
Bruton’s Tyrosine Kinase Inhibitor Zanubrutinib Effectively Modulates Cancer Resistance by Inhibiting Anthracycline Metabolism and Efflux

 , , and
, , and
Abstract
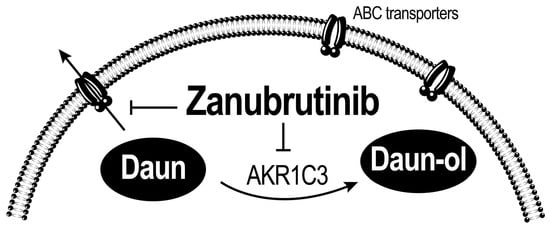
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Material
2.2. Cell Culture
2.3. Cloning, Overexpression, and Purification of Recombinant Enzymes
2.4. Inhibitory Assays
2.5. Inhibition of AKR1C3 in Transiently Transfected HCT116 Cells
2.6. UHPLC
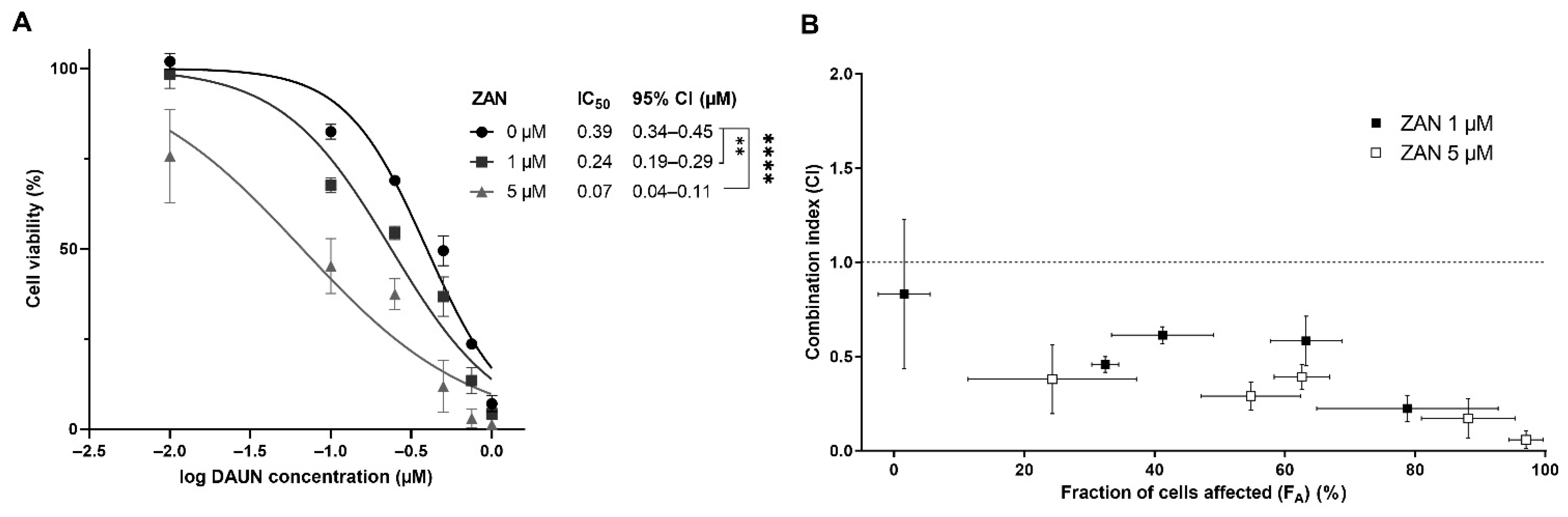
2.7. Drug Combination Assays
2.8. XTT Cell Proliferation Assay
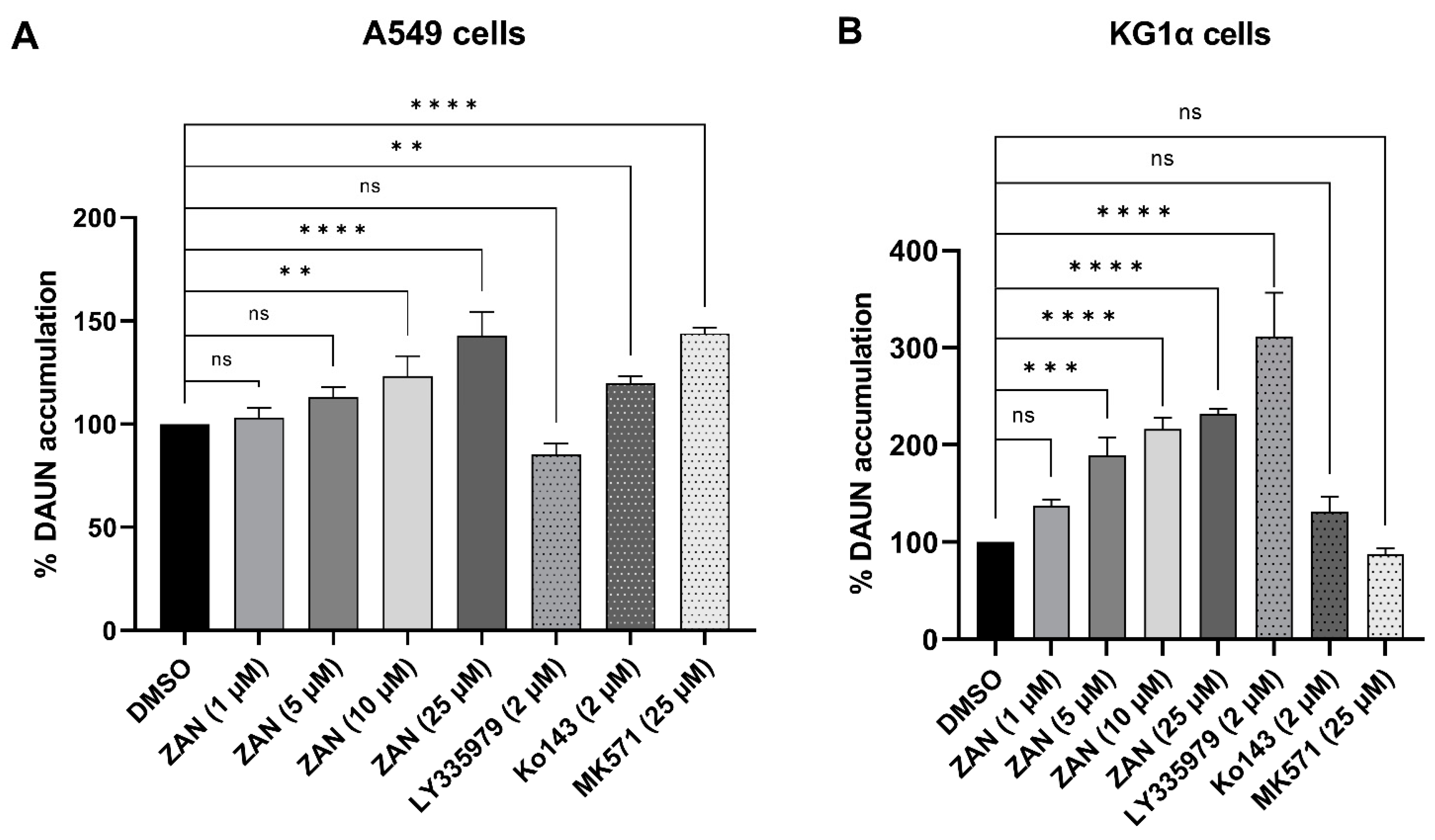
2.9. Accumulation Studies in KG1α and A549 Cells
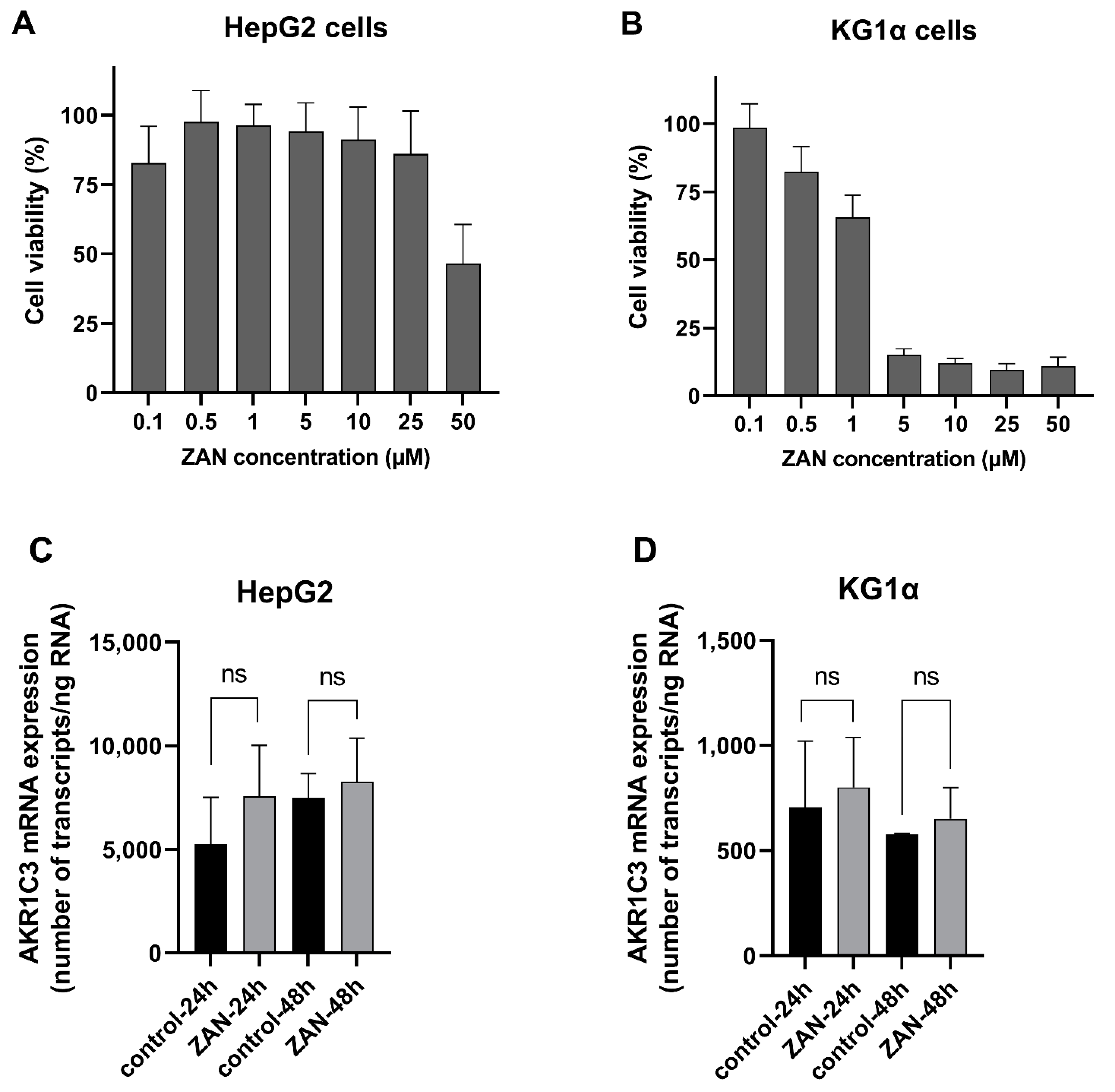
2.10. Determination of ZAN Cytotoxicity in HepG2 and KG1α Cells
2.11. Induction Studies in HepG2 and KG1α Cells
2.12. Statistical Analysis
3. Results
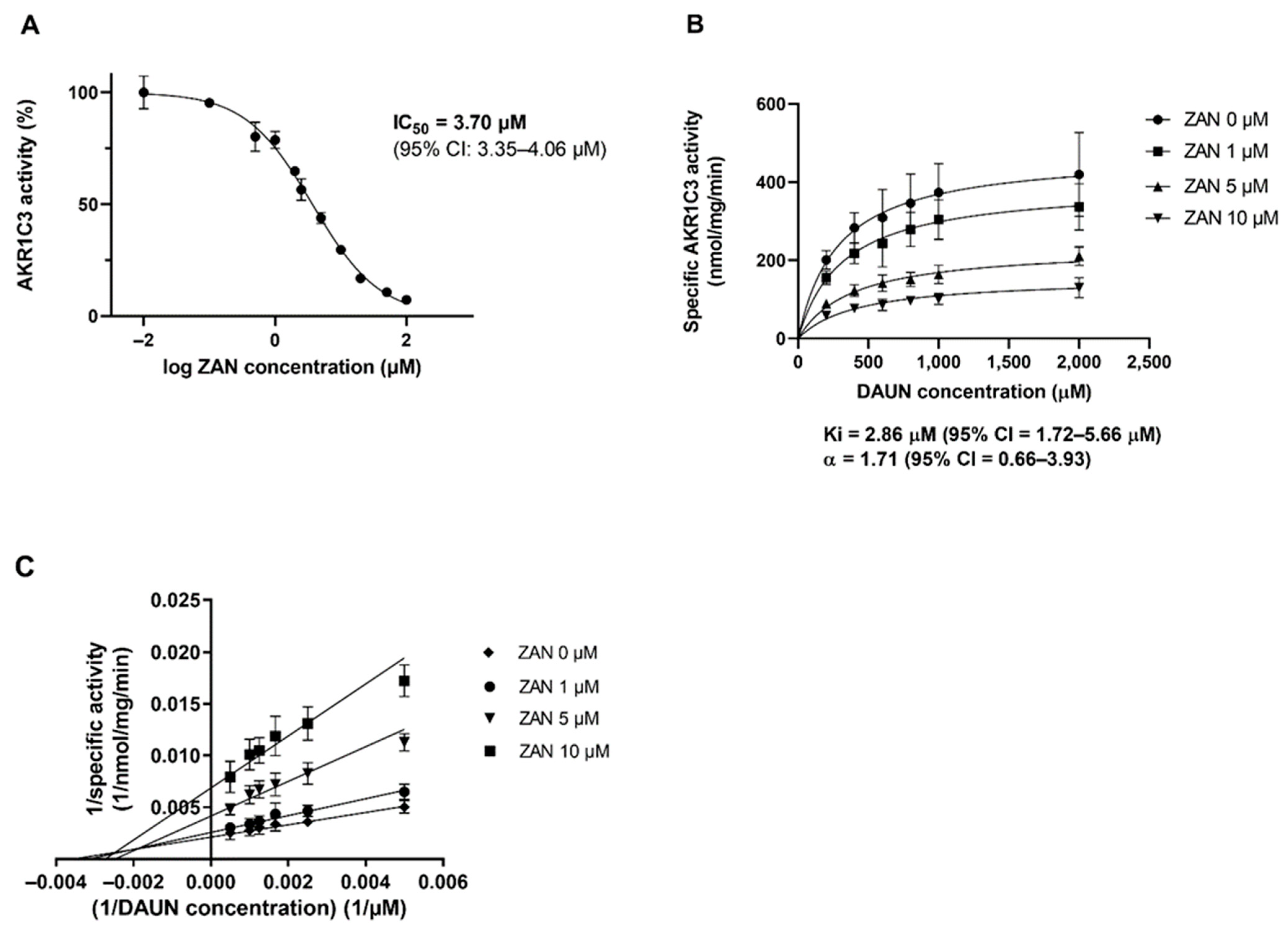
3.1. ZAN Inhibits the Recombinant AKR1C3-Mediated DAUN Reduction In Vitro
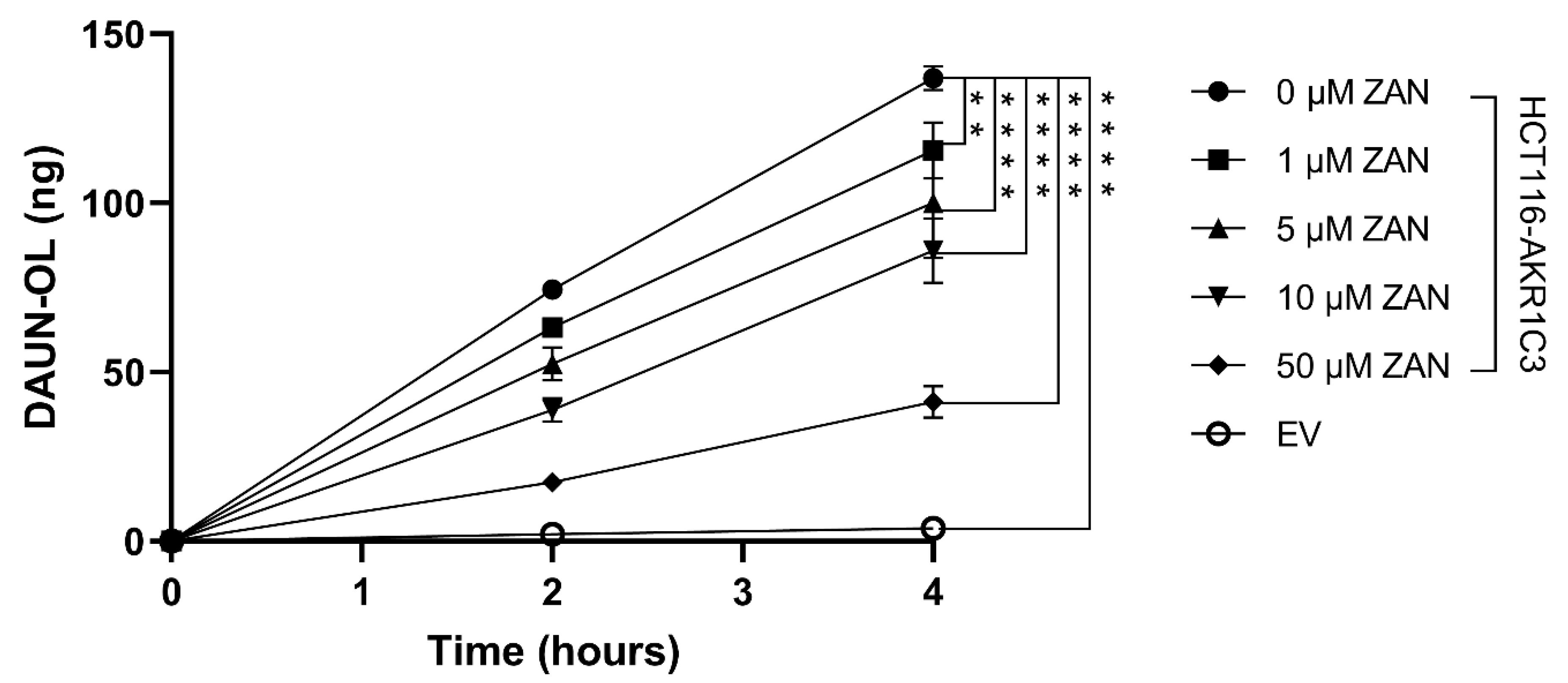
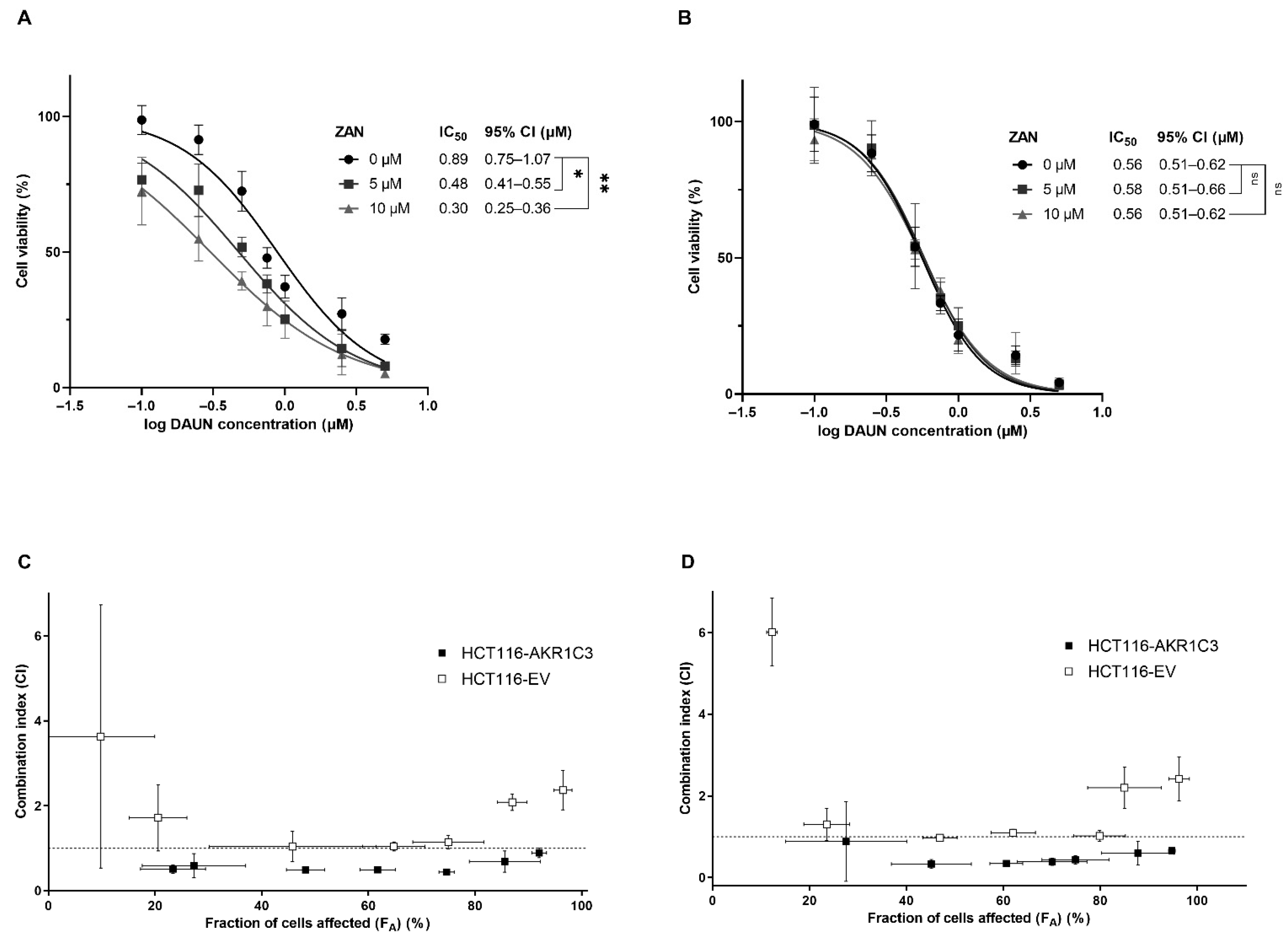
3.2. Effect of ZAN on AKR1C3-Mediated DAUN Metabolism in Transfected HCT116 Cells
3.3. ZAN Overcomes DAUN Resistance in A549 and Transfected HCT116 Cells
3.4. ZAN Increases DAUN Accumulation by Inhibiting the Efflux Activity of ABC Transporters
3.5. ZAN Does Not Affect AKR1C3 Expression
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Uckun, F.M.; Tibbles, H.E.; Vassilev, A.O. Bruton’s tyrosine kinase as a new therapeutic target. Anticancer Agents Med. Chem. 2007, 7, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Mano, H. Tec family of protein-tyrosine kinases: An overview of their structure and function. Cytokine Growth Factor Rev. 1999, 10, 267–280. [Google Scholar] [CrossRef]
- Niemann, C.U.; Wiestner, A. B-cell receptor signaling as a driver of lymphoma development and evolution. Semin. Cancer Biol. 2013, 23, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Buggy, J.J.; Elias, L. Bruton tyrosine kinase (BTK) and its role in B-cell malignancy. Int. Rev. Immunol. 2012, 31, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.E.; Ngo, V.N.; Lenz, G.; Tolar, P.; Young, R.M.; Romesser, P.B.; Kohlhammer, H.; Lamy, L.; Zhao, H.; Yang, Y.; et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature 2010, 463, 88–92. [Google Scholar] [CrossRef]
- Honigberg, L.A.; Smith, A.M.; Sirisawad, M.; Verner, E.; Loury, D.; Chang, B.; Li, S.; Pan, Z.; Thamm, D.H.; Miller, R.A.; et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc. Natl. Acad. Sci. USA 2010, 107, 13075–13080. [Google Scholar] [CrossRef]
- Herman, S.E.; Gordon, A.L.; Hertlein, E.; Ramanunni, A.; Zhang, X.; Jaglowski, S.; Flynn, J.; Jones, J.; Blum, K.A.; Buggy, J.J.; et al. Bruton tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI-32765. Blood 2011, 117, 6287–6296. [Google Scholar] [CrossRef]
- FDA (US Food and Drug Administration). FDA Approves Imbruvica. Available online: https://www.drugs.com/newdrugs/fda-approves-imbruvica-ibrutinib-mantle-cell-lymphoma-3960.html (accessed on 8 July 2022).
- FDA (US Food and Drug Administration). FDA Approves New Treatment for Adults with Mantle Cell Lymphoma. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-new-treatment-adults-mantle-cell-lymphoma (accessed on 8 July 2022).
- Byrd, J.C.; Furman, R.R.; Coutre, S.E.; Burger, J.A.; Blum, K.A.; Coleman, M.; Wierda, W.G.; Jones, J.A.; Zhao, W.; Heerema, N.A.; et al. Three-year follow-up of treatment-naive and previously treated patients with CLL and SLL receiving single-agent ibrutinib. Blood 2015, 125, 2497–2506. [Google Scholar] [CrossRef]
- Castillo, J.J.; Meid, K.; Gustine, J.N.; Leventoff, C.; White, T.; Flynn, C.A.; Sarosiek, S.; Demos, M.G.; Guerrera, M.L.; Kofides, A.; et al. Long-term follow-up of ibrutinib monotherapy in treatment-naive patients with Waldenstrom macroglobulinemia. Leukemia 2022, 36, 532–539. [Google Scholar] [CrossRef]
- Rusconi, C.; Cheah, C.Y.; Eyre, T.A.; Tucker, D.L.; Klener, P.; Gine, E.; Crucitti, L.; Muzi, C.; Iadecola, S.; Infante, G.; et al. Ibrutinib improves survival compared to chemotherapy in mantle cell lymphoma with central nervous system relapse. Blood 2022, in press. [CrossRef]
- Noy, A.; de Vos, S.; Coleman, M.; Martin, P.; Flowers, C.R.; Thieblemont, C.; Morschhauser, F.; Collins, G.P.; Ma, S.; Peles, S.; et al. Durable ibrutinib responses in relapsed/refractory marginal zone lymphoma: Long-term follow-up and biomarker analysis. Blood Adv. 2020, 4, 5773–5784. [Google Scholar] [CrossRef] [PubMed]
- Byrd, J.C.; Hillmen, P.; O’Brien, S.; Barrientos, J.C.; Reddy, N.M.; Coutre, S.; Tam, C.S.; Mulligan, S.P.; Jaeger, U.; Barr, P.M.; et al. Long-term follow-up of the RESONATE phase 3 trial of ibrutinib vs ofatumumab. Blood 2019, 133, 2031–2042. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Scheerens, H.; Li, S.J.; Schultz, B.E.; Sprengeler, P.A.; Burrill, L.C.; Mendonca, R.V.; Sweeney, M.D.; Scott, K.C.; Grothaus, P.G.; et al. Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase. ChemMedChem 2007, 2, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Barf, T.; Covey, T.; Izumi, R.; van de Kar, B.; Gulrajani, M.; van Lith, B.; van Hoek, M.; de Zwart, E.; Mittag, D.; Demont, D.; et al. Acalabrutinib (ACP-196): A Covalent Bruton Tyrosine Kinase Inhibitor with a Differentiated Selectivity and In Vivo Potency Profile. J. Pharmacol. Exp. Ther. 2017, 363, 240–252. [Google Scholar] [CrossRef]
- Von Hundelshausen, P.; Siess, W. Bleeding by Bruton Tyrosine Kinase-Inhibitors: Dependency on Drug Type and Disease. Cancers 2021, 13, 1103. [Google Scholar] [CrossRef]
- Ahn, I.E. Cardiovascular adverse events of ibrutinib. Blood 2019, 134, 1881–1882. [Google Scholar] [CrossRef]
- Woyach, J.A.; Furman, R.R.; Liu, T.M.; Ozer, H.G.; Zapatka, M.; Ruppert, A.S.; Xue, L.; Li, D.H.; Steggerda, S.M.; Versele, M.; et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N. Engl. J. Med. 2014, 370, 2286–2294. [Google Scholar] [CrossRef]
- Burger, J.A.; Landau, D.A.; Taylor-Weiner, A.; Bozic, I.; Zhang, H.; Sarosiek, K.; Wang, L.; Stewart, C.; Fan, J.; Hoellenriegel, J.; et al. Clonal evolution in patients with chronic lymphocytic leukaemia developing resistance to BTK inhibition. Nat. Commun. 2016, 7, 11589. [Google Scholar] [CrossRef]
- Bond, D.A.; Woyach, J.A. Targeting BTK in CLL: Beyond Ibrutinib. Curr. Hematol. Malig. Rep. 2019, 14, 197–205. [Google Scholar] [CrossRef]
- Byrd, J.C.; Smith, S.; Wagner-Johnston, N.; Sharman, J.; Chen, A.I.; Advani, R.; Augustson, B.; Marlton, P.; Renee Commerford, S.; Okrah, K.; et al. First-in-human phase 1 study of the BTK inhibitor GDC-0853 in relapsed or refractory B-cell NHL and CLL. Oncotarget 2018, 9, 13023–13035. [Google Scholar] [CrossRef]
- FDA (US Food and Drug Administration). FDA Approves Therapy to Treat Patients with Relapsed and Refractory Mantle Cell Lymphoma Supported by Clinical Trial Results Showing High Response Rate of Tumor Shrinkage. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-therapy-treat-patients-relapsed-and-refractory-mantle-cell-lymphoma-supported-clinical (accessed on 8 July 2022).
- FDA (US Food and Drug Administration). FDA Approves Zanubrutinib for Waldenström’s Macroglobulinemia. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-zanubrutinib-waldenstroms-macroglobulinemia (accessed on 8 July 2022).
- FDA (US Food and Drug Administration). FDA Grants Accelerated Approval to Zanubrutinib for Marginal Zone Lymphoma. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-zanubrutinib-marginal-zone-lymphoma (accessed on 8 July 2022).
- Younes, A.; Thieblemont, C.; Morschhauser, F.; Flinn, I.; Friedberg, J.W.; Amorim, S.; Hivert, B.; Westin, J.; Vermeulen, J.; Bandyopadhyay, N.; et al. Combination of ibrutinib with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) for treatment-naive patients with CD20-positive B-cell non-Hodgkin lymphoma: A non-randomised, phase 1b study. Lancet Oncol. 2014, 15, 1019–1026. [Google Scholar] [CrossRef]
- Younes, A.; Sehn, L.H.; Johnson, P.; Zinzani, P.L.; Hong, X.; Zhu, J.; Patti, C.; Belada, D.; Samoilova, O.; Suh, C.; et al. Randomized Phase III Trial of Ibrutinib and Rituximab Plus Cyclophosphamide, Doxorubicin, Vincristine, and Prednisone in Non-Germinal Center B-Cell Diffuse Large B-Cell Lymphoma. J. Clin. Oncol. 2019, 37, 1285–1295. [Google Scholar] [CrossRef] [PubMed]
- Davies, A.; Caddy, J.; Mercer, K.; Saunders, G.N.; Stanton, L.; Collins, G.P.; Cummin, T.E.C.; Schuh, A.; Ardeshna, K.M.; McMillan, A.; et al. Acalabrutinib in Combination with Rituximab, Cyclophosphamide, Doxorubicin, Vincristine and Prednisolone (R-CHOP) As First Line Therapy for Patients with Diffuse Large B-Cell Lymphoma (DLBCL): The Accept Phase Ib/II Single Arm Study. Blood 2020, 136, 38–39. [Google Scholar] [CrossRef]
- Zhu, H.; Sha, Y.; Wu, W.; Chen, R.; Yang, Y.; Qiu, J.; Mi, H.; Peng, C.; Ding, C.; Wang, Z.; et al. Zanubrutinib, Lenalidomide Plus R-CHOP (ZR2-CHOP) as the Treatment for Diffused Large B-Cell Lymphoma (DLBCL). Hematol. Oncol. 2021, 39, 425–427. [Google Scholar] [CrossRef]
- Xiao, H.; Zheng, Y.; Ma, L.; Tian, L.; Sun, Q. Clinically-Relevant ABC Transporter for Anti-Cancer Drug Resistance. Front. Pharmacol. 2021, 12, 648407. [Google Scholar] [CrossRef] [PubMed]
- Bains, O.S.; Grigliatti, T.A.; Reid, R.E.; Riggs, K.W. Naturally occurring variants of human aldo-keto reductases with reduced in vitro metabolism of daunorubicin and doxorubicin. J. Pharmacol. Exp. Ther. 2010, 335, 533–545. [Google Scholar] [CrossRef]
- Bains, O.S.; Szeitz, A.; Lubieniecka, J.M.; Cragg, G.E.; Grigliatti, T.A.; Riggs, K.W.; Reid, R.E. A correlation between cytotoxicity and reductase-mediated metabolism in cell lines treated with doxorubicin and daunorubicin. J. Pharmacol. Exp. Ther. 2013, 347, 375–387. [Google Scholar] [CrossRef]
- Penning, T.M.; Jonnalagadda, S.; Trippier, P.C.; Rizner, T.L. Aldo-Keto Reductases and Cancer Drug Resistance. Pharmacol. Rev. 2021, 73, 1150–1171. [Google Scholar] [CrossRef]
- Morell, A.; Cermakova, L.; Novotna, E.; Lastovickova, L.; Haddad, M.; Haddad, A.; Portillo, R.; Wsol, V. Bruton’s Tyrosine Kinase Inhibitors Ibrutinib and Acalabrutinib Counteract Anthracycline Resistance in Cancer Cells Expressing AKR1C3. Cancers 2020, 12, 3731. [Google Scholar] [CrossRef]
- Wang, B.; Gu, Y.; Hui, K.; Huang, J.; Xu, S.; Wu, S.; Li, L.; Fan, J.; Wang, X.; Hsieh, J.T.; et al. AKR1C3, a crucial androgenic enzyme in prostate cancer, promotes epithelial-mesenchymal transition and metastasis through activating ERK signaling. Urol. Oncol. 2018, 36, 472.e411–472.e420. [Google Scholar] [CrossRef]
- Yamashita, N.; Kanno, Y.; Saito, N.; Terai, K.; Sanada, N.; Kizu, R.; Hiruta, N.; Park, Y.; Bujo, H.; Nemoto, K. Aryl hydrocarbon receptor counteracts pharmacological efficacy of doxorubicin via enhanced AKR1C3 expression in triple negative breast cancer cells. Biochem. Biophys. Res. Commun. 2019, 516, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Miller, V.L.; Lin, H.K.; Murugan, P.; Fan, M.; Penning, T.M.; Brame, L.S.; Yang, Q.; Fung, K.M. Aldo-keto reductase family 1 member C3 (AKR1C3) is expressed in adenocarcinoma and squamous cell carcinoma but not small cell carcinoma. Int. J. Clin. Exp. Pathol. 2012, 5, 278–289. [Google Scholar] [PubMed]
- Bortolozzi, R.; Bresolin, S.; Rampazzo, E.; Paganin, M.; Maule, F.; Mariotto, E.; Boso, D.; Minuzzo, S.; Agnusdei, V.; Viola, G.; et al. AKR1C enzymes sustain therapy resistance in paediatric T-ALL. Br. J. Cancer 2018, 118, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Bernardini, N.; Giannessi, F.; Bianchi, F.; Dolfi, A.; Lupetti, M.; Zaccaro, L.; Malvaldi, G.; Del Tacca, M. Comparative activity of doxorubicin and its major metabolite, doxorubicinol, on fibroblasts: A morphofunctional study. Exp. Mol. Pathol. 1991, 55, 238–250. [Google Scholar] [CrossRef]
- Heibein, A.D.; Guo, B.; Sprowl, J.A.; Maclean, D.A.; Parissenti, A.M. Role of aldo-keto reductases and other doxorubicin pharmacokinetic genes in doxorubicin resistance, DNA binding, and subcellular localization. BMC Cancer 2012, 12, 381. [Google Scholar] [CrossRef]
- Zhang, H.; Patel, A.; Ma, S.L.; Li, X.J.; Zhang, Y.K.; Yang, P.Q.; Kathawala, R.J.; Wang, Y.J.; Anreddy, N.; Fu, L.W.; et al. In vitro, in vivo and ex vivo characterization of ibrutinib: A potent inhibitor of the efflux function of the transporter MRP1. Br. J. Pharmacol. 2014, 171, 5845–5857. [Google Scholar] [CrossRef]
- Kosztyu, P.; Bukvova, R.; Dolezel, P.; Mlejnek, P. Resistance to daunorubicin, imatinib, or nilotinib depends on expression levels of ABCB1 and ABCG2 in human leukemia cells. Chem. Biol. Interact. 2014, 219, 203–210. [Google Scholar] [CrossRef]
- Hofman, J.; Malcekova, B.; Skarka, A.; Novotna, E.; Wsol, V. Anthracycline resistance mediated by reductive metabolism in cancer cells: The role of aldo-keto reductase 1C3. Toxicol. Appl. Pharmacol. 2014, 278, 238–248. [Google Scholar] [CrossRef]
- Skarydova, L.; Tomanova, R.; Havlikova, L.; Stambergova, H.; Solich, P.; Wsol, V. Deeper insight into the reducing biotransformation of bupropion in the human liver. Drug Metab. Pharmacokinet. 2014, 29, 177–184. [Google Scholar] [CrossRef]
- Bukum, N.; Novotna, E.; Morell, A.; Zelazkova, J.; Lastovickova, L.; Cermakova, L.; Portillo, R.; Solich, P.; Wsol, V. Inhibition of AKR1B10-mediated metabolism of daunorubicin as a novel off-target effect for the Bcr-Abl tyrosine kinase inhibitor dasatinib. Biochem. Pharmacol. 2021, 192, 114710. [Google Scholar] [CrossRef]
- Tavares, T.S.; Hofman, J.; Lekesova, A.; Zelazkova, J.; Wsol, V. Olaparib Synergizes the Anticancer Activity of Daunorubicin via Interaction with AKR1C3. Cancers 2020, 12, 3127. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Patel, A.; Wang, Y.J.; Zhang, Y.K.; Kathawala, R.J.; Qiu, L.H.; Patel, B.A.; Huang, L.H.; Shukla, S.; Yang, D.H.; et al. The BTK Inhibitor Ibrutinib (PCI-32765) Overcomes Paclitaxel Resistance in ABCB1- and ABCC10-Overexpressing Cells and Tumors. Mol. Cancer Ther. 2017, 16, 1021–1030. [Google Scholar] [CrossRef] [PubMed]
- Juan-Carlos, P.M.; Perla-Lidia, P.P.; Stephanie-Talia, M.M.; Monica-Griselda, A.M.; Luz-Maria, T.E. ABC transporter superfamily. An updated overview, relevance in cancer multidrug resistance and perspectives with personalized medicine. Mol. Biol. Rep. 2021, 48, 1883–1901. [Google Scholar] [CrossRef] [PubMed]
- Shi, B.; Xu, F.F.; Xiang, C.P.; Jia, R.; Yan, C.H.; Ma, S.Q.; Wang, N.; Wang, A.J.; Fan, P. Effect of sodium butyrate on ABC transporters in lung cancer A549 and colorectal cancer HCT116 cells. Oncol. Lett. 2020, 20, 148. [Google Scholar] [CrossRef]
- Lainey, E.; Sebert, M.; Thepot, S.; Scoazec, M.; Bouteloup, C.; Leroy, C.; De Botton, S.; Galluzzi, L.; Fenaux, P.; Kroemer, G. Erlotinib antagonizes ABC transporters in acute myeloid leukemia. Cell Cycle 2012, 11, 4079–4092. [Google Scholar] [CrossRef]
- Grundy, M.; Seedhouse, C.; Russell, N.H.; Pallis, M. P-glycoprotein and breast cancer resistance protein in acute myeloid leukaemia cells treated with the aurora-B kinase inhibitor barasertib-hQPA. BMC Cancer 2011, 11, 254. [Google Scholar] [CrossRef][Green Version]
- Hauswald, S.; Duque-Afonso, J.; Wagner, M.M.; Schertl, F.M.; Lubbert, M.; Peschel, C.; Keller, U.; Licht, T. Histone deacetylase inhibitors induce a very broad, pleiotropic anticancer drug resistance phenotype in acute myeloid leukemia cells by modulation of multiple ABC transporter genes. Clin. Cancer Res. 2009, 15, 3705–3715. [Google Scholar] [CrossRef]
- Tam, C.S.; Trotman, J.; Opat, S.; Burger, J.A.; Cull, G.; Gottlieb, D.; Harrup, R.; Johnston, P.B.; Marlton, P.; Munoz, J.; et al. Phase 1 study of the selective BTK inhibitor zanubrutinib in B-cell malignancies and safety and efficacy evaluation in CLL. Blood 2019, 134, 851–859. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/home (accessed on 21 July 2022).
- Hillmen, P.; Brown, J.R.; Eichhorst, B.F.; Lamanna, N.; O’Brien, S.M.; Qiu, L.; Salmi, T.; Hilger, J.; Wu, K.; Cohen, A.; et al. ALPINE: Zanubrutinib versus ibrutinib in relapsed/refractory chronic lymphocytic leukemia/small lymphocytic lymphoma. Future Oncol. 2020, 16, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Tam, C.S.; Giannopoulos, K.; Jurczak, W.; Šimkovič, M.; Shadman, M.; Österborg, A.; Laurenti, L.; Walker, P.; Opat, S.; Chan, H.; et al. SEQUOIA: Results of a Phase 3 Randomized Study of Zanubrutinib versus Bendamustine + Rituximab (BR) in Patients with Treatment-Naïve (TN) Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma (CLL/SLL). Blood 2021, 138, 396. [Google Scholar] [CrossRef]
- Yang, H.; Xiang, B.; Song, Y.; Zhang, H.; Zhao, W.; Zou, D.; Lv, F.; Guo, W.; Liu, A.; Li, C.; et al. Zanubrutinib monotherapy for relapsed or refractory non-germinal center diffuse large B-cell lymphoma. Blood Adv. 2022, 6, 1629–1636. [Google Scholar] [CrossRef] [PubMed]
- Phillips, T.; Chan, H.; Tam, C.S.; Tedeschi, A.; Johnston, P.; Oh, S.Y.; Opat, S.; Eom, H.S.; Allewelt, H.; Stern, J.C.; et al. Zanubrutinib monotherapy in relapsed/refractory indolent non-Hodgkin lymphoma. Blood Adv. 2022, 6, 3472–3479. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Y.; Zhuang, Z.; Wang, W.; Wei, C.; Zhao, D.; Zhou, D.; Zhang, W. Preliminary Evaluation of Zanubrutinib-Containing Regimens in DLBCL and the Cerebrospinal Fluid Distribution of Zanubrutinib: A 13-Case Series. Front. Oncol. 2021, 11, 760405. [Google Scholar] [CrossRef]
- Armitage, J.O.; Longo, D.L. Is watch and wait still acceptable for patients with low-grade follicular lymphoma? Blood 2016, 127, 2804–2808. [Google Scholar] [CrossRef]
- Brieghel, C.; Galle, V.; Agius, R.; da Cunha-Bang, C.; Andersen, M.A.; Vlummens, P.; Mattsson, M.; Rosenquist, R.; Smedby, K.E.; Herling, C.D.; et al. Identifying patients with chronic lymphocytic leukemia without need of treatment: End of endless watch and wait? Eur. J. Haematol. 2022, 108, 369–378. [Google Scholar] [CrossRef]
- Sindel, A.; Al-Juhaishi, T.; Yazbeck, V. Marginal Zone Lymphoma: State-of-the-Art Treatment. Curr. Treat. Options Oncol. 2019, 20, 90. [Google Scholar] [CrossRef]
- Durot, E.; Delmer, A. Watch and wait in Waldenstrom macroglobulinaemia: Looking for who to watch carefully and who can wait without worrying. Is it that simple? Br. J. Haematol. 2021, 195, 155–157. [Google Scholar] [CrossRef]
- Rossi, D.; Spina, V.; Gaidano, G. Biology and treatment of Richter syndrome. Blood 2018, 131, 2761–2772. [Google Scholar] [CrossRef]
- Gonzalez-Rincon, J.; Mendez, M.; Gomez, S.; Garcia, J.F.; Martin, P.; Bellas, C.; Pedrosa, L.; Rodriguez-Pinilla, S.M.; Camacho, F.I.; Quero, C.; et al. Unraveling transformation of follicular lymphoma to diffuse large B-cell lymphoma. PLoS ONE 2019, 14, e0212813. [Google Scholar] [CrossRef] [PubMed]
- Coiffier, B.; Sarkozy, C. Diffuse large B-cell lymphoma: R-CHOP failure-what to do? Hematol. Am. Soc. Hematol. Educ. Program 2016, 2016, 366–378. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, H.L.; Yao, S.; Goldman, B.H.; Lee, K.; Spier, C.M.; LeBlanc, M.L.; Rimsza, L.M.; Cerhan, J.R.; Habermann, T.M.; Link, B.K.; et al. Genetic polymorphisms in oxidative stress-related genes are associated with outcomes following treatment for aggressive B-cell non-Hodgkin lymphoma. Am. J. Hematol. 2014, 89, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Wu, N.; Sun, H.; Dong, M.; Guo, T.; Chi, P.; Li, G.; Sun, D.; Jin, Y. ABCG2 and NCF4 polymorphisms are associated with clinical outcomes in diffuse large B-cell lymphoma patients treated with R-CHOP. Oncotarget 2017, 8, 58292–58303. [Google Scholar] [CrossRef] [PubMed]
- Takimoto-Shimomura, T.; Nagoshi, H.; Maegawa, S.; Fujibayashi, Y.; Tsukamoto, T.; Matsumura-Kimoto, Y.; Mizuno, Y.; Chinen, Y.; Mizutani, S.; Shimura, Y.; et al. Establishment and Characteristics of a Novel Mantle Cell Lymphoma-derived Cell Line and a Bendamustine-resistant Subline. Cancer Genom. Proteom. 2018, 15, 213–223. [Google Scholar] [CrossRef]
- Tabata, M.; Tsubaki, M.; Takeda, T.; Tateishi, K.; Tsurushima, K.; Imano, M.; Satou, T.; Ishizaka, T.; Nishida, S. Dasatinib reverses drug resistance by downregulating MDR1 and Survivin in Burkitt lymphoma cells. BMC Complement Med. Ther. 2020, 20, 84. [Google Scholar] [CrossRef]
- Molina-Cerrillo, J.; Alonso-Gordoa, T.; Gajate, P.; Grande, E. Bruton’s tyrosine kinase (BTK) as a promising target in solid tumors. Cancer Treat. Rev. 2017, 58, 41–50. [Google Scholar] [CrossRef]
- Szklener, K.; Michalski, A.; Zak, K.; Piwonski, M.; Mandziuk, S. Ibrutinib in the Treatment of Solid Tumors: Current State of Knowledge and Future Directions. Cells 2022, 11, 1338. [Google Scholar] [CrossRef]
- Rushworth, S.A.; Murray, M.Y.; Zaitseva, L.; Bowles, K.M.; MacEwan, D.J. Identification of Bruton’s tyrosine kinase as a therapeutic target in acute myeloid leukemia. Blood 2014, 123, 1229–1238. [Google Scholar] [CrossRef]
- Yeh, C.T.; Chen, T.T.; Satriyo, P.B.; Wang, C.H.; Wu, A.T.H.; Chao, T.Y.; Lee, K.Y.; Hsiao, M.; Wang, L.S.; Kuo, K.T. Bruton’s tyrosine kinase (BTK) mediates resistance to EGFR inhibition in non-small-cell lung carcinoma. Oncogenesis 2021, 10, 56. [Google Scholar] [CrossRef]
- Rotin, L.E.; Gronda, M.; Hurren, R.; Wang, X.; Minden, M.D.; Slassi, M.; Schimmer, A.D. Investigating the synergistic mechanism between ibrutinib and daunorubicin in acute myeloid leukemia cells. Leuk. Lymphoma 2016, 57, 2432–2436. [Google Scholar] [CrossRef]
- Birtwistle, J.; Hayden, R.E.; Khanim, F.L.; Green, R.M.; Pearce, C.; Davies, N.J.; Wake, N.; Schrewe, H.; Ride, J.P.; Chipman, J.K.; et al. The aldo-keto reductase AKR1C3 contributes to 7,12-dimethylbenz(a)anthracene-3,4-dihydrodiol mediated oxidative DNA damage in myeloid cells: Implications for leukemogenesis. Mutat. Res. 2009, 662, 67–74. [Google Scholar] [CrossRef]
- Byrns, M.C.; Duan, L.; Lee, S.H.; Blair, I.A.; Penning, T.M. Aldo-keto reductase 1C3 expression in MCF-7 cells reveals roles in steroid hormone and prostaglandin metabolism that may explain its over-expression in breast cancer. J. Steroid Biochem. Mol. Biol. 2010, 118, 177–187. [Google Scholar] [CrossRef]
- Desmond, J.C.; Mountford, J.C.; Drayson, M.T.; Walker, E.A.; Hewison, M.; Ride, J.P.; Luong, Q.T.; Hayden, R.E.; Vanin, E.F.; Bunce, C.M. The aldo-keto reductase AKR1C3 is a novel suppressor of cell differentiation that provides a plausible target for the non-cyclooxygenase-dependent antineoplastic actions of nonsteroidal anti-inflammatory drugs. Cancer Res. 2003, 63, 505–512. [Google Scholar]
- Oduwole, O.O.; Li, Y.; Isomaa, V.V.; Mantyniemi, A.; Pulkka, A.E.; Soini, Y.; Vihko, P.T. 17beta-hydroxysteroid dehydrogenase type 1 is an independent prognostic marker in breast cancer. Cancer Res. 2004, 64, 7604–7609. [Google Scholar] [CrossRef]
- Zhu, P.; Feng, R.; Lu, X.; Liao, Y.; Du, Z.; Zhai, W.; Chen, K. Diagnostic and prognostic values of AKR1C3 and AKR1D1 in hepatocellular carcinoma. Aging 2021, 13, 4138–4156. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.F.; Wang, S.G.; Zhao, Z.Y.; Li, W.L. AKR1C1-3, notably AKR1C3, are distinct biomarkers for liver cancer diagnosis and prognosis: Database mining in malignancies. Oncol. Lett. 2019, 18, 4515–4522. [Google Scholar] [CrossRef]
- Mahadevan, D.; DiMento, J.; Croce, K.D.; Riley, C.; George, B.; Fuchs, D.; Mathews, T.; Wilson, C.; Lobell, M. Transcriptosome and serum cytokine profiling of an atypical case of myelodysplastic syndrome with progression to acute myelogenous leukemia. Am. J. Hematol. 2006, 81, 779–786. [Google Scholar] [CrossRef]
- Liu, C.Y.; Hsu, Y.H.; Pan, P.C.; Wu, M.T.; Ho, C.K.; Su, L.; Xu, X.; Li, Y.; Christiani, D.C.; Kaohsiung Leukemia Research, G. Maternal and offspring genetic variants of AKR1C3 and the risk of childhood leukemia. Carcinogenesis 2008, 29, 984–990. [Google Scholar] [CrossRef]
- FDA (US Food and Drug Administration). U.S. Food & Drug Administration: List of the Relevant Molecular Targets in Paediatric Oncology. Available online: https://www.fda.gov/about-fda/oncology-center-excellence/pediatric-oncology (accessed on 16 July 2022).
- FDA (US Food and Drug Administration). U.S. Food & Drug Administration: List of the Relevant Molecular Targets in Paediatric Oncology. Available online: https://www.fda.gov/media/120332/download (accessed on 16 July 2022).






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Čermáková, L.; Hofman, J.; Laštovičková, L.; Havlíčková, L.; Špringrová, I.; Novotná, E.; Wsól, V. Bruton’s Tyrosine Kinase Inhibitor Zanubrutinib Effectively Modulates Cancer Resistance by Inhibiting Anthracycline Metabolism and Efflux. Pharmaceutics 2022, 14, 1994. https://doi.org/10.3390/pharmaceutics14101994
Čermáková L, Hofman J, Laštovičková L, Havlíčková L, Špringrová I, Novotná E, Wsól V. Bruton’s Tyrosine Kinase Inhibitor Zanubrutinib Effectively Modulates Cancer Resistance by Inhibiting Anthracycline Metabolism and Efflux. Pharmaceutics. 2022; 14(10):1994. https://doi.org/10.3390/pharmaceutics14101994
Chicago/Turabian StyleČermáková, Lucie, Jakub Hofman, Lenka Laštovičková, Lucie Havlíčková, Ivona Špringrová, Eva Novotná, and Vladimír Wsól. 2022. "Bruton’s Tyrosine Kinase Inhibitor Zanubrutinib Effectively Modulates Cancer Resistance by Inhibiting Anthracycline Metabolism and Efflux" Pharmaceutics 14, no. 10: 1994. https://doi.org/10.3390/pharmaceutics14101994
APA StyleČermáková, L., Hofman, J., Laštovičková, L., Havlíčková, L., Špringrová, I., Novotná, E., & Wsól, V. (2022). Bruton’s Tyrosine Kinase Inhibitor Zanubrutinib Effectively Modulates Cancer Resistance by Inhibiting Anthracycline Metabolism and Efflux. Pharmaceutics, 14(10), 1994. https://doi.org/10.3390/pharmaceutics14101994