kowarsch, A.; Schmidl, D.; Buggenthin, F.; Brauner, B.; Dunger, I.; Fobo, G.; Frishman, G.; Montrone, C.; Theis, F.J. PhenomiR: A knowledgebase for microRNA expression in diseases and biological processes. Genome Biol. 2010, 11, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Cho, K.; Van Merriënboer, B.; Gulcehre, C.; Bahdanau, D.; Bougares, F.; Schwenk, H.; Bengio, Y. Learning phrase representations using RNN encoder-decoder for statistical machine translation. arXiv 2014, arXiv:1406.1078. [Google Scholar]
- Lipton, Z.C.; Berkowitz, J.; Elkan, C. A critical review of recurrent neural networks for sequence learning. arXiv 2015, arXiv:1506.00019. [Google Scholar]
- Wang, X.D.; Huang, J.L.; Yang, L.; Wei, D.Q.; Qi, Y.X.; Jiang, Z.L. Identification of human disease genes from interactome network using graphlet interaction. PLoS ONE 2014, 9, e86142. [Google Scholar] [CrossRef]
- Pržulj, N. Biological network comparison using graphlet degree distribution. Bioinformatics 2007, 23, e177–e183. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Percent | Guan et al. | 64-d Features | 128-d Features |
|---|---|---|---|
| 0.01 | 0.26 | 0.26 | 0.27 |
| 0.02 | 0.40 | 0.39 | 0.37 |
| 0.05 | 0.66 | 0.62 | 0.62 |
| 0.10 | 0.83 | 0.83 | 0.80 |
| 0.15 | 0.88 | 0.88 | 0.87 |
| 0.20 | 0.90 | 0.89 | 0.89 |
| 0.25 | 0.92 | 0.92 | 0.92 |
| 0.30 | 0.94 | 0.94 | 0.94 |
| 0.40 | 0.96 | 0.96 | 0.96 |
| 0.50 | 1.00 | 1.00 | 1.00 |
| Percent | Guan et al. | 64-d Features | 128-d Features |
|---|---|---|---|
| 0.01 | 0.04 | 0.05 | 0.05 |
| 0.02 | 0.07 | 0.10 | 0.08 |
| 0.05 | 0.16 | 0.20 | 0.17 |
| 0.10 | 0.25 | 0.31 | 0.32 |
| 0.15 | 0.35 | 0.45 | 0.42 |
| 0.20 | 0.49 | 0.53 | 0.52 |
| 0.25 | 0.56 | 0.57 | 0.57 |
| 0.30 | 0.59 | 0.62 | 0.63 |
| 0.40 | 0.70 | 0.71 | 0.69 |
| 0.50 | 0.74 | 0.73 | 0.72 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdelbaky, I.; Tayara, H.; Chong, K.T. Identification of miRNA-Small Molecule Associations by Continuous Feature Representation Using Auto-Encoders. Pharmaceutics 2022, 14, 3. https://doi.org/10.3390/pharmaceutics14010003
Abdelbaky I, Tayara H, Chong KT. Identification of miRNA-Small Molecule Associations by Continuous Feature Representation Using Auto-Encoders. Pharmaceutics. 2022; 14(1):3. https://doi.org/10.3390/pharmaceutics14010003
Chicago/Turabian StyleAbdelbaky, Ibrahim, Hilal Tayara, and Kil To Chong. 2022. "Identification of miRNA-Small Molecule Associations by Continuous Feature Representation Using Auto-Encoders" Pharmaceutics 14, no. 1: 3. https://doi.org/10.3390/pharmaceutics14010003
APA StyleAbdelbaky, I., Tayara, H., & Chong, K. T. (2022). Identification of miRNA-Small Molecule Associations by Continuous Feature Representation Using Auto-Encoders. Pharmaceutics, 14(1), 3. https://doi.org/10.3390/pharmaceutics14010003