
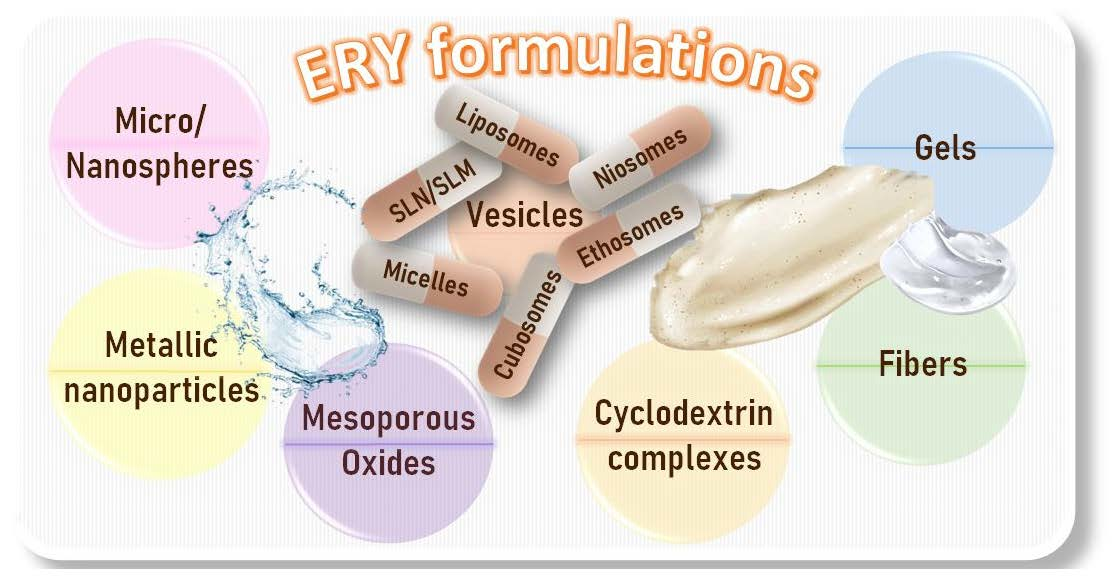
Erythromycin Formulations—A Journey to Advanced Drug Delivery



Abstract
:
1. Introduction
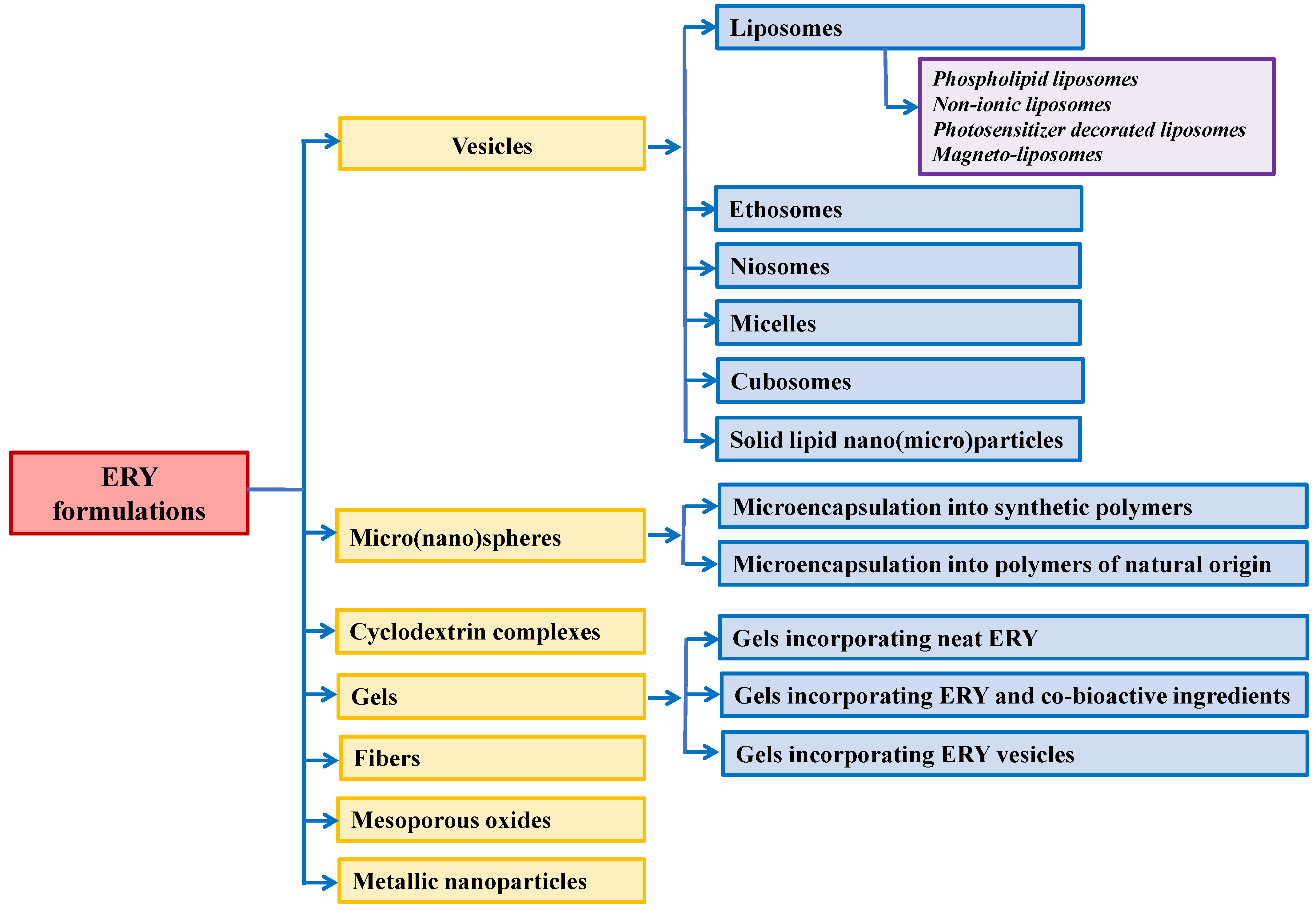
2. Vesicles
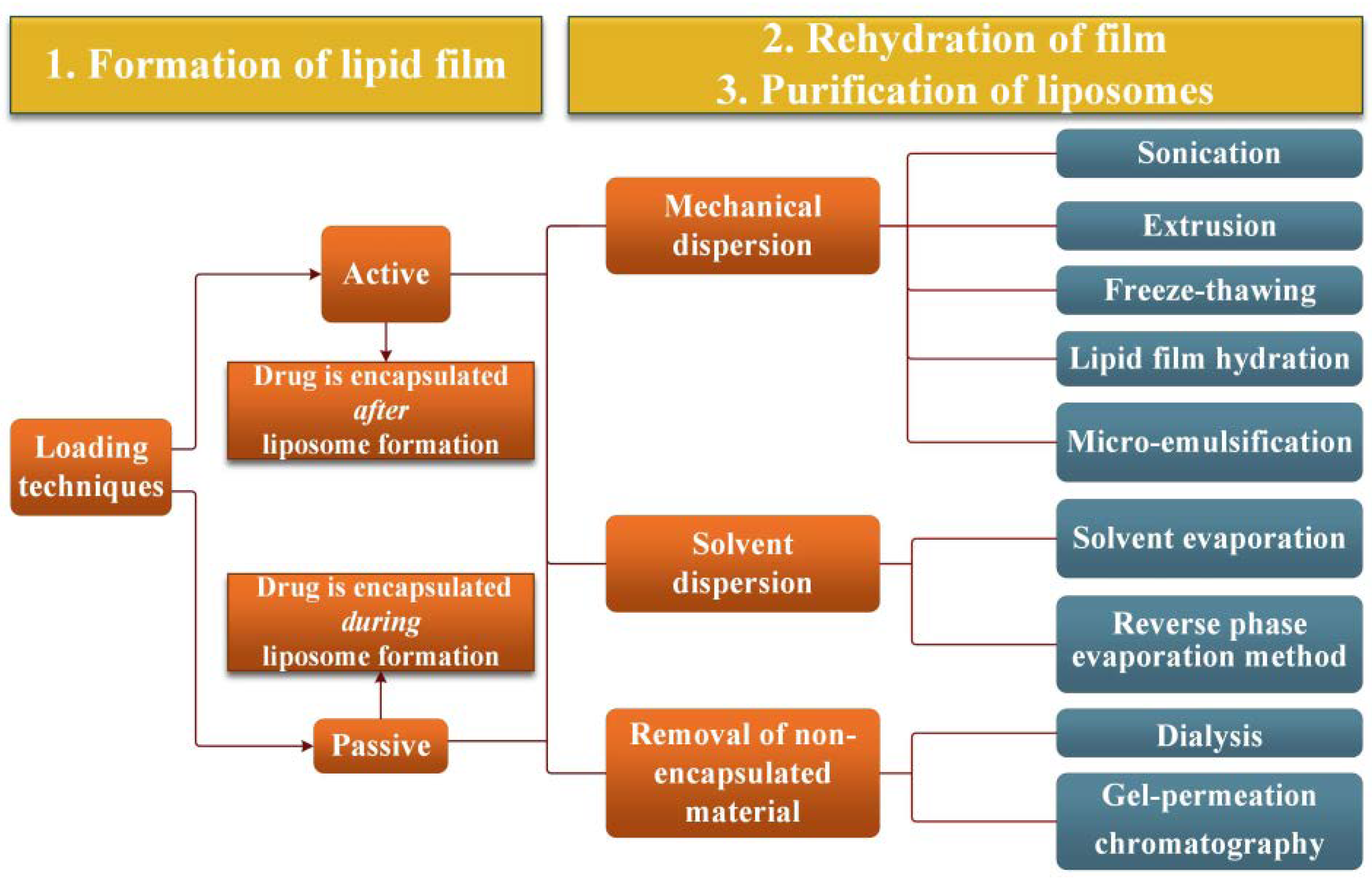
2.1. Liposomes
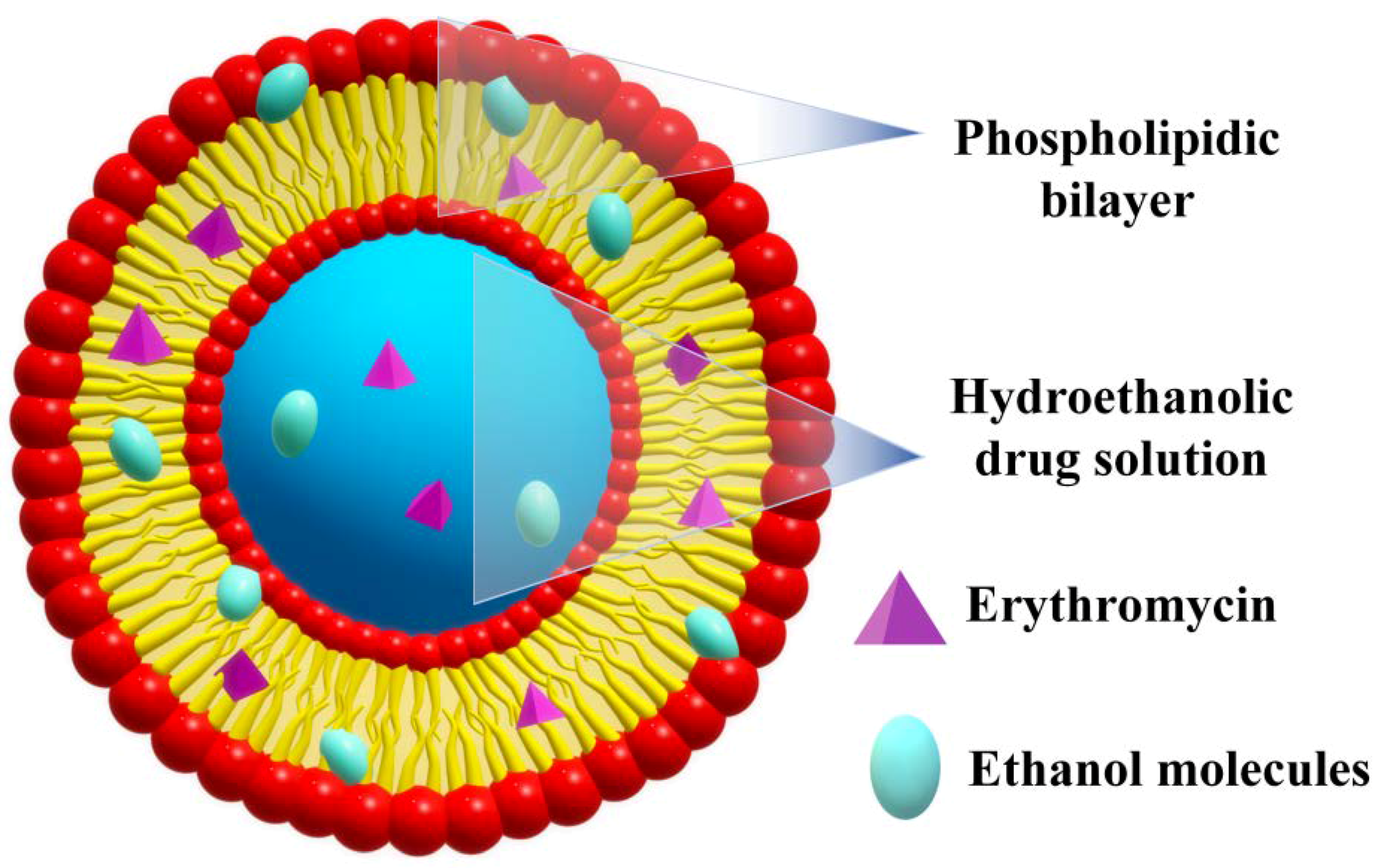
2.2. Ethosomes
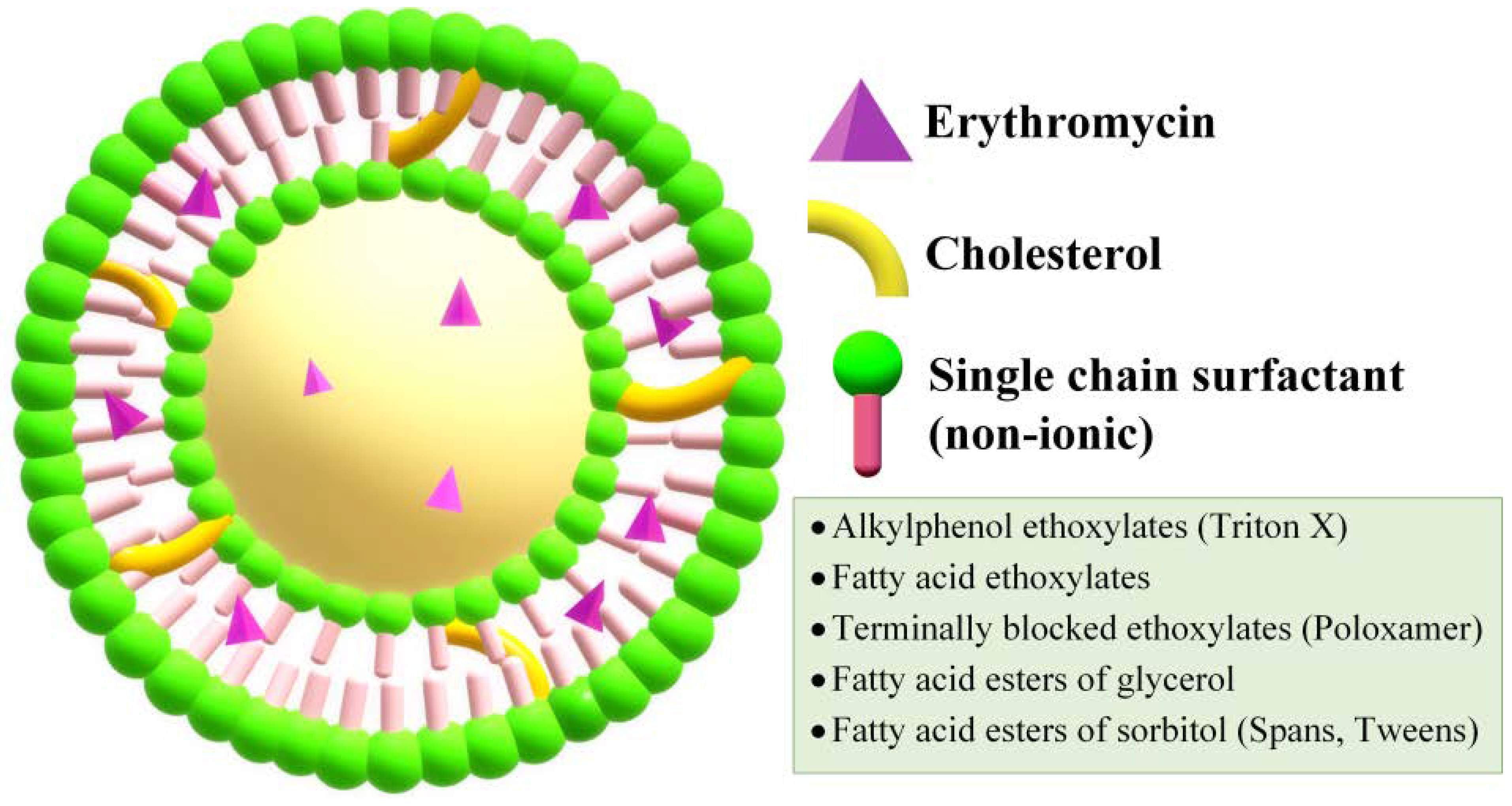
2.3. Niosomes
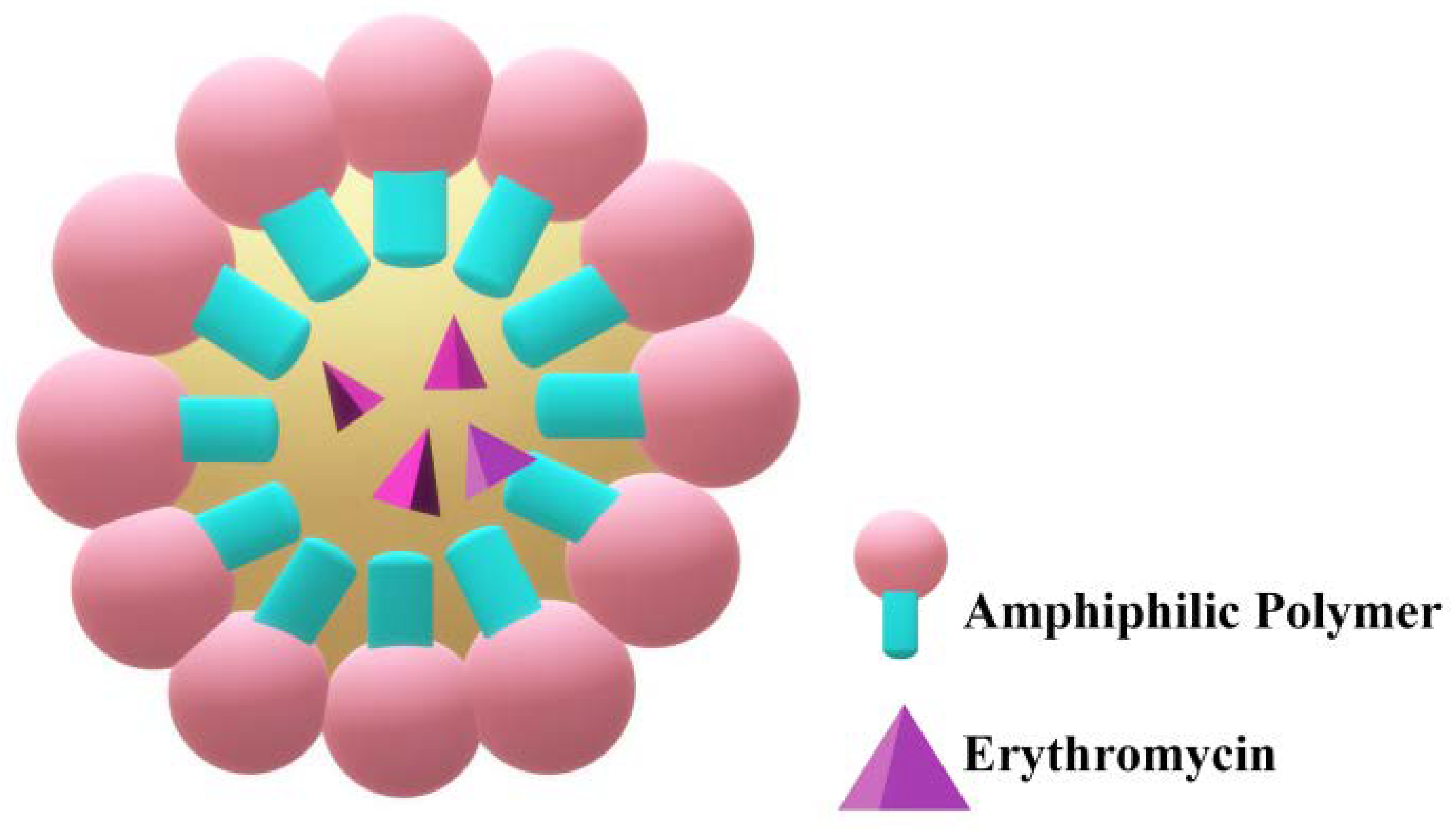
2.4. Micelles
2.5. Cubosomes
2.6. Solid Lipid Nano(Micro)Particles
2.6.1. Solid Lipid Nanoparticles
2.6.2. Solid Lipid Microparticles
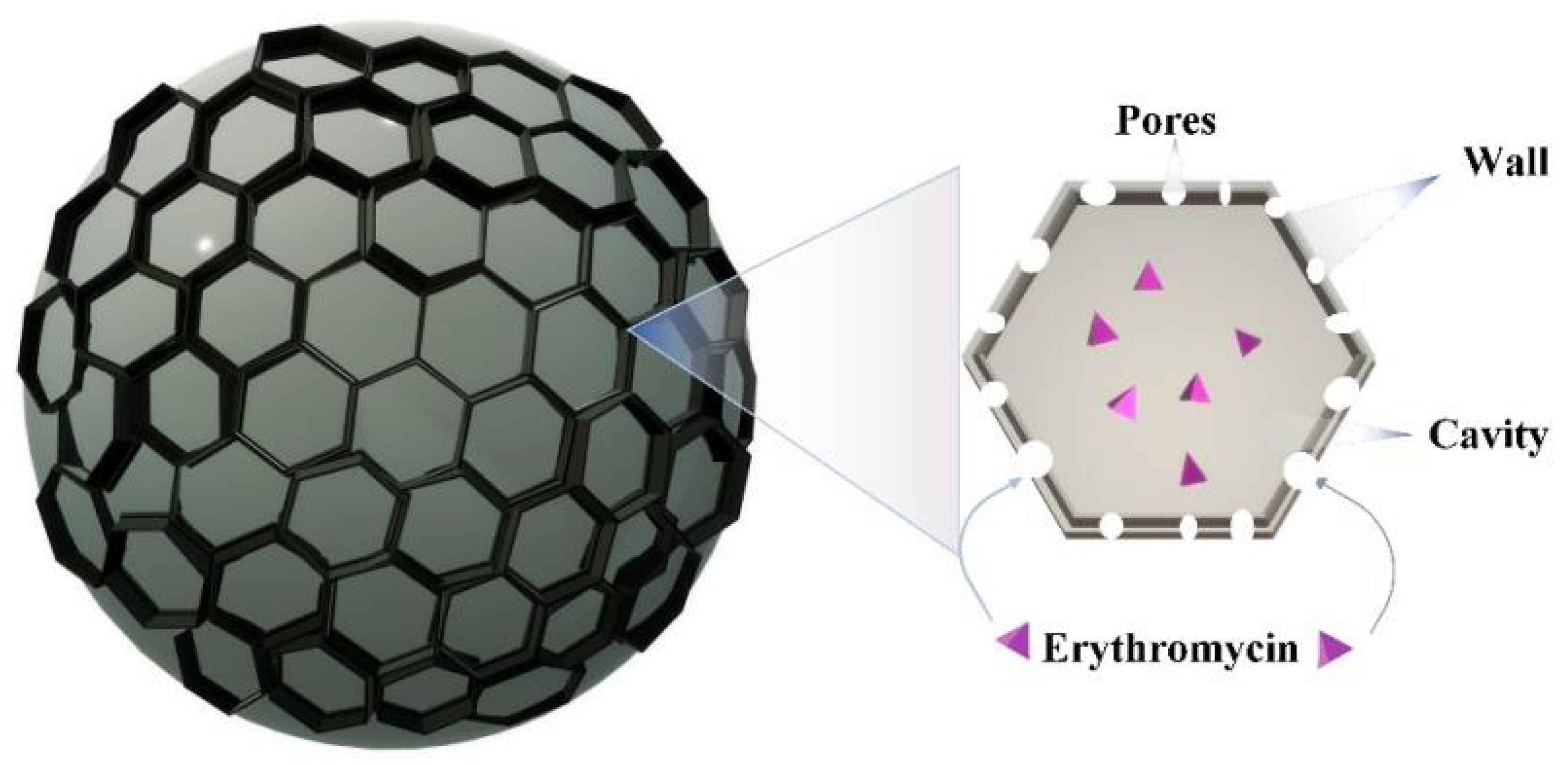
3. Polymer Micro(Nano)Spheres
3.1. Microencapsulation in Synthetic Polymers
3.2. Microencapsulation into Polymers of Natural Origin
4. ERY–Cyclodextrin Complexes
5. Gels
5.1. Gels Encapsulating Neat ERY
5.2. Gels Incorporating ERY and Co-Bioactive Ingredients
5.3. Gels Incorporating ERY Vesicles
6. Fibres
7. ERY Loading into Mesoporous Oxides
8. Metallic-Based Nanoparticles as ERY Carriers/Cofactors
9. Overview of Current Progress in ERY Formulations
10. Quantification of ERY
10.1. Ultraviolet Spectroscopy
10.2. High-Pressure Liquid Chromatography
10.3. Thermogravimetry
10.4. Near Infrared Spectroscopy
10.5. Microbiological Assay
11. Conclusions
Funding
Informed Consent Statement
Conflicts of Interest
References
- Bunch, R.L.; Mcguire, J.M. Erythromycin, Its Salts, and Method of Preparation. U.S. Patent 2,653,899, 29 September 1952. [Google Scholar]
- Oliynyk, M.; Samborskyy, M.; Lester, J.B.; Mironenko, T.; Scott, N.; Dickens, S.; Haydock, S.F.; Leadlay, P.F. Complete Genome Sequence of the Erythromycin-Producing Bacterium Saccharopolyspora Erythraea NRRL23338. Nat. Biotechnol. 2007, 25, 447–453. [Google Scholar] [CrossRef]
- Dehouck, P.; van der Heyden, Y.; Smeyers-Verbeke, J.; Massart, D.L.; Marini, R.D.; Chiap, P.; Hubert, P.; Crommen, J.; van de Wauw, W.; de Beer, J.; et al. Interlaboratory Study of a Liquid Chromatography Method for Erythromycin: Determination of Uncertainty. J. Chromatogr. A 2003, 1010, 63–74. [Google Scholar] [CrossRef]
- Heilman, F.R.; Herrell, W.E.; Wellman, W.E.; Geraci, J.E. Some Laboratory and Clinical Observations on a New Antibiotic, Erythromycin (Ilotycin). Proc. Staff Meet. Mayo Clin. 1952, 27, 285–304. [Google Scholar] [PubMed]
- Pendela, M.; van den Bossche, L.; Hoogmartens, J.; van Schepdael, A.; Adams, E. Combination of a Liquid Chromatography–Ultraviolet Method with a Non-Volatile Eluent, Peak Trapping and a Liquid Chromatography–Mass Spectrometry Method with a Volatile Eluent to Characterise Erythromycin Related Substances. J. Chromatogr. A 2008, 1180, 108–121. [Google Scholar] [CrossRef]
- Hoyt, J.C.; Robbins, R.A. Macrolide Antibiotics and Pulmonary Inflammation. FEMS Microbiol. Lett. 2001, 205, 1–7. [Google Scholar] [CrossRef]
- Block, S.; Hedrick, J.; Hammerschlag, M.R.; Cassell, G.H.; Craft, C.J. Mycoplasma Pneumoniae and Chlamydia Pneumoniae in Pediatric Community-Acquired Pneumonia. Pediatr. Infect. Dis. J. 1995, 14, 471–477. [Google Scholar] [CrossRef]
- Horwitz, M.A.; Silverstein, S.C. Intracellular Multiplication of Legionnaires’ Disease Bacteria (Legionella Pneumophila) in Human Monocytes Is Reversibly Inhibited by Erythromycin and Rifampin. J. Clin. Investig. 1983, 71, 15–26. [Google Scholar] [CrossRef]
- Kneen, R.; Pham, N.G.; Solomon, T.; Tran, T.M.; Nguyen, T.T.; Tran, B.L.; Wain, J.; Day, N.P.; Tran, T.H.; Parry, C.M.; et al. Penicillin vs. Erythromycin in the Treatment of Diphtheria. Clin. Infect. Dis. 1998, 27, 845–850. [Google Scholar] [CrossRef] [Green Version]
- Rolain, J.M.; Brouqui, P.; Koehler, J.E.; Maguina, C.; Dolan, M.J.; Raoult, D. Recommendations for Treatment of Human Infections Caused by Bartonella Species. Antimicrob. Agents Chemother. 2004, 48, 1921–1933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salazar-Lindo, E.; Sack, R.B.; Chea-Woo, E.; Kay, B.A.; Piscoya, Z.A.; Leon-Barua, R.; Yi, A. Early Treatment with Erythromycin of Campylobacter Jejuni-Associated Dysentery in Children. J. Pediatr. 1986, 109, 355–360. [Google Scholar] [CrossRef]
- Slaney, L.; Chubb, H.; Ronald, A.; Brunham, R. In-Vitro Activity of Azithromycin, Erythromycin, Ciprofloxacin and Norfloxacin against Neisseria Gonorrhoeae, Haemophilus Ducreyi, and Chlamydia Trachomatis. J. Antimicrob. Chemother. 1990, 25, 1–5. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, Z.; Luan, Y.; Li, Y.; Liu, X.; Peng, X.; Octavia, S.; Payne, M.; Lan, R. Genomic Epidemiology of Erythromycin-Resistant Bordetella Pertussis in China. Emerg. Microbes Infect. 2019, 8, 461–470. [Google Scholar] [CrossRef] [Green Version]
- WEBSTER, G.F.; McGINLEY, K.J.; LEYDEN, J.J. Inhibition of Lipase Production in Propionibacterium Acnes by Sub-Minimal-Inhibitory Concentrations of Tetracycline and Erythromycin. Br. J. Dermatol. 1981, 104, 453–457. [Google Scholar] [CrossRef]
- Tamura, H.; Maekawa, T.; Domon, H.; Hiyoshi, T.; Hirayama, S.; Isono, T.; Sasagawa, K.; Yonezawa, D.; Takahashi, N.; Oda, M.; et al. Effects of Erythromycin on Osteoclasts and Bone Resorption via DEL-1 Induction in Mice. Antibiotics 2021, 10, 312. [Google Scholar] [CrossRef] [PubMed]
- Nandi, S.K.; Mukherjee, P.; Roy, S.; Kundu, B.; De, D.K.; Basu, D. Local Antibiotic Delivery Systems for the Treatment of Osteomyelitis—A Review. Mater. Sci. Eng. C 2009, 29, 2478–2485. [Google Scholar] [CrossRef]
- Weber, F.H.; Richards, R.D.; McCallum, R.W. Erythromycin: A Motilin Agonist and Gastrointestinal Prokinetic Agent. Am. J. Gastroenterol. 1993, 88, 485–490. [Google Scholar]
- Sanger, G.J.; Wang, Y.; Hobson, A.; Broad, J. Motilin: Towards a New Understanding of the Gastrointestinal Neuropharmacology and Therapeutic Use of Motilin Receptor Agonists. Br. J. Pharmacol. 2013, 170, 1323–1332. [Google Scholar] [CrossRef]
- Yang, F.; Wu, S.-G.; Pan, Y.-F.; Song, F.-L.; Tao, L. Preparation and Characteristics of Erythromycin Microspheres for Lung Targeting. Drug Dev. Ind. Pharm. 2009, 35, 639–645. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, J.; Zhang, M.; Dang, L. Solubility of Erythromycin A Dihydrate in Different Pure Solvents and Acetone + Water Binary Mixtures between 293 K and 323 K. J. Chem. Eng. Data 2006, 51, 1062–1065. [Google Scholar] [CrossRef]
- Carter, B.L.; Woodhead, J.C.; Cole, K.J.; Milavetz, G. Gastrointestinal Side Effects with Erythromycin Preparations. Drug Intell. Clin. Pharm. 1987, 21, 734–738. [Google Scholar] [CrossRef]
- Baguley, D.; Lim, E.; Bevan, A.; Pallet, A.; Faust, S.N. Prescribing for Children—Taste and Palatability Affect Adherence to Antibiotics: A Review. Arch. Dis. Child. 2012, 97, 293–297. [Google Scholar] [CrossRef]
- Kanfer, I.; Skinner, M.F.; Walker, R.B. Analysis of Macrolide Antibiotics. J. Chromatogr. A 1998, 812, 255–286. [Google Scholar] [CrossRef]
- Jadhav, S.M.; Morey, P.; Karpe, M.M.; Kadam, V. Novel Vesicular System: An Overview. J. Appl. Pharm. Sci. 2012, 2, 193–202. [Google Scholar]
- Amarandi, R.-M.; Ibanescu, A.; Carasevici, E.; Marin, L.; Dragoi, B. Liposomal-Based Formulations: A Path from Basic Research to Temozolomide Delivery Inside Glioblastoma Tissue. Pharmaceutics 2022, 14, 308. [Google Scholar] [CrossRef] [PubMed]
- Briuglia, M.-L.; Rotella, C.; McFarlane, A.; Lamprou, D.A. Influence of Cholesterol on Liposome Stability and on in Vitro Drug Release. Drug Deliv. Transl. Res. 2015, 5, 231–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sułkowski, W.W.; Pentak, D.; Nowak, K.; Sułkowska, A. The Influence of Temperature, Cholesterol Content and PH on Liposome Stability. J. Mol. Struct. 2005, 744–747, 737–747. [Google Scholar] [CrossRef]
- Vemuri, S.; Rhodes, C.T. Preparation and Characterization of Liposomes as Therapeutic Delivery Systems: A Review. Pharm. Acta Helv. 1995, 70, 95–111. [Google Scholar] [CrossRef]
- Allen, T.M.; Cullis, P.R. Liposomal Drug Delivery Systems: From Concept to Clinical Applications. Adv. Drug Deliv. Rev. 2013, 65, 36–48. [Google Scholar] [CrossRef]
- Bangham, A.D.; Standish, M.M.; Watkins, J.C. Diffusion of Univalent Ions across the Lamellae of Swollen Phospholipids. J. Mol. Biol. 1965, 13, 238–252. [Google Scholar] [CrossRef]
- Rajabi, O.; Layegh, P.; Hashemzadeh, S.; Khoddami, M. Topical Liposomal Azithromycin in the Treatment of Acute Cutaneous Leishmaniasis. Dermatol. Ther. 2016, 29, 358–363. [Google Scholar] [CrossRef]
- Abtahi-Naeini, B.; Hadian, S.; Sokhanvari, F.; Hariri, A.; Varshosaz, J.; Shahmoradi, Z.; Feizi, A.; Khorvash, F.; Hakamifard, A. Effect of Adjunctive Topical Liposomal Azithromycin on Systemic Azithromycin on Old World Cutaneous Leishmaniasis: A Pilot Clinical Study. Iran. J. Pharm. Res. 2021, 20, 383–389. [Google Scholar] [CrossRef]
- Stuhne-Sekalec, L.; Stanacev, N.Z.; Djokic, S. Liposomes as Carriers of Macrolides: Preferential Association of Erythromycin A and Azithromycin with Liposomes of Phosphatidylglycerol Containing Unsaturated Fatty Acid(s). J. Microencapsul. 1991, 8, 171–183. [Google Scholar] [CrossRef]
- Mugabe, C.; Azghani, A.O.; Omri, A. Preparation and Characterization of Dehydration–Rehydration Vesicles Loaded with Aminoglycoside and Macrolide Antibiotics. Int. J. Pharm. 2006, 307, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Mugabe, C.; Azghani, A.O.; Omri, A. Liposome-Mediated Gentamicin Delivery: Development and Activity against Resistant Strains of Pseudomonas Aeruginosa Isolated from Cystic Fibrosis Patients. J. Antimicrob. Chemother. 2005, 55, 269–271. [Google Scholar] [CrossRef] [Green Version]
- Shah, S.P.; Misra, A. Liposomal Amikacin Dry Powder Inhaler: Effect of Fines on in Vitro Performance. AAPS Pharm. Sci. Tech. 2004, 5, 107–113. [Google Scholar] [CrossRef] [Green Version]
- Gbian, D.L.; Omri, A. The Impact of an Efflux Pump Inhibitor on the Activity of Free and Liposomal Antibiotics against Pseudomonas Aeruginosa. Pharmaceutics 2021, 13, 577. [Google Scholar] [CrossRef]
- de Meyer, F.; Smit, B. Effect of Cholesterol on the Structure of a Phospholipid Bilayer. Proc. Natl. Acad. Sci. USA 2009, 106, 3654–3658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, H.; Tao, H.; Wan, J.; Lee, K.Y.; Zheng, Z.; Leung, S.S.Y. Preparation of Drug-Loaded Liposomes with Multi-Inlet Vortex Mixers. Pharmaceutics 2022, 14, 1223. [Google Scholar] [CrossRef] [PubMed]
- Akbarzadeh, A.; Rezaei-Sadabady, R.; Davaran, S.; Joo, S.W.; Zarghami, N.; Hanifehpour, Y.; Samiei, M.; Kouhi, M.; Nejati-Koshki, K. Liposome: Classification, Preparation, and Applications. Nanoscale Res. Lett. 2013, 8, 102. [Google Scholar] [CrossRef] [Green Version]
- Jayaraman, S.C.; Ramachandran, C.; Weiner, N. Topical Delivery of Erythromycin from Various Formulations: An in Vivo Hairless Mouse Study. J. Pharm. Sci. 1996, 85, 1082–1084. [Google Scholar] [CrossRef]
- Majack, T.J.; Rust, M.B.; Massee, K.C.; Kissil, G.W.; Hardy, R.W.; Peterson, M.E. Bioencapsulation of Erythromycin Using Adult Brine Shrimp, Artemia Franciscana (Latreille). J. Fish Dis. 2000, 23, 71–76. [Google Scholar] [CrossRef]
- Hilițanu, L.N.; Mititelu-Tarțău, L.; Popa, G.E.; Buca, B.R.; Pavel, L.L.; Pelin, A.-M.; Meca, A.-D.; Bogdan, M.; Pricop, D.A. The Analysis of Chitosan-Coated Nanovesicles Containing Erythromycin—Characterization and Biocompatibility in Mice. Antibiotics 2021, 10, 1471. [Google Scholar] [CrossRef]
- Kulkarni, P.; Yadav, J.; Vaidya, K. Liposomes: A Novel Drug Delivery System. Int. J. Curr. Pharm. Rev. Res. 2011, 3, 10–18. [Google Scholar]
- Filipczak, N.; Pan, J.; Yalamarty, S.S.K.; Torchilin, V.P. Recent Advancements in Liposome Technology. Adv. Drug Deliv. Rev. 2020, 156, 4–22. [Google Scholar] [CrossRef]
- Jeong, S.; Lee, J.; Im, B.N.; Park, H.; Na, K. Combined Photodynamic and Antibiotic Therapy for Skin Disorder via Lipase-Sensitive Liposomes with Enhanced Antimicrobial Performance. Biomaterials 2017, 141, 243–250. [Google Scholar] [CrossRef]
- Park, H.; Park, H.; Na, K. Dual Propionibacterium Acnes Therapy Using Skin Penetration-Enhanced Liposomes Loaded with a Photosensitizer and an Antibiotic. J. Porphyr. Phthalocyanines 2015, 19, 956–966. [Google Scholar] [CrossRef]
- Akbarzadeh, A.; Samiei, M.; Davaran, S. Magnetic Nanoparticles: Preparation, Physical Properties, and Applications in Biomedicine. Nanoscale Res. Lett. 2012, 7, 144. [Google Scholar] [CrossRef] [Green Version]
- Salah, B.M.; Rady, M.; Abdel-Halim, M.; Fahmy, H.M.; El-Din, N.S.; Gaber, M.H. Alternating Magnetic Field Induced Membrane Permeability in Erythromycin Magneto-Liposomes A Potential Solution to Antibiotic Resistance. Biophysics 2021, 66, 264–272. [Google Scholar] [CrossRef]
- Estelrich, J.; Escribano, E.; Queralt, J.; Busquets, M. Iron Oxide Nanoparticles for Magnetically-Guided and Magnetically-Responsive Drug Delivery. Int. J. Mol. Sci. 2015, 16, 8070–8101. [Google Scholar] [CrossRef] [Green Version]
- Fifere, N.; Airinei, A.; Timpu, D.; Rotaru, A.; Sacarescu, L.; Ursu, L. New Insights into Structural and Magnetic Properties of Ce Doped ZnO Nanoparticles. J. Alloys Compd. 2018, 757, 60–69. [Google Scholar] [CrossRef]
- Montenez, J.-P.; van Bambeke, F.; Piret, J.; Brasseur, R.; Tulkens, P.M.; Mingeot-Leclercq, M.-P. Interactions of Macrolide Antibiotics (Erythromycin A, Roxithromycin, Erythromycylamine [Dirithromycin], and Azithromycin) with Phospholipids: Computer-Aided Conformational Analysis and Studies on Acellular and Cell Culture Models. Toxicol. Appl. Pharmacol. 1999, 156, 129–140. [Google Scholar] [CrossRef]
- Abdulbaqi, I.M.; Darwis, Y.; Abdul Karim Khan, N.; Abou Assi, R.; Ali Khan, A. Ethosomal Nanocarriers: The Impact of Constituents and Formulation Techniques on Ethosomal Properties, in Vivo Studies, and Clinical Trials. Int. J. Nanomed. 2016, 2016, 2279–2304. [Google Scholar] [CrossRef] [Green Version]
- Touitou, E.; Natsheh, H. Topical Administration of Drugs Incorporated in Carriers Containing Phospholipid Soft Vesicles for the Treatment of Skin Medical Conditions. Pharmaceutics 2021, 13, 2129. [Google Scholar] [CrossRef]
- Natsheh, H.; Touitou, E. Phospholipid Vesicles for Dermal/Transdermal and Nasal Administration of Active Molecules: The Effect of Surfactants and Alcohols on the Fluidity of Their Lipid Bilayers and Penetration Enhancement Properties. Molecules 2020, 25, 2959. [Google Scholar] [CrossRef]
- Godin, B.; Touitou, E. Erythromycin Ethosomal Systems: Physicochemical Characterization and Enhanced Antibacterial Activity. Curr. Drug Deliv. 2005, 2, 269–275. [Google Scholar] [CrossRef]
- Godin, B.; Touitou, E.; Rubinstein, E.; Athamna, A.; Athamna, M. A New Approach for Treatment of Deep Skin Infections by an Ethosomal Antibiotic Preparation: An in Vivo Study. J. Antimicrob. Chemother. 2005, 55, 989–994. [Google Scholar] [CrossRef] [Green Version]
- Kazi, K.M.; Mandal, A.S.; Biswas, N.; Guha, A.; Chatterjee, S.; Behera, M.; Kuotsu, K. Niosome: A Future of Targeted Drug Delivery Systems. J. Adv. Pharm. Technol. Res. 2010, 1, 374–380. [Google Scholar] [CrossRef] [Green Version]
- Vyas, J.; Puja, V.; Sawant, K. Formulation and Evaluation of Topical Niosomal Gel of Erythromycin. Int. J. Pharm. Pharm. Sci. 2011, 3, 123–126. [Google Scholar]
- Vyas, J.; Vishal, G.; Tejas, G.; Vishal, C.; Umesh, U. Formulation and Characterization of Topical Gel of Erythromycin Entrapped into Niosomes. Int. J. Pharmtech. Res. 2011, 3, 1714–1718. [Google Scholar]
- Mohammadi, S.; Farajzadeh, S.; Pardakhty, A.; Khalili Meybodi, M.; Mohebbi, A.; Yousefian, M.R.; Aflatoonian, M. A Survey to Compare the Efficacy of Niosomal Erythromycin Alone versus Combination of Erythromycin and Zinc Acetate in the Treatment of Acne Vulgaris. J. Kerman Univ. Med. Sci. 2017, 24, 420–430. [Google Scholar]
- Liu, T.; Wu, T.; Liu, H.; Ke, B.; Huang, H.; Jiang, Z.; Xie, M. Ultraviolet-Crosslinked Hydrogel Sustained-Release Hydrophobic Antibiotics with Long-Term Antibacterial Activity and Limited Cytotoxicity. J. Appl. Polym. Sci. 2014, 131, 40438. [Google Scholar] [CrossRef]
- Basak, R.; Bandyopadhyay, R. Encapsulation of Hydrophobic Drugs in Pluronic F127 Micelles: Effects of Drug Hydrophobicity, Solution Temperature, and PH. Langmuir 2013, 29, 4350–4356. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Sun, Y.; Wang, Q. Encapsulation and in Vitro Release of Erythromycin Using Biopolymer Micelle. Cell Mol. Biol. 2015, 61, 60–64. [Google Scholar]
- Song, H.; Yin, Y.; Peng, J.; Du, Z.; Bao, W. Preparation, Characteristics, and Controlled Release Efficiency of the Novel PCL-PEG/EM Rod Micelles. J. Nanomater. 2021, 2021, 8132868. [Google Scholar] [CrossRef]
- Barriga, H.M.G.; Holme, M.N.; Stevens, M.M. Cubosomes: The Next Generation of Smart Lipid Nanoparticles? Angew. Chem. Int. Ed. 2019, 58, 2958–2978. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.; Jain, P.; Jain, S.; Jain, R.; Bhargava, S.; Jain, A. Topical Delivery of Erythromycin Through Cubosomes for Acne. Pharm. Nanotechnol. 2018, 6, 38–47. [Google Scholar] [CrossRef]
- Jaspart, S.; Piel, G.; Delattre, L.; Evrard, B. Solid Lipid Microparticles: Formulation, Preparation, Characterisation, Drug Release and Applications. Expert Opin. Drug Deliv. 2005, 2, 75–87. [Google Scholar] [CrossRef]
- Joachim, C. To Be Nano or Not to Be Nano? Nat. Mater. 2005, 4, 107–109. [Google Scholar] [CrossRef]
- Strambeanu, N.; Demetrovici, L.; Dragos, D.; Lungu, M. Nanoparticles: Definition, Classification and General Physical Properties. In Nanoparticles’ Promises and Risks: Characterization, Manipulation, and Potential Hazards to Humanity and the Environment; Lungu, M., Neculae, A., Bunoiu, M., Biris, C., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 3–8. ISBN 978-3-319-11728-7. [Google Scholar]
- Desai, P.P.; Date, A.A.; Patravale, V.B. Overcoming Poor Oral Bioavailability Using Nanoparticle Formulations—Opportunities and Limitations. Drug Discov. Today Technol. 2012, 9, e87–e95. [Google Scholar] [CrossRef]
- Mehnert, W.; Mäder, K. Solid Lipid Nanoparticles: Production, Characterization and Applications. Adv. Drug Deliv. Rev. 2012, 64, 83–101. [Google Scholar] [CrossRef]
- Scioli Montoto, S.; Muraca, G.; Ruiz, M.E. Solid Lipid Nanoparticles for Drug Delivery: Pharmacological and Biopharmaceutical Aspects. Front. Mol. Biosci. 2020, 7, 587997. [Google Scholar] [CrossRef]
- Sahu, A.K.; Kumar, T.; Jain, V. Formulation Optimization of Erythromycin Solid Lipid Nanocarrier Using Response Surface Methodology. Biomed. Res. Int. 2014, 2014, 689391. [Google Scholar] [CrossRef]
- Dhillon, P.; Mirza, M.A.; Anwer, M.K.; Alshetaili, A.S.; Alshahrani, S.M.; Iqbal, Z. Development and Optimization of Erythromycin-Loaded Lipid-Based Gel by Taguchi Design: In Vitro Characterization and Antimicrobial Evaluation. Brazilian J. Pharm. Sci. 2019, 55, e17395. [Google Scholar] [CrossRef] [Green Version]
- Pignatello, R.; Fuochi, V.; Petronio Petronio, G.; Greco, A.; Furneri, P.M. Formulation and Characterization of Erythromycin–Loaded Solid Lipid Nanoparticles. Biointerface Res. Appl. Chem. 2017, 7, 2145–2150. [Google Scholar]
- Momoh, M.A.; Ossai, E.C.; Chidozie, O.E.; Precscila, O.O.; Kenechukwu, F.C.; Ofokansi, K.O.; Attama, A.A.; Olobayo, K.O. A New Lipid-Based Oral Delivery System of Erythromycin for Prolong Sustain Release Activity. Mater. Sci. Eng. C 2019, 97, 245–253. [Google Scholar] [CrossRef]
- Nzekwe, I.T.; Okere, A.C.; Okoye, I.E.; Ekere, K.E.; Ezenwa, A.A.; Agubata, C.O. Bioassay-Guided Optimization of Lipid-Based Erythromycin Microparticles. Trop. J. Pharm. Res. 2020, 19, 1351–1358. [Google Scholar] [CrossRef]
- Alvarez-Elcoro, S.; Enzler, M.J. The Macrolides: Erythromycin, Clarithromycin, and Azithromycin. Mayo Clin. Proc. 1999, 74, 613–634. [Google Scholar] [CrossRef]
- Choudhury, N.; Meghwal, M.; Das, K. Microencapsulation: An Overview on Concepts, Methods, Properties and Applications in Foods. Food Front. 2021, 2, 426–442. [Google Scholar] [CrossRef]
- Singh, M.N.; Hemant, K.S.Y.; Ram, M.; Shivakumar, H.G. Microencapsulation: A Promising Technique for Controlled Drug Delivery. Res. Pharm. Sci. 2010, 5, 65–77. [Google Scholar]
- Jurić, S.; Stracenski, K.; Król-Kilińska, Ż.; Zutic, I.; Fabek, S.; Edyta, Đ.; Topolovec-Pintaric, S.; Vinceković, M. The Enhancement of Plant Secondary Metabolites Content in Lactuca Sativa L. by Encapsulated Bioactive Agents. Sci. Rep. 2020, 10, 3737. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, A.; Yadav, T.; Sharma, S.; Nayak, A.; Akanksha Kumari, A.; Mishra, N. Polymers in Drug Delivery. J. Biosci. Med. 2016, 4, 69–84. [Google Scholar] [CrossRef] [Green Version]
- Park, S.-J.; Kim, S.-H. Preparation and Characterization of Biodegradable Poly(l-Lactide)/Poly(Ethylene Glycol) Microcapsules Containing Erythromycin by Emulsion Solvent Evaporation Technique. J. Colloid Interface Sci. 2004, 271, 336–341. [Google Scholar] [CrossRef]
- Cibotaru, S.; Sandu, A.-I.; Belei, D.; Marin, L. Water Soluble PEGylated Phenothiazines as Valuable Building Blocks for Bio-Materials. Mater. Sci. Eng. C 2020, 116, 111216. [Google Scholar] [CrossRef]
- Park, S.J.; Kim, S.H.; Lee, J.R.; Lee, H.B.; Hong, S.K. Preparation and characterization of biodegradable poly({epsilon}-caprolactone) microcapsules containing erythromycin by emulsion solvent evaporation technique. Polymer 2002, 26, 326–334. [Google Scholar]
- Park, S.-J.; Kim, S.-H.; Lee, J.-R.; Lee, H.B.; Hong, S.-K. Preparation and Characterization of Biodegradable Poly(ε-Caprolactone)/Poly(Ethylene Oxide) Microcapsules Containing Erythromycin. Polymer 2003, 27, 449–457. [Google Scholar]
- Park, S.-J.; Kim, K.-S.; Kim, S.-H. Effect of Poly(Ethylene Oxide) on the Release Behaviors of Poly(ɛ-Caprolactone) Microcapsules Containing Erythromycin. Colloids Surf B Biointerfaces 2005, 43, 238–244. [Google Scholar] [CrossRef]
- Grizić, D.; Lamprecht, A. Predictability of Drug Encapsulation and Release from Propylene Carbonate/PLGA Microparticles. Int. J. Pharm. 2020, 586, 119601. [Google Scholar] [CrossRef]
- Bennabi, L.; Abedlmalek, I.; Ammari, A.; Sediri, K.; Bennabi, F.; Guemra, K. Synthesis and Characterization of Erythromycin Loaded PLGA and PCL Microspheres: Antimicrobial Application. Microbial. Biosyst. 2022, 6, 43–52. [Google Scholar] [CrossRef]
- Zgoulli, S.; Grek, V.; Barre, G.; Goffinet, G.; Thonart, P.H.; Zinner, S. Microencapsulation of Erythromycin and Clarithromycin Using a Spray-Drying Technique. J. Microencapsul. 1999, 16, 565–571. [Google Scholar] [CrossRef]
- Kurmi, B.D.; Kayat, J.; Gajbhiye, V.; Tekade, R.K.; Jain, N.K. Micro- and Nanocarrier-Mediated Lung Targeting. Expert Opin. Drug Deliv. 2010, 7, 781–794. [Google Scholar] [CrossRef]
- Woodruff, M.A.; Hutmacher, D.W. The Return of a Forgotten Polymer—Polycaprolactone in the 21st Century. Prog. Polym. Sci. 2010, 35, 1217–1256. [Google Scholar] [CrossRef]
- Li, T.; Wen, J.; Jiang, W.; Gong, M.; Yang, Z.; Xiao, H. [Antimicrobial effects of erythromycin microspheres against Mycoplasma Pneumoniae in rats]. J. South. Med. Univ. 2008, 28, 1875–1877. [Google Scholar]
- Fathollahipour, S.; Abouei Mehrizi, A.; Ghaee, A.; Koosha, M. Electrospinning of PVA/Chitosan Nanocomposite Nanofibers Containing Gelatin Nanoparticles as a Dual Drug Delivery System. J. Biomed. Mater. Res. A 2015, 103, 3852–3862. [Google Scholar] [CrossRef]
- al Qushawi, A.A.H.; Al-Ruaby, K.J. Antibacterial Activity Study of Erythromycin-Loaded Chitosan Nanoparticles and Chitosan Nanoparticle against Identified MRSA and MSSA Bacteria Isolated from Mastitis Cow Milk. Eurasian J. Biosci. 2020, 14, 7397–7405. [Google Scholar]
- Pelin, I.M.; Suflet, D.M. Mucoadhesive Buccal Drug Delivery Systems Containing Polysaccharides. Cellul. Chem. Technol. 2020, 54, 889–902. [Google Scholar] [CrossRef]
- Doostan, M.; Maleki, H.; Doostan, M.; Khoshnevisan, K.; Faridi-Majidi, R.; Arkan, E. Effective Antibacterial Electrospun Cellulose Acetate Nanofibrous Patches Containing Chitosan/Erythromycin Nanoparticles. Int. J. Biol. Macromol. 2021, 168, 464–473. [Google Scholar] [CrossRef]
- Han, K.; Sathiyaseelan, A.; Saravanakumar, K.; Park, S.; Shin, S.; Choi, H.b.; Naveen, K.V.; Wang, M.-H. Biomimetic Hydroxyapatite-Chitosan Nanoparticles Deliver the Erythromycin for Improved Antibacterial Activity. J. Drug Deliv. Sci. Technol. 2022, 72, 103374. [Google Scholar] [CrossRef]
- Zhang, H.; Wu, H.; Fan, L.; Li, F.; Gu, C.; Jia, M. Preparation and Characteristics of PH-Sensitive Derivated Dextran Hydrogel Nanoparticles. Polym. Compos. 2009, 30, 1243–1250. [Google Scholar] [CrossRef]
- Dyab, A.K.F.; Mohamed, M.A.; Meligi, N.M.; Mohamed, S.K. Encapsulation of Erythromycin and Bacitracin Antibiotics into Natural Sporopollenin Microcapsules: Antibacterial, Cytotoxicity, in Vitro and in Vivo Release Studies for Enhanced Bioavailability. RSC Adv. 2018, 8, 33432–33444. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Bai, T.; Chi, Q.; Wang, Z.; Xu, S.; Liu, Q.; Wang, Q. How to Make a Fast, Efficient Bubble-Driven Micromotor: A Mechanical View. Micromachines 2017, 8, 267. [Google Scholar] [CrossRef]
- Yang, S.; Ren, J.; Wang, H. Injectable Micromotor@Hydrogel System for Antibacterial Therapy. Chem.—A Eur. J. 2022, 28, e202103867. [Google Scholar] [CrossRef]
- Sivakumar, K.; Parinamachivayam, G. Review on β-Cyclodextrin Inclusion Complex Based Chemosensors for Heavy Metals. J. Incl. Phenom. Macrocycl. Chem. 2022, 102, 603–618. [Google Scholar] [CrossRef]
- Song, W.; Yu, X.; Wang, S.; Blasier, R.; Markel, D.C.; Mao, G.; Shi, T.; Ren, W. Cyclodextrin-Erythromycin Complexes as a Drug Delivery Device for Orthopedic Application. Int. J. Nanomed. 2011, 6, 3173–3186. [Google Scholar] [CrossRef]
- Thanh Ha, P.T.; van Schepdael, A.; Roets, E.; Hoogmartens, J. Investigating the Potential of Erythromycin and Derivatives as Chiral Selector in Capillary Electrophoresis. J. Pharm. Biomed. Anal. 2004, 34, 861–870. [Google Scholar] [CrossRef]
- Marian, E.; Tünde, J.; Vicas, L.; Kacso, I.; Miclaus, M.O.; Bratu, I. Inclusion Compounds of Erythromycin with β-Cyclodextrin. Rev. De Chim. 2011, 62, 1065–1068. [Google Scholar]
- Marian, E.; Mureşan, M.; Tünde, J.; Vicas, L. Evaluation of Antimicrobial Activity of Some Types of Inclusion Complexes of Erythromycin with β-Cyclodextrin on Staphylococcus Aureus. Farmacia 2013, 61, 518–525. [Google Scholar]
- Li, M.; Neoh, K.G.; Xu, L.; Yuan, L.; Leong, D.T.; Kang, E.-T.; Chua, K.L.; Hsu, L.Y. Sugar-Grafted Cyclodextrin Nanocarrier as a “Trojan Horse” for Potentiating Antibiotic Activity. Pharm. Res. 2016, 33, 1161–1174. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Kang, E.-T.; Chua, K.L.; Neoh, K.G. Sugar-Powered Nanoantimicrobials for Combating Bacterial Biofilms. Biomater. Sci. 2019, 7, 2961–2974. [Google Scholar] [CrossRef] [PubMed]
- Ochiuz, L.; CRETEANU, A.; HORTOLOMEI, M.; PEPTU, C.; Stoleriu, I.; POPA, M.; Peptu, C. Development Study of New Topical Formulations with Erythromycin. Rom. J. Pharm. Pract. 2017, 10, 109–115. [Google Scholar] [CrossRef]
- Yamamura, H.; Hagiwara, T.; Hayashi, Y.; Osawa, K.; Kato, H.; Katsu, T.; Masuda, K.; Sumino, A.; Yamashita, H.; Jinno, R.; et al. Antibacterial Activity of Membrane-Permeabilizing Bactericidal Cyclodextrin Derivatives. ACS Omega 2021, 6, 31831–31842. [Google Scholar] [CrossRef]
- Cyphert, E.; Wallat, J.; Pokorski, J.; von Recum, H. Erythromycin Modification That Improves Its Acidic Stability While Optimizing It for Local Drug Delivery. Antibiotics 2017, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Dreno, B. Topical Antibacterial Therapy for Acne Vulgaris. Drugs 2004, 64, 2389–2397. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration Approved Drug Products with Therapeutic Equivalence Evaluations|Orange Book. Available online: https://www.fda.gov/drugs/drug-approvals-and-databases/approved-drug-products-therapeutic-equivalence-evaluations-orange-book (accessed on 26 July 2022).
- Broniarczyk-Dyla, G.; Arkuszewska, C. [Local Treatment of Acne Vulgaris with Erythromycin]. Dermatol. Monatsschr. 1989, 175, 40–43. [Google Scholar]
- Aburideh, H.; Tigrine, Z.; Aoudjit, L.; Belgroun, Z.; Redjimi, K.; Tassalit, D. Development of acid modified cellulose acetate membranes for salt water treatment. Cellul. Chem. Technol. 2021, 55, 1153–1161. [Google Scholar] [CrossRef]
- Burkhart, C.N.; Specht, K.; Neckers, D. Synergistic Activity of Benzoyl Peroxide and Erythromycin. Skin Pharmacol. Physiol. 2000, 13, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Cunha, J.P. BENZAMYCIN. Available online: https://www.rxlist.com/benzamycin-drug.htm (accessed on 26 July 2022).
- Vermeulen, B.; Remon, J.P.; Nelis, H. The Formulation and Stability of Erythromycin–Benzoyl Peroxide in a Topical Gel. Int. J. Pharm. 1999, 178, 137–141. [Google Scholar] [CrossRef]
- Vandenbossche, G.M.R.; Vanhaecke, E.; de Muynck, C.; Remon, J.P. Stability of Topical Erythromycin Formulations. Int. J. Pharm. 1991, 67, 195–199. [Google Scholar] [CrossRef]
- Wróblewska, M.; Winnicka, K. The Effect of Cationic Polyamidoamine Dendrimers on Physicochemical Characteristics of Hydrogels with Erythromycin. Int. J. Mol. Sci. 2015, 16, 20277–20289. [Google Scholar] [CrossRef] [Green Version]
- Chehreghanianzabi, Y.; Barua, R.; Shi, T.; Yurgelevic, S.; Auner, G.; Markel, D.C.; Ren, W. Comparing the Release of Erythromycin and Vancomycin from Calcium Polyphosphate Hydrogel Using Different Drug Loading Methods. J. Biomed. Mater. Res. B Appl. Biomater. 2020, 108, 475–483. [Google Scholar] [CrossRef]
- Alavi, T.; Rezvanian, M.; Ahmad, N.; Mohamad, N.; Ng, S.-F. Pluronic-F127 Composite Film Loaded with Erythromycin for Wound Application: Formulation, Physicomechanical and in Vitro Evaluations. Drug Deliv. Transl. Res. 2019, 9, 508–519. [Google Scholar] [CrossRef]
- Kwon, J.S.; Kim, D.Y.; Seo, H.W.; Jeong, S.H.; Kim, J.H.; Kim, M.S. Preparation of Erythromycin-Loaded Poly(Vinylalcohol) Film and Investigation of Its Feasibility as a Transdermal Delivery Carrier. Tissue Eng. Regen. Med. 2014, 11, 211–216. [Google Scholar] [CrossRef]
- Ochiuz, L.; Hortolomei, M.; Stoleriu, I.; Bercea, M. Dermatocosmetics based on hydroxypropyl cellulose for acne treatment. Rheological and drug delivery behaviour. Cellul. Chem. Technol. 2016, 50, 569–575. [Google Scholar]
- Fathollahipour, S.; Koosha, M.; Tavakoli, J.; Maziarfar, S.; Fallah Mehrabadi, J. Erythromycin Releasing PVA/Sucrose and PVA/Honey Hydrogels as Wound Dressings with Antibacterial Activity and Enhanced Bio-Adhesion. Iran. J. Pharm. Res. 2020, 19, 448–464. [Google Scholar] [CrossRef]
- Movassaghi, S.; Nadia Sharifi, Z.; Koosha, M.; Abdollahifar, M.A.; Fathollahipour, S.; Tavakoli, J.; Abdi, S. Effect of Honey/PVA Hydrogel Loaded by Erythromycin on Full-Thickness Skin Wound Healing in Rats; Stereological Study. Galen Med. J. 2019, 8, e1362. [Google Scholar] [CrossRef]
- Tavakoli, J.; Tang, Y. Honey/PVA Hybrid Wound Dressings with Controlled Release of Antibiotics: Structural, Physico-Mechanical and in-Vitro Biomedical Studies. Mater. Sci. Eng. C 2017, 77, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Markel, D.C.; Jin, X.; Shi, T.; Ren, W. Poly(Vinyl Alcohol)/Collagen/Hydroxyapatite Hydrogel: Properties and in Vitro Cellular Response. J. Biomed. Mater. Res. A 2012, 100A, 3071–3079. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Ren, W.; Wan, C.; Esquivel, A.O.; Shi, T.; Blasier, R.; Markel, D.C. A Novel Strontium-Doped Calcium Polyphosphate/Erythromycin/Poly(Vinyl Alcohol) Composite for Bone Tissue Engineering. J. Biomed. Mater. Res. A 2011, 98A, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Perchyonok, V.; Zhang, S.; Basson, N. Insights into Functional Erythromycin/Antioxidant Containing Chitosan Hydrogels as Potential Bio-Active Restorative Materials: Structure, Function and Antimicrobial Activity. Adv. Tech. Biol. Med. 2014, 2, 10001156. [Google Scholar] [CrossRef] [Green Version]
- Sayyafan, M.S.; Ramzi, M.; Salmanpour, R. Clinical Assessment of Topical Erythromycin Gel with and without Zinc Acetate for Treating Mild-to-Moderate Acne Vulgaris. J. Dermatol. Treat. 2019, 31, 730–733. [Google Scholar] [CrossRef]
- Habbema, L.; Koopmans, B.; Menke, H.E.; Doornweerd, S.; de Boulle, K. A 4% Erythromycin and Zinc Combination (Zineryt) versus 2% Erythromycin (Eryderm) in Acne Vulgaris: A Randomized, Double-Blind Comparative Study. Br. J. Dermatol. 1989, 121, 497–502. [Google Scholar] [CrossRef]
- Marin, L.; Ailincai, D.; Mares, M.; Paslaru, E.; Cristea, M.; Nica, V.; Simionescu, B.C. Imino-Chitosan Biopolymeric Films. Obtaining, Self-Assembling, Surface and Antimicrobial Properties. Carbohydr. Polym. 2015, 117, 762–770. [Google Scholar] [CrossRef] [PubMed]
- Craciun, A.-M.; Mititelu-Tartau, L.; Gavril, G.; Marin, L. Chitosan Crosslinking with Pyridoxal 5-Phosphate Vitamer toward Biocompatible Hydrogels for in Vivo Applications. Int. J. Biol. Macromol. 2021, 193, 1734–1743. [Google Scholar] [CrossRef] [PubMed]
- Anisiei, A.; Rosca, I.; Sandu, A.-I.; Bele, A.; Cheng, X.; Marin, L. Imination of Microporous Chitosan Fibers—A Route to Biomaterials with “on Demand” Antimicrobial Activity and Biodegradation for Wound Dressings. Pharmaceutics 2022, 14, 117. [Google Scholar] [CrossRef]
- Craciun, A.M.; Morariu, S.; Marin, L. Self-Healing Chitosan Hydrogels: Preparation and Rheological Characterization. Polymers 2022, 14, 2570. [Google Scholar] [CrossRef]
- Lungu, R.; Paun, M.-A.; Peptanariu, D.; Ailincai, D.; Marin, L.; Nichita, M.-V.; Paun, V.-A.; Paun, V.-P. Biocompatible Chitosan-Based Hydrogels for Bioabsorbable Wound Dressings. Gels 2022, 8, 107. [Google Scholar] [CrossRef]
- Lardner, A. The Effects of Extracellular PH on Immune Function. J. Leukoc. Biol. 2001, 69, 522–530. [Google Scholar] [CrossRef]
- Alam, A.; Mustafa, G.; Agrawal, G.P.; Hashmi, S.; Khan, R.A.; Aba Alkhayl, F.F.; Ullah, Z.; Ali, M.S.; Elkirdasy, A.F.; Khan, S. A Microemulsion-Based Gel of Isotretinoin and Erythromycin Estolate for the Management of Acne. J. Drug Deliv. Sci. Technol. 2022, 71, 103277. [Google Scholar] [CrossRef]
- Liu, M.; Duan, X.P.; Li, Y.M.; Yang, D.P.; Long, Y.Z. Electrospun nanofibers for wound healing. Mater. Sci. Eng. C 2017, 76, 1413–1423. [Google Scholar] [CrossRef]
- Anisiei, A.; Bostanaru, A.-C.; Mares, M.; Marin, L. Imination of chitosan nanofibers in a heterogeneous system. Synthesis optimization and impact on fiber morphology. Cellul. Chem. Technol. 2021, 55, 785–793. [Google Scholar] [CrossRef]
- Wang, M.; Wang, L.; Huang, Y. Electrospun Hydroxypropyl Methyl Cellulose Phthalate (HPMCP)/Erythromycin Fibers for Targeted Release in Intestine. J. Appl. Polym. Sci. 2007, 106, 2177–2184. [Google Scholar] [CrossRef]
- Darbasizadeh, B.; Fatahi, Y.; Feyzi-barnaji, B.; Arabi, M.; Motasadizadeh, H.; Farhadnejad, H.; Moraffah, F.; Rabiee, N. Crosslinked-Polyvinyl Alcohol-Carboxymethyl Cellulose/ZnO Nanocomposite Fibrous Mats Containing Erythromycin (PVA-CMC/ZnO-EM): Fabrication, Characterization and in-Vitro Release and Anti-Bacterial Properties. Int. J. Biol. Macromol. 2019, 141, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Baghali, M.; Ziyadi, H.; Faridi-Majidi, R. Fabrication and Characterization of Core–Shell TiO2-Containing Nanofibers of PCL-Zein by Coaxial Electrospinning Method as an Erythromycin Drug Carrier. Polym. Bull. 2022, 79, 1729–1749. [Google Scholar] [CrossRef]
- Anisiei, A.; Oancea, F.; Marin, L. Electrospinning of Chitosan-Based Nanofibers: From Design to Prospective Applications. Rev. Chem. Eng. 2021. [Google Scholar] [CrossRef]
- Ren, W.; Yu, X.; Chen, L.; Shi, T.; Bou-Akl, T.; Markel, D.C. Osteoblastic Differentiation and Bactericidal Activity Are Enhanced by Erythromycin Released from PCL/PLGA-PVA Coaxial Nanofibers. J. Biomater. Appl. 2022, 37, 712–723. [Google Scholar] [CrossRef]
- Khoshbakht, S.; Asghari Sana, F.; Fathi-Azarbayjani, A.; Sharifi, Y. Fabrication and Characterization of Tretinoin-Loaded Nanofiber for Topical Skin Delivery. Biomater. Res. 2020, 24, 8. [Google Scholar] [CrossRef] [Green Version]
- Vallet-Regí, M.; Balas, F.; Arcos, D. Mesoporous Materials for Drug Delivery. Angew. Chem. Int. Ed. 2007, 46, 7548–7558. [Google Scholar] [CrossRef]
- Niculescu, V.-C. Mesoporous Silica Nanoparticles for Bio-Applications. Front Mater 2020, 7, 36. [Google Scholar] [CrossRef]
- Doadrio, J.C.; Sousa, E.M.B.; Izquierdo-Barba, I.; Doadrio, A.L.; Perez-Pariente, J.; Vallet-Regí, M. Functionalization of Mesoporous Materials with Long Alkyl Chains as a Strategy for Controlling Drug Delivery Pattern. J. Mater. Chem. 2006, 16, 462–466. [Google Scholar] [CrossRef]
- Izquierdo-Barba, I.; Martinez, Á.; Doadrio, A.L.; Pérez-Pariente, J.; Vallet-Regí, M. Release Evaluation of Drugs from Ordered Three-Dimensional Silica Structures. Eur. J. Pharm. Sci. 2005, 26, 365–373. [Google Scholar] [CrossRef]
- Pathan, S.; Solanki, P.; Patel, A. Functionalized SBA-15 for Controlled Release of Poorly Soluble Drug, Erythromycin. Microporous Mesoporous Mater. 2018, 258, 114–121. [Google Scholar] [CrossRef]
- Pourjavadi, A.; Tehrani, Z.M. Mesoporous Silica Nanoparticles (MCM-41) Coated PEGylated Chitosan as a PH-Responsive Nanocarrier for Triggered Release of Erythromycin. Int. J. Polym. Mater. Polym. Biomater. 2014, 63, 692–697. [Google Scholar] [CrossRef]
- Wang, N.; Xu, H.; Sun, S.; Guo, P.; Wang, Y.; Qian, C.; Zhong, Y.; Yang, D. Wound Therapy via a Photo-Responsively Antibacterial Nano-Graphene Quantum Dots Conjugate. J. Photochem. Photobiol. B 2020, 210, 111978. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, D.; Cao, L.; Li, Y.; Boyd, B.J.; Caruso, R.A. Mesoporous Titanium Zirconium Oxide Nanospheres with Potential for Drug Delivery Applications. ACS Appl. Mater. Interfaces 2013, 5, 10926–10932. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Du, L.; Zheng, X.; Yi, C.; Niu, W.; Yang, S.; Yin, Y. Smart Eryc@mZnO Nanoparticles with Enhanced Antibacterial Activity Under Ultraviolet and Prolonged Antibacterial Activity Without Ultraviolet. Nanosci. Nanotechnol. Lett. 2018, 10, 1572–1577. [Google Scholar] [CrossRef]
- Anderson, S.D.; Gwenin, V.; Gwenin, C.D. Magnetic Functionalized Nanoparticles for Biomedical, Drug Delivery and Imaging Applications. Nanoscale Res. Lett. 2019, 14, 188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holban, A.M.; Grumezescu, V.; Ficai, A.; Alexandru, M.M.; Grumezescu; Chifiriuc, M.C.; Iordache, F.; Andronescu, E. Highly biocompatible magnetite nanoparticles functionalized with chitosan for improving the efficiency of antibiotics. Proc. U.P.B. Sci. Bull. Ser. B 2016, 78, 48–58. [Google Scholar]
- Caamano, M.A.; Carrillo Morales, M.; José De Jesús Olivares-Trejo, J. Iron Oxide Nanoparticle Improve the Antibacterial Activity of Erythromycin. J. Bacteriol. Parasitol. 2016, 7, 2. [Google Scholar] [CrossRef]
- Safdar, M.; Ozaslan, M. Enhanced Catalytic, Antibacterial and Anti-Cancer Activities of Erythromycin Capped Gold Nanoparticles. J. Inorg. Organomet. Polym. Mater. 2022, 32, 1819–1827. [Google Scholar] [CrossRef]
- Wu, V.M.; Tang, S.; Uskoković, V. Calcium Phosphate Nanoparticles as Intrinsic Inorganic Antimicrobials: The Antibacterial Effect. ACS Appl. Mater. Interfaces 2018, 10, 34013–34028. [Google Scholar] [CrossRef]
- Hussein, K.A.M.; Khalaf, A.A. Preparation, Diagnosis and Study of the Inhibitory Effect of Copper Nanoparticles before and after Erythromycin Loading on Pseudomonas Aeruginosa. In Proceedings of the IOP Conference Series: Materials Science and Engineering, Ulaanbaatar, Mongolia, 9 January 2020. [Google Scholar]
- Princy, G.; Saravanan, P.; Gandhi, N. A Novel Approach for Studying the Combined Antimicrobial Effects of Silver Nanoparticles and Antibiotics through Agar over Layer Method and Disk Diffusion Method. Dig. J. Nanomater. Biostruct. 2011, 6, 1557–1565. [Google Scholar]
- Gad El-Rab, S.M.F.; Halawani, E.M.; Alzahrani, S.S.S. Biosynthesis of Silver Nano-Drug Using Juniperus Excelsa and Its Synergistic Antibacterial Activity against Multidrug-Resistant Bacteria for Wound Dressing Applications. 3 Biotech 2021, 11, 255. [Google Scholar] [CrossRef] [PubMed]
- Hassan, K.T.; Ibraheem, I.J.; Hassan, O.M.; Obaid, A.S.; Ali, H.H.; Salih, T.A.; Kadhim, M.S. Facile Green Synthesis of Ag/AgCl Nanoparticles Derived from Chara Algae Extract and Evaluating Their Antibacterial Activity and Synergistic Effect with Antibiotics. J. Environ. Chem. Eng. 2021, 9, 105359. [Google Scholar] [CrossRef]
- Ghosh, S.; Patil, S.; Ahire, M.; Kitture, R.; Kale, S.; Pardesi, K.; Cameotra, S.S.; Bellare, J.; Dhavale, D.D.; Jabgunde, A.; et al. Synthesis of Silver Nanoparticles Using Dioscorea Bulbifera Tuber Extract and Evaluation of Its Synergistic Potential in Combination with Antimicrobial Agents. Int. J. Nanomed. 2012, 7, 483–496. [Google Scholar] [CrossRef] [Green Version]
- Joshi, N.; Kaviratna, A.; Banerjee, R. Multi Trigger Responsive, Surface Active Lipid Nanovesicle Aerosols for Improved Efficacy of Paclitaxel in Lung Cancer. Integr. Biol. 2013, 5, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Ford, J.H.; Prescott, G.C.; Hinman, J.W.; Caron, E.L. Colorimetric Determination of Erythromycin. Anal. Chem. 1953, 25, 1195–1197. [Google Scholar] [CrossRef]
- Tepe, J.B.; John, C. Determination of Erythromycin by Ultraviolet Spectrophotometry. Anal. Chem. 1955, 27, 744–746. [Google Scholar] [CrossRef]
- Wankhade, R.; Bhalerao, S.; Panchory, H.; Pundir, A.; Pradhan, R. Analysis of Erythromycin and Benzoyl Peroxide in Combined Dosage Form by UV-Visible Spectrophotometry. Int. J. Pharm. Pharm. Sci. 2012, 4, 527–531. [Google Scholar]
- Koch, W.L. Erythromycin. In Analytical Profiles of Drug Substances and Excipients; Academic Press: London, UK, 1979; Volume 8, pp. 159–177. [Google Scholar]
- Chehreghanianzabi, Y.; Auner, G.; Shi, T.; Dietz, P.; Bou-akl, T.; Markel, D.C.; Ren, W. Impacts of Compacting Methods on the Delivery of Erythromycin and Vancomycin from Calcium Polyphosphate Hydrogel Matrices. J. Biomed. Mater. Res. B Appl. Biomater. 2022, 110, 412–421. [Google Scholar] [CrossRef]
- McPolin, O. An Introduction to HPLC for Pharmaceutical Analysis; Mourne Trading Services: Northern Ireland, UK, 2009. [Google Scholar]
- Dafale, N.A.; Semwal, U.P.; Rajput, R.K.; Singh, G.N. Selection of Appropriate Analytical Tools to Determine the Potency and Bioactivity of Antibiotics and Antibiotic Resistance. J. Pharm. Anal. 2016, 6, 207–213. [Google Scholar] [CrossRef] [Green Version]
- Hekster, Y.A.; Baars, A.M.; Vree, T.B.; van Klingeren, B.; Rutgers, A. Comparison of High Performance Liquid Chromatography and Microbiological Assay in the Determination of Plasma Cefuroxime Concentrations in Rabbits. J. Antimicrob. Chemother. 1980, 6, 65–71. [Google Scholar] [CrossRef]
- Coats, A.W.; Redfern, J.P. Thermogravimetric Analysis. A Review. Analyst 1963, 88, 906. [Google Scholar] [CrossRef]
- Marian, E.; Tiţa, B.; Tünde, J.; Fulias, A.; Vicas, L.; Tiţa, D. Thermal Behaviour of Erythromycin-Active Substance and Tablets. J. Therm. Anal. Calorim. 2013, 111, 1025–1031. [Google Scholar] [CrossRef]
- Schlegel, L.B.; Schubert-Zsilavecz, M.; Abdel-Tawab, M. Quantification of Active Ingredients in Semi-Solid Pharmaceutical Formulations by near Infrared Spectroscopy. J. Pharm. Biomed. Anal. 2017, 142, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Zuluaga, A.F.; Agudelo, M.; Rodriguez, C.A.; Vesga, O. Application of Microbiological Assay to Determine Pharmaceutical Equivalence of Generic Intravenous Antibiotics. BMC Clin. Pharmacol. 2009, 9, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, K.E. The Theory of Antibiotic Inhibition Zones. In Analytical Microbiology; Elsevier: Amsterdam, The Netherlands, 1963; pp. 1–86. [Google Scholar]
- Cooper, K.E.; Linton, A.H. The Importance of the Temperature during the Early Hours of Incubation of Agar Plates in Assays. J. Gen. Microbiol. 1952, 7, 8–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conte, J.E.; Golden, J.A.; Duncan, S.; McKenna, E.; Zurlinden, E. Intrapulmonary Pharmacokinetics of Clarithromycin and of Erythromycin. Antimicrob. Agents Chemother 1995, 39, 334–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uskoković, V.; Huynh, E.; Wu, V.M. Mimicking the Transit of Nanoparticles through the Body: When the Path Determines Properties at the Destination. J. Nanoparticle Res. 2020, 22, 184. [Google Scholar] [CrossRef]
- Farrant, R.D.; Hollerton, J.C.; Lynn, S.M.; Provera, S.; Sidebottom, P.J.; Upton, R.J. NMR Quantification Using an Artificial Signal. Magn. Reson. Chem. 2010, 48, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S. How Efficient Are the Efficiency Terms of Encapsulation? Ther. Deliv. 2018, 9, 237–239. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ERY Formulation | Route of Administration | In Vitro Studies | In Vivo Studies | Clinical Trials | Reference |
|---|---|---|---|---|---|
| Liposomes | n.m. | + | − | − | [37] |
| n.m. | + | − | − | [34] | |
| topical | − | + | − | [41] | |
| oral | + | + | − | [43] | |
| topical | + | + | − | [46] | |
| topical | + | − | − | [47] | |
| Magneto-Liposomes | n.m. | + | − | − | [49] |
| Ethosomes | topical | + | + | − | [56] |
| topical | + | + | − | [57] | |
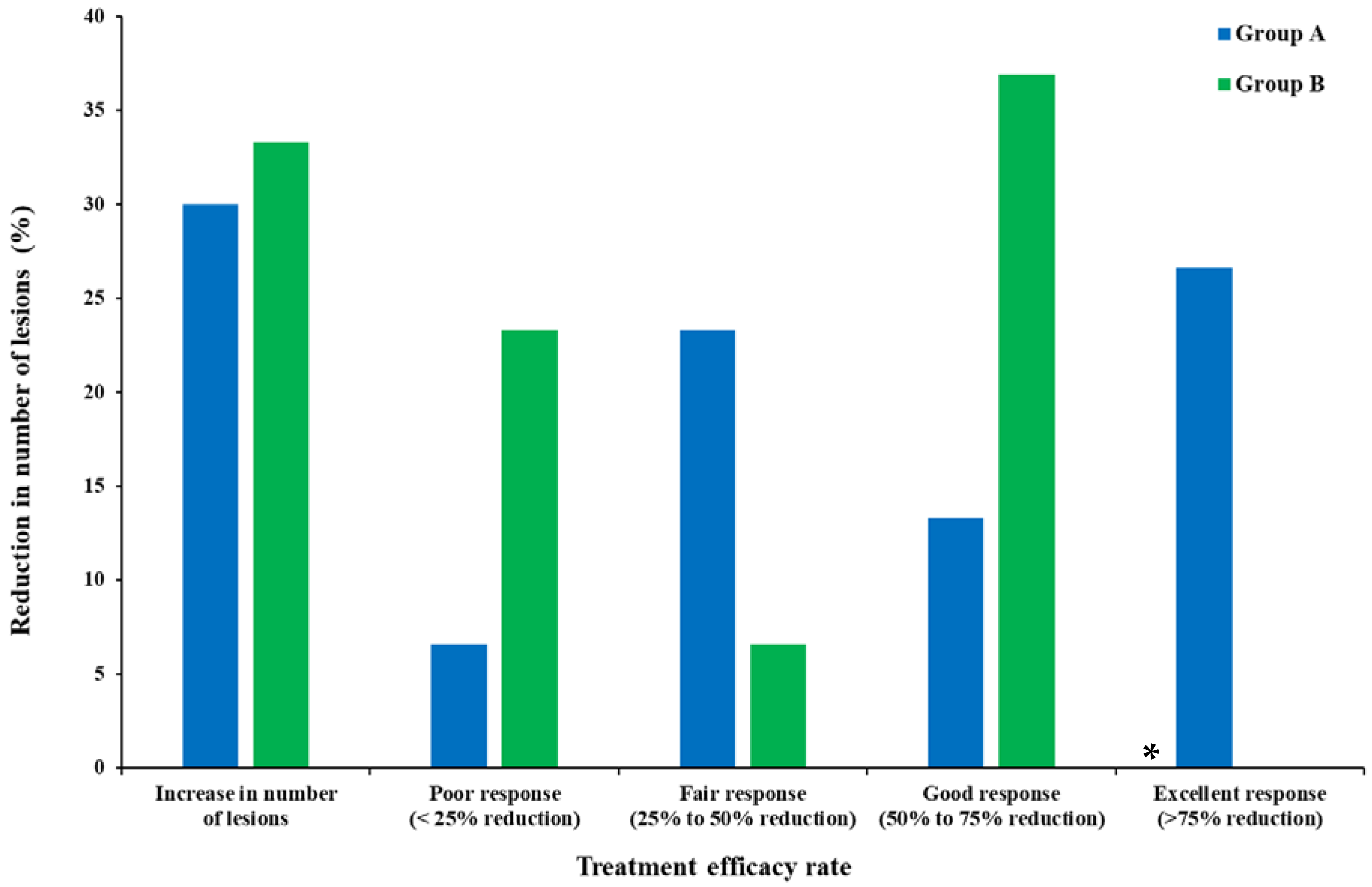
| Niosomes | topical | − | − | + | [61] |
| Biopolymeric micelles | n.m. | + | − | − | [64] |
| Polymeric micelles | topical | + | − | − | [65] |
| SLN | n.m. | + | − | − | [74] |
| n.m. | + | − | − | [76] | |
| SLM | oral | + | + | − | [77] |
| n.m. | + | + | − | [78] | |
| Microspheres | n.m. | + | − | − | [89] |
| oral | + | − | − | [90] | |
| oral | + | − | − | [91] | |
| intragastrical | − | + | − | [94] | |
| oral | + | + | − | [101] | |
| Nanospheres | n.m. | + | − | − | [96] |
| n.m. | + | + | − | [99] | |
| topical | + | − | − | [158] | |
| n.m. | + | − | − | [163] | |
| Cyclodextrin complex | n.m. | + | − | − | [105] |
| n.m. | + | − | − | [108] | |
| n.m. | + | − | − | [109] | |
| n.m. | + | − | − | [110] | |
| Film | topical | + | − | − | [124] |
| topical | + | − | − | [125] | |
| Composite | bone scaffolds | + | − | − | [131] |
| Fibres | oral | + | − | − | [144] |
| topical | + | − | − | [145] | |
| topical | + | − | − | [169] | |
| bone scaffolds | + | * | − | [148] | |
| topical | + | − | − | [149] | |
| Nanoparticle nanofibre | topical | + | − | − | [95] |
| topical | + | − | − | [98] | |
| n.m. | + | − | − | [100] | |
| Gel | topical | + | − | − | [123] |
| topical | + | − | − | [126] | |
| topical | + | − | − | [127] | |
| topical | − | + | − | [128] | |
| topical | + | − | − | [129] | |
| topical | + | − | − | [130] | |
| topical | + | − | − | [132] | |
| topical | − | − | + | [133] | |
| Microemulsion gel | topical | + | − | − | [141] |
| Niosomal gel | topical | + | − | − | [60] |
| SLN gel | topical | + | − | − | [75] |
| Polymeric micelle hydrogel | topical | + | − | − | [62] |
| Micromotor hydrogel | n.m. | + | − | − | [103] |
| Cubosomal gel | topical | + | − | − | [67] |
| Lotion | topical | − | − | + | [134] |
| Mesoporous silica nanoparticles | oral | + | − | − | [154] |
| n.m. | + | − | − | [155] | |
| topical | + | − | − | [156] | |
| Mesoporous nanospheres | n.m. | + | − | − | [157] |
| Magnetite nanoparticles | n.m. | + | − | − | [160] |
| Iron oxide nanoparticles | n.m. | + | − | − | [161] |
| Gold nanoparticles | n.m. | + | − | − | [162] |
| Copper nanoparticles | n.m. | + | − | − | [164] |
| Silver nanoparticles | n.m. | + | − | − | [165] |
| n.m. | + | − | − | [167] | |
| n.m. | + | − | − | [168] |
| Formulation | λmax (nm) | Determination | Method | ERY Content | Reference |
|---|---|---|---|---|---|
| Liposomes | 215 | Quantification of ERY/Release profile | − a | − b,c | [46] |
| Liposomes | 210 | Loading efficiency/ Loading contents | − a | 87.6 ± 1.7% 7.3 ± 0.2% | [47] |
| Niosomes | − b | Percentage drug entrapment (PDE) | Colorimetric method: PDAB | 88.2 ± 3.64% | [60] |
| Structural modification of ERY | 480 | Release profile | NaOH H2SO4 | − c | [113] |
| Cyclodextrin complex | 235 | Erythromycin concentration | sodium phosphate NaOH; 60 °C, 15 min | 1.6 mg/mL | [105] |
| Microcapsules | 280 | Release profile | − a | − c | [88] |
| Mesoporous nanoparticles | 280 | Release profile | − a | − c | [155] |
| Microspheres | 620 | Drug content/ Yield of encapsulation | − d | 22% 62.5% | [90] |
| Microspheres | 482 | Drug loading Encapsulation efficiency | H2SO4 (75% v/v) V = 5 mL | 13.56 ± 0.25% 55.82 ± 2.23% | [19] |
| Nanoparticles | 257 | Entrapment efficiency | − a | 82.45 ± 2.33% | [96] |
| Micromotor hydrogel | 288 | Drug loading efficiency | − a | Microcapsule: 40% MM hydrogel: 53% | [103] |
| Encapsulation efficiency | Microcapsule: 42% MM Hydrogel: 48% | ||||
| Nanospheres | 236 | Drug Loading Capacity | HCl (37%) | 37 ± 11% | [157] |
| Hydrogel | 485 | Release profile | 0.1 mL of H2SO4 3M, 0.1 mL supernatant 50 °C, 30 min | − c | [123] |
| Hydrogel | 486 | Release profile | Microplate reader H2SO4, 50 °C,30 min | − c | [175] |
| Bone scaffolds | 235 | Release profile | sodium phosphate NaOH, 60 °C, 15′ | − c | [131] |
| SLN gel | 483.5 | Drug entrapment/ Release profile | 1 mL conc. H2SO4 50 °C, 30 min | 78.1 ± 0.74% − c | [75] |
| Cubosomes | 214 | Entrapment Efficiency | − a | 95.29 ± 1.32% | [67] |
| SLN | 236 | Entrapment Efficiency/ Drug Loading | − a | 94.27% 30.57% | [74] |
| SLM | 214 | Encapsulation efficiency/ Loading capacity | − a | 95.11 ± 0.3% 43.22 ± 0.1% | [77] |
| SLM | 320 | Release profile | − a | − c | [78] |
| Fibres | 408 | Release profile | Colorimetric method: bromocresol purple, CHCl3 | − c | [144] |
| Fibres | 285 | Release profile | − a | − c | [145] |
| Fibres | 280 285 e | Encapsulation efficiency | − a | 94% | [169] |
| Fibres | 285 | Drug loading efficiency f | Detected in release media after 7 days | 89.34% | [95] |
| Nanofibrous patches | 205 | Encapsulation Efficiency/ Drug Loading | − a | 95.3 ± 3.1% 41.14 ± 0.05% | [98] |
| Formulation | Measurement Conditions | ERY Content | Reference |
|---|---|---|---|
| Gel | Temperature: 45 °C Mobile phase: mixture of 0.001M disodium phosphate solution: acetonitrile (20:80) Volume of sample injected: 20 μL Injection rate: 1 mL/min λ = 200 nm | Final concentration: 2% ERY | [111] |
| Nanocarrier | Temperature: 45 °C Mobile phase: 15 wt% methanol, 45 wt% acetonitrile and 40 wt% ammonium phosphate 0.01 M solution Volume of sample injected: 50 μL Injection rate: 1 mL/min λ = 215 nm | EE: 15.02–23.09% LC: 48–78% | [109] |
| Microcapsules | Temperature: 45 °C Mobile phase: acetonitrile–methanol–water (39:9:52, v/v) containing 0.04 M NaH2PO4 Volume of sample injected: 40 μL Injection rate: 0.8 mL/min λ = 211 nm | Yield of encapsulation (YE): 82 ± 19% | [91] |
| Hydrogel | Mobile phase: acetonitrile–phosphate buffer pH 7.0 (65:35, v/v) Injection rate: 1 mL/min λ = 195 nm | Drug content: 98.00 ± 1.30–102.25 ± 0.23% | [122] |
| Micelles | Temperature: 30 °C Mobile phase: 700 mL of acetonitrile + 300 mL of 0.05 M solution of KH2PO4 in water, adjusted to pH 7.5 with a 10% sodium hydroxide solution λ = 220 nm | Loading amount: 38.6 μg/mL EE: 28.3% | [62] |
| Formulation | Bacterial Strain | Method | ERY Content | Reference |
|---|---|---|---|---|
| Liposomes | B. subtilis (ATCC 6633) | Agar diffusion assay | EE: 32.06 ± 0.82% LC: − | [34] |
| Liposomes | B. subtilis (ATCC 6633) | Agar diffusion assay | EE: 51.58 ± 2.846% EC: 10.32 ± 0.571 mg/mL | [37] |
| Bioencapsulation | M. luteus (ATCC 9341) | Modified cylinder plate method | 177.0 ± 30.8 µg a | [42] |
| Computational analysis | B. subtilis (ATCC 6633) | Disc-plate method | − b | [52] |
| Ethosomes | B. subtilis (ATCC 6633) | − c | EE: 78.6 ± 4.8% | [56] |
| Classic oral formulation—pharmacokinetics study | M. luteus (ATCC 9341) | Agar diffusion assay | 0.8 ± 1.3 mg/mL in alveolar cells b | [185] |
| Nanoparticles | P. aeruginosa | LB agar plates with standard curve | 34.8 μg/mL loaded onto 5 mg/mL of HAp nanoparticles | [186] |
| Gel | M. luteus (ATCC 9341) | Agar diffusion assay | 100.9% of theoretical level | [120] |
| W/O and O/W emulsion Gel | M. luteus (ATCC 9341) | Agar diffusion assay | Study of the decrease in antibiotic activity under various conditions d | [121] |
| SLM | S. aureus | Agar diffusion assay followed by UV–VIS at 320 nm | Bioactive drug loading (BDL): 7.73 ± 2.66% Bioactive encapsulation efficiency (BEE): 11.59% | [78] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Platon, V.-M.; Dragoi, B.; Marin, L. Erythromycin Formulations—A Journey to Advanced Drug Delivery. Pharmaceutics 2022, 14, 2180. https://doi.org/10.3390/pharmaceutics14102180
Platon V-M, Dragoi B, Marin L. Erythromycin Formulations—A Journey to Advanced Drug Delivery. Pharmaceutics. 2022; 14(10):2180. https://doi.org/10.3390/pharmaceutics14102180
Chicago/Turabian StylePlaton, Vera-Maria, Brindusa Dragoi, and Luminita Marin. 2022. "Erythromycin Formulations—A Journey to Advanced Drug Delivery" Pharmaceutics 14, no. 10: 2180. https://doi.org/10.3390/pharmaceutics14102180
APA StylePlaton, V.-M., Dragoi, B., & Marin, L. (2022). Erythromycin Formulations—A Journey to Advanced Drug Delivery. Pharmaceutics, 14(10), 2180. https://doi.org/10.3390/pharmaceutics14102180