3.1. Evaluation of Alternative Mobile Phase Eluents
In the RPLC of proteins, acetonitrile remains the most widely used mobile phase component. However, as already demonstrated elsewhere [
9], the chromatographic selectivity can possibly be altered by changing the nature of the organic modifier. This behavior could be attributed either to the modification of weak chemical interaction (acetonitrile is aprotic, while alcohol-based modifiers are protic, so H-bond interaction may be strongly modified) or to the fact that mobile phase viscosity could vary significantly when changing the nature of the organic solvent. Indeed, it has been recently shown that the selectivity of proteins can be significantly modified with pressure changes [
10]. A change from acetonitrile to an alcoholic mobile phase system might also bring to effect π-π electron interactions between a phenyl-bonded stationary phase and protein amino acid side chains, such as tyrosine, tryptophan, and phenylalanine [
10].
In the present work, ultra-short columns were employed, generating reasonable backpressure. Therefore, it becomes possible to use isopropanol (IPA), which is known to be highly viscous, as an eluent instead of acetonitrile, without instrumental limitations. Besides pure IPA, a mixture of 50:50 IPA:ACN was also tested in this work. In each case, these mobile phases were acidified with 0.05% of difluoroacetic acid (DFA).
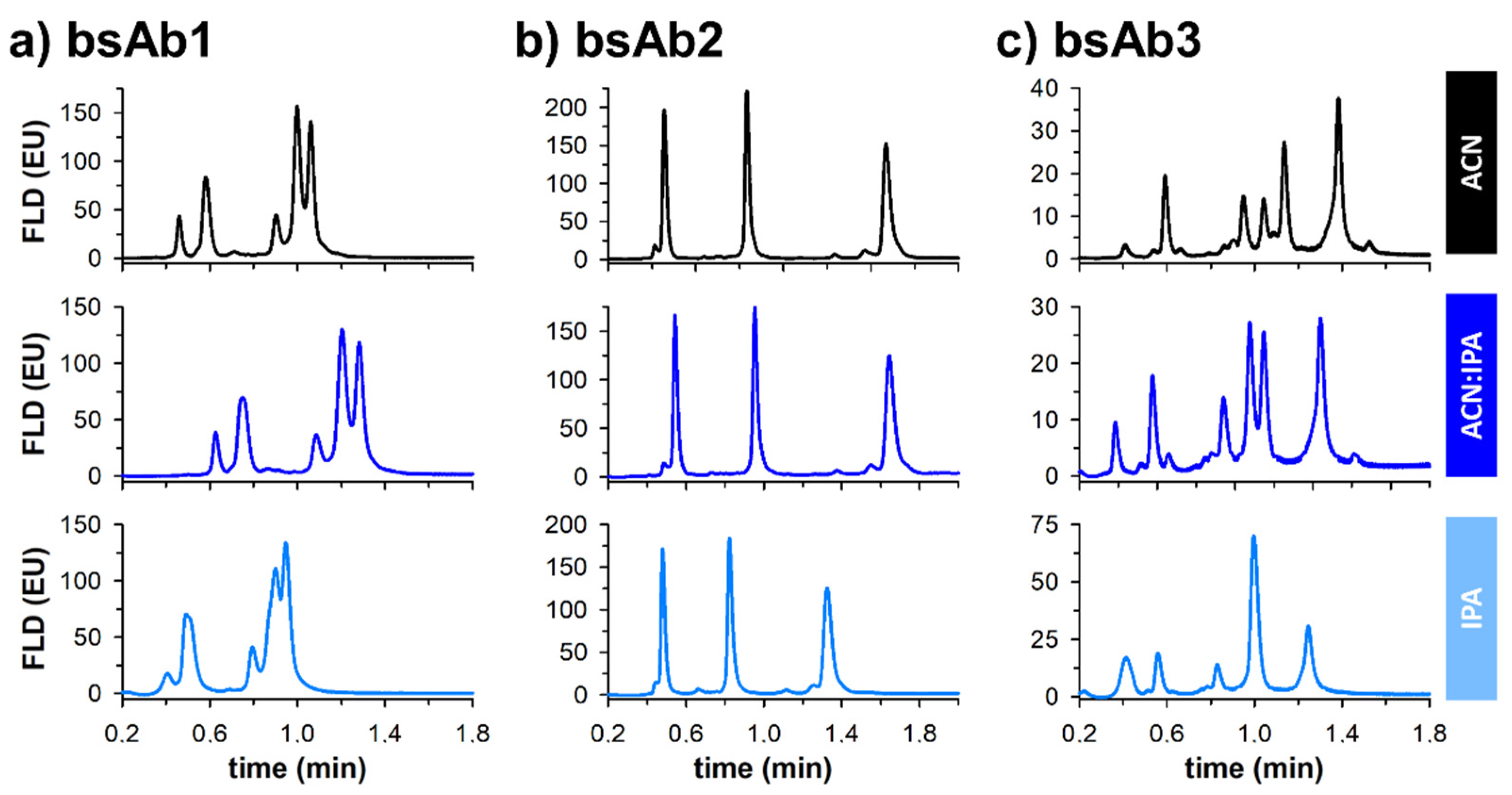
Figure 2 shows the corresponding fast separations obtained for three reduced bispecific mAb products using these three different types of mobile phases. First, it is important to note that pressure drop remained acceptable whatever the conditions. Indeed, ΔP was equal to 110, 160, and 280 bar when using ACN, ACN:IPA, and IPA as the eluent, respectively. However, even if the pressure remained within acceptable limits, the added value of IPA was found to be limited in the three selected examples.
Indeed, the separation performance was highly comparable for bsAb1 when using pure ACN or the ACN:IPA 50:50 mixture, but the overall performance was strongly reduced with pure IPA, with a severe loss of selectivity between the most critical peak pair, and a reduction of retention. For the second example (bsAb2), selectivity between the three major species and the minor variants remains identical whatever the mobile phase components, and only the retention was reduced with IPA. Finally, for the third example (bsAb3), the selectivity was significantly modified depending on the nature of the eluent. The separation carried out with IPA was clearly the worst one, with a loss of selectivity between two major peaks. Even if no selectivity improvement was noticed with IPA for the selected examples,
Figure 2 proves that selectivity can be tuned by modifying the solvent composition of the eluent. Interestingly, the use of ultra-short columns for protein analysis under RPLC conditions provides more flexibility in terms of organic solvent because it is unlikely to run into the pressure limits of the LC instrumentation.
3.2. Ultra-Fast Separations of Complex Mab Products
A clear benefit of using ultra-short columns for the analysis of therapeutic proteins is the possibility to reduce analysis time, with limited impact on overall kinetic performance. Such a behavior is demonstrated in
Figure 3.
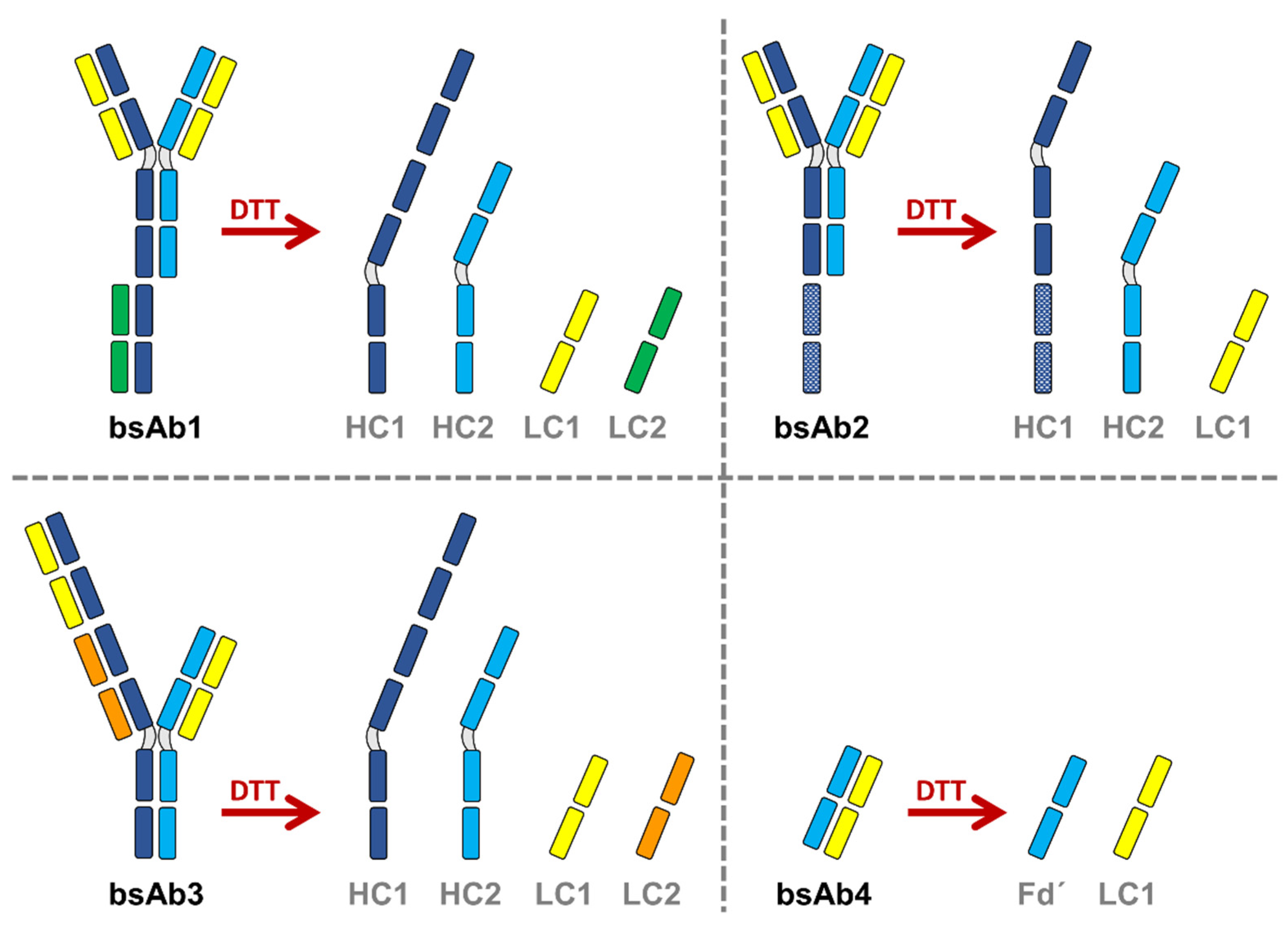
Various sub-1 min separations of complex biopharmaceutical samples are displayed as detected by native protein fluorescence. In this case, both the reference samples and various thermally stressed samples (from 1 to 12 weeks at 40 °C) were analyzed. Only the samples reduced with DTT were analyzed (sub-units described in
Figure 1), as the chromatograms were found to be much more informative than the ones obtained at the intact level. For comparison purpose, the same samples were already analyzed using multi-isocratic elution mode [
11]. To maximize separation power, the composition ranges were adjusted depending on the product hydrophobicity, while the gradient time and flow rate were constant, 1 min and 1 mL/min, respectively. This ensured to have the highest possible k* value (k* corresponds to the gradient retention factor which has a similar significance in gradient elution as the retention factor k in isocratic mode). In the present case, k* values ranged from 0.72 to 3.38 for the four different samples. In addition, no equilibration time was added at the end of the run, since the volume of the 10 × 2.1 mm I.D. column was equal to about 21 µL, corresponding to a column dead time of only 1.3 s when working at a flow rate of 1 mL/min. So, the injection time (about 30 s) was already sufficient to equilibrate the column between each run, as it corresponds to more than 20 column volumes. It is also important to mention that all system dispersion effects were reduced as much as possible to limit band broadening. To do this, we limited the injection volume to only 0.5 µL, used 220 mm × 65 µm tubing between the column outlet and detector inlet, and employed a commercial 2 µL fluorescence flow cell. Despite these extra considerations and taking into account the k* value, performance achieved on the 10 × 2.1 mm column was slightly lower than expected, and additional band broadening was observed due to the presence of both extra-column volume and extra-bed volume. [
8]
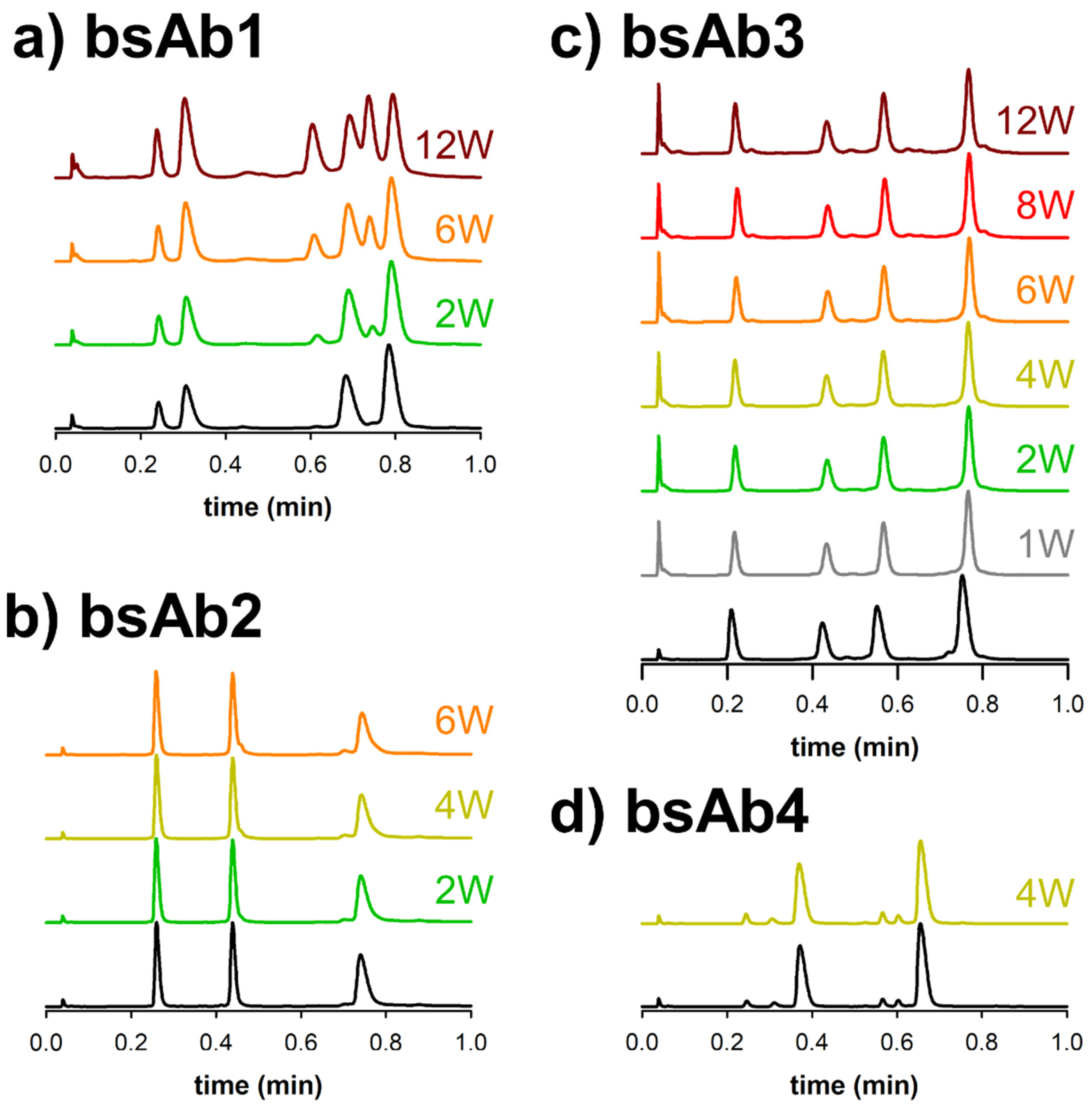
Figure 3a shows the ultra-fast separation of bsAb1 before and after a thermal stress of 2, 6 and 12 weeks. As expected, four main species were observed on the reference chromatogram (no stress), with the first two peaks corresponding to the two different light chains (LC1 and LC2), and the last two peaks corresponding to the two heavy chains (HC1 and HC2). After the thermal stress, two additional major peaks were observed in the elution region corresponding to the HC, and a minor species was observed between the elution zones of the LC and HC species. These extra species were already present in the reference sample, but in very low amounts (<1%). The chromatograms reported in
Figure 3a clearly show that the amount of the two main impurities was increasing significantly with stress time, and this amount can be easily quantified with the fluorescence detector, based on peak areas (yet this is out of the scope of the present work).
Similar conclusions were obtained for the three other biopharmaceutical products (i.e. bsAb2, bsAb3 and bsAb4). For bsAb2, LC1 and HC2 were seen to closely elute as the first and second peaks, while HC1 was more hydrophobic and eluted later. Here again, two additional species were observed on the stressed sample, including one that eluted just after HC2 and another that eluted before HC1. The amount of impurities related to the degradation from thermal stress (degradation related impurities) increased very slightly with stress time, even after 6 weeks at 40 °C. For bsAb3, four species were expected according to
Figure 1. Based on their hydrophobicity, the two LC species would be predicted to elute first, and before the molecule’s two HC subunits. Here again, we observed some differences in terms of minor species. Several various additional species were introduced to the sample over time and found to elute between the main species. Nevertheless, the amount of impurities was found to be limited even after 12 weeks of thermal stress. For bsAb4, only two samples were available, namely the reference sample and the sample exposed at 40 °C for 4 weeks. The subunit species predicted for this molecule is shown in
Figure 1. In the chromatogram, only two main species were observed: LC1 eluted as the first peak, while Fd’ eluted as the second. Four additional species were already observed in minor quantities and separated on the reference sample. Interestingly, the amount of these four impurities did not increase through thermal stress.
As illustrated in this section, ultra-fast analytical methods are very helpful for rapidly checking a sample for additional variants that are induced by stress, in biopharmaceutical samples over time.
3.3. Coupling Ultra-Fast Separations with MS
In order to obtain more information on the peaks observed in
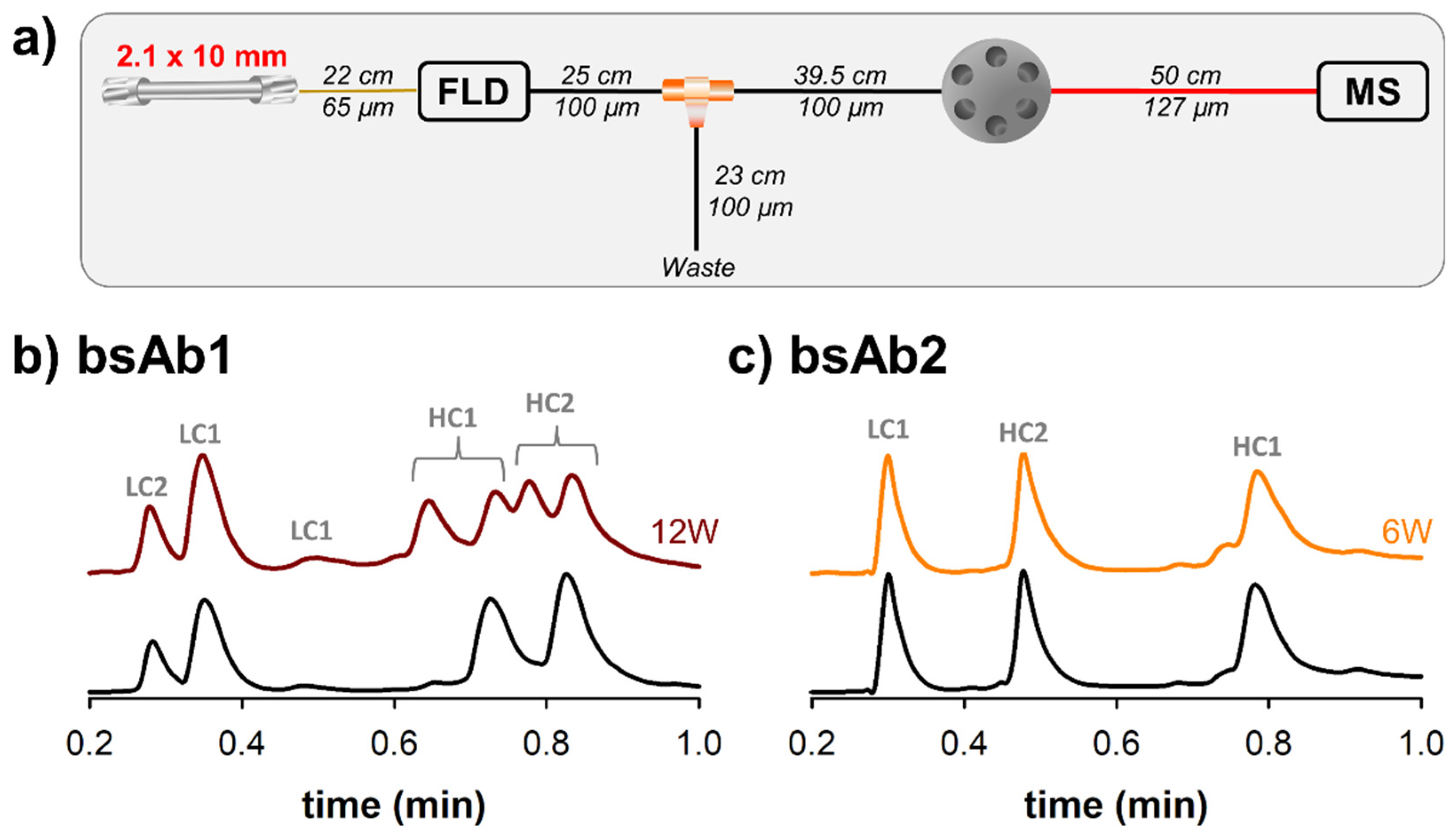
Figure 3, we have also performed some experiments with ultra-short columns using both fluorescence (quantitative detector) and MS (qualitative detector) detectors. The experimental setup employed for this part of the work has been described in
Figure 4a. Next to the fluorescence detector, a flow splitter was added to provide an optimal flow rate to the ESI-MS source. A flow rate of 275 µL/min was split to the MS detector, while the remaining flow (725 µL/min) was directed to waste. In addition, it was mandatory to add a switching valve before the ESI inlet, allowing a calibrant solution to be periodically infused for real-time lock mass adjustment. In the end, based on the minimal distances between the different parts of the MS device, three tubes of 250 mm × 100 µm, 395 mm × 100 µm and 500 mm × 127 µm had to be added between the fluorescence detector and ESI-MS source. It has already been demonstrated elsewhere that the tubes located between the ultra high performance liquid chromatography (UHPLC) system and the ionization source are particularly critical for minimizing band broadening [
12]. In the present case, the added tubing corresponds to a total volume of more than 11 µL, so band broadening may be quite severe when using ultra-short column with a volume of only 21 µL. Unfortunately, thinner tubes cannot be used to reduce the volume, as the fluorescence flow cell has an upper pressure limit of only 35 bar and thinner tubes would generate a pressure higher than this cut-off value. As an alternative, UV detection (the UV cell has a higher upper pressure limit of 70 bar, for the instrument used in this work) could be envisaged to provide more flexibility to the instrumental setup, but sensitivity would then be reduced.
Total ion chromatograms (TICs) were recorded for all four samples using the conditions described in
Section 3.2, but with the addition of MS detection. Only the traces corresponding to the unstressed and stressed bsAb1 and bsAb2 samples were reported in
Figure 4. A similar behavior was observed for bsAb3 and bsAb4.
The two chromatograms reported in
Figure 4b should be compared with the ones reported in
Figure 3a using the same color code. As expected, the retention and selectivity were comparable between these two separations, as only the detector changed. However, the peaks were much broader when using MS detection (at least 2-fold broader), leading to a non-negligible loss of resolution. To better understand the reason for this loss in performance, we have tried to modify the MS settings. Various scan rates ranging from 2 to 10 Hz were tested, but unfortunately, only sensitivity was altered with the higher data acquisition rate, while the peak broadening remained comparable. Next, we also tried to bypass the fluorescence detector, but here again, the impact on final separation was minor (less than 10% improvement in peak widths). Based on these observations, it is clear that the severe band broadening observed with MS detection was almost exclusively due to the tubing added between the fluorescence detector outlet and ESI-MS inlet. It is indeed important to keep in mind that extra column band broadening in a tube (σ
2tubing) is inversely proportional to the diffusion coefficient of the analyzed molecule (D
m), as reported elsewhere [
13]. In the present case, the analyzed species have sizes varying between 25 and 75 kDa, corresponding to calculated diffusion coefficients (using the Wilke-Chang equation, which is known to be quite precise [
14]) comprised between 4.1 and 8 × 10
−11 m²/s under the mobile phase conditions employed in this work. These values are in average 10 to 20-fold higher than the standard value considered for small molecules (D
m = 1 × 10
−9 m
2/s), thus explaining the significant contribution of the tubing to band broadening.
Similar chromatography was observed in
Figure 4c, which presents LC-MS chromatograms of bsAb2. These separations should be compared with the ones reported in
Figure 3b. In this example, the loss in performance due to MS was even more severe than for bsAb1, since a 3-fold loss in peak capacity was noticed. One possible explanation is related to the gradient span. Indeed, bsAb1 and bsAb2 were analyzed with a gradient from 30 to 36% B, and from 26 to 54% B, respectively. Therefore, the k* values were almost 5-times lower for bsAb2 vs. bsAb1 analysis (k* was equal to 0.72 for bsAb2 and 3.38 for bsAb1), thus reducing the column variance and increasing the impact of extra-column variance (it remains comparable between the two examples) on peak broadening.
In the end, an interesting solution to limit band broadening with MS detection would be to discard the fluorescence detector from the experimental setup or to have it plumbed against a splitter so that it can receive column effluent in parallel with the MS detector. In either case, thinner tubing could be employed prior to the ESI-MS inlet, without the risk to destroy the fluorescence flow cell. Special considerations would need to be made with the ToF instrument employed here, because it uses a fused silica transfer line that is directly integrated and crimped onto the electrospray capillary of the mass spectrometer.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}



