
Alternative Methotrexate Oral Formulation: Enhanced Aqueous Solubility, Bioavailability, Photostability, and Permeability

 ,
,  , ,
, ,
Abstract
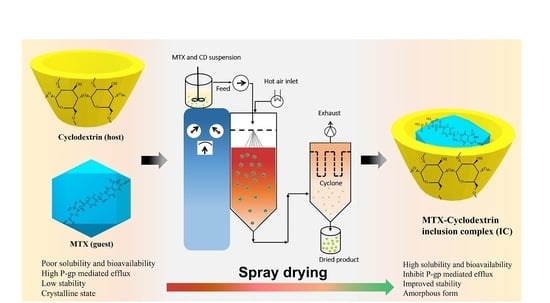
1. Introduction
2. Material and Methods
2.1. Materials
2.2. Phase Solubility Studies
2.3. HPLC System
2.4. Preparation of the Binary Inclusion Complex and Solubilization Test
2.5. In Vitro Dissolution Study
2.6. The Physicochemical and Morphological Characterization
2.6.1. Particle Size and Morphology Analysis
2.6.2. Differential Scanning Calorimetry (DSC) and Powder X-ray Diffraction (PXRD)
2.6.3. Fourier-Transform Infrared Spectroscopy (FTIR)
2.6.4. 1H NMR Studies
2.7. In Vivo Pharmacokinetic Studies
Sample Preparation and Chromatographic Analysis
2.8. Photostability Assessments
2.9. Permeation Experiments through Caco-2 Cells
2.9.1. In Vitro Cytotoxicity Study
2.9.2. In Vitro Caco-2 Permeation Experiment
3. Results and Discussion
3.1. Phase Solubility Studies
3.2. Solubility and In-Vitro Dissolution Studies
3.3. Physicochemical Characterization
3.3.1. SEM Analysis
3.3.2. Differential Scanning Calorimetry (DSC)
3.3.3. Powder X-ray Diffraction (PXRD)
3.3.4. FT-IR Spectroscopy
3.3.5. 1H NMR and 2D NMR Studies
3.4. In Vivo Pharmacokinetic Studies
3.5. Photostability Study
3.6. Influence of DM-β-CD on Permeation Behavior of MTX
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bleyer, W.A. The clinical pharmacology of methotrexate: New applications of an old drug. Cancer 1978, 41, 36–51. [Google Scholar] [CrossRef]
- Giletti, A.; Vital, M.; Lorenzo, M.; Cardozo, P.; Borelli, G.; Gabus, R.; Martínez, L.; Díaz, L.; Assar, R.; Rodriguez, M.N.; et al. Methotrexate pharmacogenetics in Uruguayan adults with hematological malignant diseases. Eur. J. Pharm. Sci. 2017, 109, 480–485. [Google Scholar] [CrossRef] [PubMed]
- Braun, J.; Rau, R. An update on methotrexate. Curr. Opin. Rheumatol. 2009, 21, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Abolmaali, S.S.; Tamaddon, A.M.; Dinarvand, R. A review of therapeutic challenges and achievements of methotrexate delivery systems for treatment of cancer and rheumatoid arthritis. Cancer Chemother. Pharmacol. 2013, 71, 1115–1130. [Google Scholar] [CrossRef]
- Kim, D.S.; Cho, J.H.; Park, J.H.; Kim, J.S.; Song, E.S.; Kwon, J.; Giri, B.R.; Jin, S.G.; Kim, K.S.; Choi, H.-G.; et al. Self-microemulsifying drug delivery system (SMEDDS) for improved oral delivery and photostability of methotrexate. Int. J. Nanomed. 2019, 14, 4949–4960. [Google Scholar] [CrossRef] [PubMed]
- Chatterji, D.C.; Gallelli, J.F. Thermal and photolytic decomposition of methotrexate in aqueous solutions. J. Pharm. Sci. 1978, 67, 526–531. [Google Scholar] [CrossRef]
- Ray, S.; Joy, M.; Sa, B.; Ghosh, S.; Chakraborty, J. pH dependent chemical stability and release of methotrexate from a novel nanoceramic carrier. RSC Adv. 2015, 5, 39482–39494. [Google Scholar] [CrossRef]
- Santos, A.C.; Costa, D.; Ferreira, L.; Guerra, C.; Pereira-Silva, M.; Pereira, I.; Peixoto, D.; Ferreira, N.R.; Veiga, F. Cyclodextrin-based delivery systems for in vivo-tested anticancer therapies. Drug Deliv. Transl. Res. 2021, 11, 49–71. [Google Scholar] [CrossRef]
- Datta, S.; Grant, D.J.W. Crystal structures of drugs: Advances in determination, prediction and engineering. Nat. Rev. Drug Discov. 2004, 3, 42–57. [Google Scholar] [CrossRef]
- Giri, B.R.; Kwon, J.; Vo, A.Q.; Bhagurkar, A.M.; Bandari, S.; Kim, D.W. Hot-melt extruded amorphous solid dispersion for solubility, stability, and bioavailability enhancement of telmisartan. Pharmaceuticals 2021, 14, 73. [Google Scholar] [CrossRef]
- Noh, G.; Keum, T.; Bashyal, S.; Seo, J.-E.; Shrawani, L.; Kim, J.H.; Lee, S. Recent progress in hydrophobic ion-pairing and lipid-based drug delivery systems for enhanced oral delivery of biopharmaceuticals. J. Pharm. Investig. 2022, 52, 75–93. [Google Scholar] [CrossRef]
- Sanches, B.M.A.; Ferreira, E.I. Is prodrug design an approach to increase water solubility? Int. J. Pharm. 2019, 568, 118498. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Park, H.; Kang, K.T.; Ha, E.S.; Kim, M.S.; Hwang, S.J. Micronization of a poorly water-soluble drug, fenofibrate, via supercritical-fluid-assisted spray-drying. J. Pharm. Investig. 2022, 52, 353–366. [Google Scholar] [CrossRef]
- Kim, D.-H.; Lee, S.-E.; Pyo, Y.-C.; Tran, P.; Park, J.-S. Solubility enhancement and application of cyclodextrins in local drug delivery. J. Pharm. Investig. 2020, 50, 17–27. [Google Scholar] [CrossRef]
- Jansook, P.; Ogawa, N.; Loftsson, T. Cyclodextrins: Structure, physicochemical properties and pharmaceutical applications. Int. J. Pharm. 2018, 535, 272–284. [Google Scholar] [CrossRef] [PubMed]
- Bilensoy, E. Cyclodextrins in Pharmaceutics, Cosmetics, and Biomedicine: Current and Future Industrial Applications; Bilensoy, E., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011; ISBN 9780470474228. [Google Scholar]
- Soni, S.S.; Alsasa, A.; Rodell, C.B. Applications of macrocyclic host molecules in immune modulation and therapeutic delivery. Front. Chem. 2021, 9, 190. [Google Scholar] [CrossRef] [PubMed]
- Jicsinszky, L.; Cravotto, G. Cyclodextrins in skin formulations and transdermal delivery. J. Ski. Stem Cell 2020, 6, e102561. [Google Scholar] [CrossRef]
- Truzzi, E.; Rustichelli, C.; de Oliveira Junior, E.R.; Ferraro, L.; Maretti, E.; Graziani, D.; Botti, G.; Beggiato, S.; Iannuccelli, V.; Lima, E.M.; et al. Nasal biocompatible powder of Geraniol oil complexed with cyclodextrins for neurodegenerative diseases: Physicochemical characterization and in vivo evidences of nose to brain delivery. J. Control. Release 2021, 335, 191–202. [Google Scholar] [CrossRef]
- Szente, L.; Singhal, A.; Domokos, A.; Song, B. Cyclodextrins: Assessing the impact of cavity size, occupancy, and substitutions on cytotoxicity and cholesterol homeostasis. Molecules 2018, 23, 1228. [Google Scholar] [CrossRef]
- Brewster, M.E.; Loftsson, T. Cyclodextrins as pharmaceutical solubilizers. Adv. Drug Deliv. Rev. 2007, 59, 645–666. [Google Scholar] [CrossRef]
- Przybyla, M.A.; Yilmaz, G.; Becer, C.R. Natural cyclodextrins and their derivatives for polymer synthesis. Polym. Chem. 2020, 11, 7582–7602. [Google Scholar] [CrossRef]
- Möller, K.; Macaulay, B.; Bein, T. Curcumin encapsulated in crosslinked cyclodextrin nanoparticles enables immediate inhibition of cell growth and efficient killing of cancer cells. Nanomaterials 2021, 11, 489. [Google Scholar] [CrossRef] [PubMed]
- Gidwani, B.; Vyas, A. A comprehensive review on cyclodextrin-based carriers for delivery of chemotherapeutic cytotoxic anticancer drugs. Biomed Res. Int. 2015, 2015, 198268. [Google Scholar] [CrossRef] [PubMed]
- Connors, K.A.; Higuchi, T. Phase solubility techniques. Adv. Anal. Chem. Instrum. 1965, 4, 117–212. [Google Scholar]
- Giri, B.R.; Kim, J.S.; Park, J.H.; Jin, S.G.; Kim, K.S.; Ud Din, F.; Choi, H.G.; Kim, D.W. Improved bioavailability and high photostability of methotrexate by spray-dried surface-attached solid dispersion with an aqueous medium. Pharmaceutics 2021, 13, 111. [Google Scholar] [CrossRef]
- Bashyal, S.; Seo, J.-E.; Keum, T.; Noh, G.; Lamichhane, S.; Kim, J.H.; Kim, C.H.; Choi, Y.W.; Lee, S. Facilitated buccal insulin delivery via hydrophobic ion-pairing approach: In vitro and ex vivo evaluation. Int. J. Nanomed. 2021, 16, 4677–4691. [Google Scholar] [CrossRef]
- Giri, B.R.; Lee, J.; Lim, D.Y.; Kim, D.W. Docetaxel/dimethyl-β-cyclodextrin inclusion complexes: Preparation, in vitro evaluation and physicochemical characterization. Drug Dev. Ind. Pharm. 2021, 47, 319–328. [Google Scholar] [CrossRef]
- Caira, M.R.; Bourne, S.A.; Samsodien, H.; Smith, V.J. Inclusion complexes of 2-methoxyestradiol with dimethylated and permethylated β-cyclodextrins: Models for cyclodextrin-steroid interaction. Beilstein J. Org. Chem. 2015, 11, 2616–2630. [Google Scholar] [CrossRef]
- Fu, Y.; Kao, W.J. Drug release kinetics and transport mechanisms of non-degradable and degradable polymeric delivery systems. Expert Opin. Drug Deliv. 2010, 7, 429–444. [Google Scholar] [CrossRef]
- Carneiro, S.; Costa Duarte, F.; Heimfarth, L.; Siqueira Quintans, J.; Quintans-Júnior, L.; Veiga Júnior, V.; Neves de Lima, Á. Cyclodextrin–drug inclusion complexes: In vivo and in vitro approaches. Int. J. Mol. Sci. 2019, 20, 642. [Google Scholar] [CrossRef]
- Loftsson, T. Drug solubilization by complexation. Int. J. Pharm. 2017, 531, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.; Giri, B.R.; Song, E.S.; Bae, J.; Lee, J.; Kim, D.W. Spray-dried amorphous solid dispersions of atorvastatin calcium for improved supersaturation and oral bioavailability. Pharmaceutics 2019, 11, 461. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Van den Mooter, G. Spray drying formulation of amorphous solid dispersions. Adv. Drug Deliv. Rev. 2016, 100, 27–50. [Google Scholar] [CrossRef]
- Periasamy, R.; Kothainayaki, S.; Sivakumar, K. Host-guest inclusion complex of β-cyclodextrin and 4,4′-(1,4-phenylenediisopropylidene)bisaniline: Spectral, structural and molecular modeling studies. J. Mol. Struct. 2021, 1224, 129050. [Google Scholar] [CrossRef]
- Geng, Q.; Li, T.; Wang, X.; Chu, W.; Cai, M.; Xie, J.; Ni, H. The mechanism of bensulfuron-methyl complexation with β-cyclodextrin and 2-hydroxypropyl-β-cyclodextrin and effect on soil adsorption and bio-activity. Sci. Rep. 2019, 9, 1–11. [Google Scholar]
- Al-Marzouqi, A.H.; Shehatta, I.; Jobe, B.; Dowaidar, A. Phase solubility and inclusion complex of itraconazole with β-cyclodextrin using supercritical carbon dioxide. J. Pharm. Sci. 2006, 95, 292–304. [Google Scholar] [CrossRef] [PubMed]
- Manne, A.S.N.; Hegde, A.R.; Raut, S.Y.; Rao, R.R.; Kulkarni, V.I.; Mutalik, S. Hot liquid extrusion assisted drug-cyclodextrin complexation: A novel continuous manufacturing method for solubility and bioavailability enhancement of drugs. Drug Deliv. Transl. Res. 2020, 11, 1273–1287. [Google Scholar] [CrossRef] [PubMed]
- Dedroog, S.; Pas, T.; Vergauwen, B.; Huygens, C.; Van den Mooter, G. Solid-state analysis of amorphous solid dispersions: Why DSC and XRPD may not be regarded as stand-alone techniques. J. Pharm. Biomed. Anal. 2020, 178, 112937. [Google Scholar] [CrossRef]
- Ghodke, D.; Ghodke, G.; Patil, K.; Nakhat, P.; Nakhat, P.; Naikwade, N.; Magdum, C. Solid state characterization of domperidone: Hydroxypropyl-b-cyclodextrin inclusion complex. Indian J. Pharm. Sci. 2010, 72, 245. [Google Scholar] [CrossRef]
- Gao, S.; Bie, C.; Ji, Q.; Ling, H.; Li, C.; Fu, Y.; Zhao, L.; Ye, F. Preparation and characterization of cyanazine-hydroxypropyl-beta-cyclodextrin inclusion complex. RSC Adv. 2019, 9, 26109–26115. [Google Scholar] [CrossRef]
- Sangwai, M.; Vavia, P. Amorphous ternary cyclodextrin nanocomposites of telmisartan for oral drug delivery: Improved solubility and reduced pharmacokinetic variability. Int. J. Pharm. 2013, 453, 423–432. [Google Scholar] [CrossRef] [PubMed]
- De Almeida Magalhães, T.S.S.; de Oliveira Macedo, P.C.; Kawashima Pacheco, S.Y.; da Silva, S.S.; Barbosa, E.G.; Pereira, R.R.; Costa, R.M.R.; Silva Junior, J.O.C.; da Silva Ferreira, M.A.; de Almeida, J.C.; et al. Development and Evaluation of Antimicrobial and Modulatory Activity of Inclusion Complex of Euterpe oleracea Mart Oil and β-Cyclodextrin or HP-β-Cyclodextrin. Int. J. Mol. Sci. 2020, 21, 942. [Google Scholar] [CrossRef] [PubMed]
- Martins, M.H.; Calderini, A.; Pessine, F.B.T. Host-guest interactions between dapsone and β-cyclodextrin (Part II): Thermal analysis, spectroscopic characterization, and solubility studies. J. Incl. Phenom. Macrocycl. Chem. 2012, 74, 109–116. [Google Scholar] [CrossRef]
- Yuan, C.; Jin, Z.; Xu, X. Inclusion complex of astaxanthin with hydroxypropyl-β-cyclodextrin: UV, FTIR, 1H NMR and molecular modeling studies. Carbohydr. Polym. 2012, 89, 492–496. [Google Scholar] [CrossRef]
- NE, P.; TV, L.; TA, K.; EO, H.; LD, K. Inclusion complexes of carotenoids with cyclodextrins: 1H NMR, EPR, and optical studies. Free Radic. Biol. Med. 2004, 36, 872–880. [Google Scholar]
- Yang, L.-J.; Ma, S.-X.; Zhou, S.-Y.; Chen, W.; Yuan, M.-W.; Yin, Y.-Q.; Yang, X.-D. Preparation and characterization of inclusion complexes of naringenin with β-cyclodextrin or its derivative. Carbohydr. Polym. 2013, 98, 861–869. [Google Scholar] [CrossRef] [PubMed]
- Hemine, K.; Skwierawska, A.; Kleist, C.; Olewniczak, M.; Szwarc-Karabyka, K.; Wyrzykowski, D.; Mieszkowska, A.; Chojnacki, J.; Czub, J.; Nierzwicki, L. Effect of chemical structure on complexation efficiency of aromatic drugs with cyclodextrins: The example of dibenzazepine derivatives. Carbohydr. Polym. 2020, 250, 116957. [Google Scholar] [CrossRef]
- Li, H.; Krstin, S.; Wang, S.; Wink, M. Capsaicin and piperine can overcome multidrug resistance in cancer cells to doxorubicin. Molecules 2018, 23, 557. [Google Scholar] [CrossRef]
- Tran, P.H.L.; Wang, T.; Yang, C.; Tran, T.T.; Duan, W. Development of conjugate-by-conjugate structured nanoparticles for oral delivery of docetaxel. Mater. Sci. Eng. C 2020, 107, 110346. [Google Scholar] [CrossRef]
- Arima, H.; Yunomae, K.; Hirayama, F.; Uekama, K. Contribution of P-glycoprotein to the enhancing effects of dimethyl-beta-cyclodextrin on oral bioavailability of tacrolimus. J. Pharmacol. Exp. Ther. 2001, 297, 547–555. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | IC1 | IC2 | IC3 | IC4 |
|---|---|---|---|---|
| MTX (g) | 1 | 1 | 1 | 1 |
| Cyclodextrin (g) | 3.08 | 2.50 | 2.71 | 2.93 |
| 0.01 M HCl ethanolic solution (mL) | 200 | 200 | 200 | 200 |
| Water (mL) | 200 | 200 | 200 | 200 |
| Cyclodextrins | Stability Constant (Kα, M−1) |
|---|---|
| HP-β-CD | 433.56 |
| β-CD | 552.25 |
| M-β-CD | 574.48 |
| DM-β-CD | 3114.69 |
| Dx (10), μm | Dx (50), μm | Dx (90), μm | |
|---|---|---|---|
| MTX | 3.60 ± 0.48 | 14.03 ± 2.97 | 103.73 ± 12.13 |
| IC1 | 0.78 ± 0.15 | 3.56 ± 0.07 | 8.88 ± 0.29 |
| IC2 | 1.20 ± 0.03 | 4.39 ± 0.12 | 13.35 ± 0.49 |
| IC3 | 0.45 ± 0.04 | 3.26 ± 0.06 | 7.26 ± 0.32 |
| IC4 | 0.38 ± 0.03 | 3.23 ± 0.05 | 7.65 ± 0.25 |
| H Protons | δ(DM-β-CD) | δ(IC) | Δδ (ppm) | H Protons | δ(MTX) | δ(IC) | Δδ (ppm) |
|---|---|---|---|---|---|---|---|
| H-1 | 4.97 | 4.97 | - | H-7 | 8.57 | 8.59 | 0.02 |
| H-2 | 3.70 | 3.70 | - | H-9 | 4.78 | 4.79 | 0.01 |
| H-3 | 3.20 | 3.26 | +0.06 | H-10 | 3.20 | ND | ND |
| H-4 | 3.56 | 3.57 | +0.01 | -NH2(2) | 7.51 | 7.92 | 0.39 |
| H-5 | 3.34 | 3.36 | +0.02 | -NH2(4) | 6.74 | 6.87 | 0.13 |
| H-6 | ND | ND | ND | H-13,15 | 7.73 | 7.73 | - |
| OCH3 | 3.50 | 3.50 | - | H-16,12 | 6.82 | 6.82 | - |
| OCH3 | 3.25 | 3.25 | - | H-18 | 8.19 | 8.19 | - |
| H-19 | 4.35 | 4.35 | - | ||||
| H-21 | 2.05 | 2.06 | 0.01 | ||||
| H-22 | 2.32 | 2.34 | 0.02 |
| Parameter | MTX | IC1 | IC2 | IC3 | IC4 |
|---|---|---|---|---|---|
| AUC (h ng/mL) | 1738.71 ± 294.65 | 3198.39 ± 226.79 * | 3016.90 ± 237.09 * | 2973.33 ± 316.89 * | 3820.27 ± 424.27 # |
| Cmax (ng/mL) | 265.63 ± 57.05 | 567.93 ± 70.55 * | 439.61 ± 155.24 * | 561.44 ± 149.83 * | 872.76 ± 62.19 # |
| Tmax (h) | 1.13 ± 0.23 | 1.03 ± 0.25 | 1.57 ± 0.40 | 1.04 ± 0.21 | 0.75 ± 0.29 |
| t1/2 (h) | 5.40 ± 0.72 | 5.24 ± 0.99 | 5.29 ± 0.70 | 4.95 ± 0.30 | 4.82 ± 0.17 |
| Kel (h−1) | 0.13 ± 0.02 | 0.14 ± 0.03 | 0.13 ± 0.02 | 0.15 ± 0.02 | 0.15 ± 0.02 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giri, B.R.; Yang, H.S.; Song, I.-S.; Choi, H.-G.; Cho, J.H.; Kim, D.W. Alternative Methotrexate Oral Formulation: Enhanced Aqueous Solubility, Bioavailability, Photostability, and Permeability. Pharmaceutics 2022, 14, 2073. https://doi.org/10.3390/pharmaceutics14102073
Giri BR, Yang HS, Song I-S, Choi H-G, Cho JH, Kim DW. Alternative Methotrexate Oral Formulation: Enhanced Aqueous Solubility, Bioavailability, Photostability, and Permeability. Pharmaceutics. 2022; 14(10):2073. https://doi.org/10.3390/pharmaceutics14102073
Chicago/Turabian StyleGiri, Bhupendra Raj, Hyun Seok Yang, Im-Sook Song, Han-Gon Choi, Jung Hyun Cho, and Dong Wuk Kim. 2022. "Alternative Methotrexate Oral Formulation: Enhanced Aqueous Solubility, Bioavailability, Photostability, and Permeability" Pharmaceutics 14, no. 10: 2073. https://doi.org/10.3390/pharmaceutics14102073
APA StyleGiri, B. R., Yang, H. S., Song, I.-S., Choi, H.-G., Cho, J. H., & Kim, D. W. (2022). Alternative Methotrexate Oral Formulation: Enhanced Aqueous Solubility, Bioavailability, Photostability, and Permeability. Pharmaceutics, 14(10), 2073. https://doi.org/10.3390/pharmaceutics14102073