3.2. Activity Scores
The first independent variable that was established in the proposed framework was the average activity score (AAS). This average score was calculated as the arithmetic mean of three scores based on the structural features of TRPV1 antagonists and agonists: Bemis–Murcko structural skeletons, plain ring systems and clustering based on flexophore descriptors. Firstly, a scaffold analysis was performed to extract Bemis–Murcko skeletons and plain ring systems. For each individual BM skeleton, the specific score (BMS, Bemis–Murcko score) was established by calculating the mean activity value (pIC50 for antagonists, pEC50 for agonists, 0 for inactive molecules) between all compounds that share the same skeleton. The same rationale was applied for plain rings (PRS, plain ring score). However, when PR scores were calculated for each compound, we took into account that some molecules contain more than one ring. Thus, for compounds with multiple rings, the mean PR score was calculated as the arithmetic mean between scores of all rings. Moreover, if one ring is present more than once in a structure (e.g., phenyl radical), then the score was calculated considering only one apparition of that ring. Lastly, the similarity cluster score (SCS, flexophore cluster score) was established by calculating the mean activity values between compounds that fall into one specific cluster. Therefore, two average activity scores were calculated, one for predicting antagonists (Score-ANT), and another for agonists (Score-AG).
After performing the scaffold analysis, a total of 591 BM structures resulted from the group of 3564 compounds (antagonists, agonists and inactive molecules). Most of the scaffolds are formed by 3 or 4 cyclic structures. For each BM skeleton the corresponding ANT and AG scores were calculated. For a total of 256 skeletons, both scores were zero. For the remaining 335 structures, we analyzed the relationship between the two scores (
Figure S1A). Interestingly, one BM scaffold ({2-[10-(4-cyclohexylbutyl)-hexadecahydro-1H-indeno[5,4-e]azulen-3b-yl]ethyl}cyclohexane) had both scores above 4, scoring high for both agonist and antagonist prediction. Moreover, 9 agonists and 26 inactive molecules did not contain any rings in their structure and BM skeletons could not be generated.
The PR analysis resulted in 271 unique rings. The most frequent ring is benzene, and it is present in 2682 compounds. Pyridine, the second most frequent ring, is present in 1207 derivatives. The structures contain between 3 and 25 heavy atoms. The corresponding ANT and AG scores were calculated for each PR structure. For a total of 34 rings both scores were zero, and the relationship between the two scores is shown in
Figure S1B. Notably, four plain rings had activity scores between 2 and 4, showing less specificity for one activity type: (7aR,11bR,13aR,13bS)-icosahydro-1H-cyclopenta[a]chrysene, (2S,10R,11S)-12,14-dioxatetracyclo[8.7.0.0²,⁶.0¹¹,¹⁵]heptadeca-3,8-dien-5-one, (1S,2R,10S,11R)-12,14,18-trioxapentacyclo[11.4.1.0¹,¹⁰.0²,⁶.0¹¹,¹⁵]octadeca-3,8-dien-5-one and 1,2-dihydroisoquinolin-1-one.
Flexophore descriptors were generated for the merged dataset containing TRPV1 antagonists, agonists and inactive compounds. A total number of 419 clusters was obtained, using a similarity threshold of 80% between flexophores. Scores based on structural similarity clustering were also calculated since some TRPV1 agonists and inactive molecules are acyclic compounds, thus lacking both BM skeletons and plain rings. As observed in
Figure S1C, several clusters had mean activity scores between 1 and 4 for both agonist and antagonist prediction (SCS-AG and SCS-ANT), thus being non-specific for either class. A map of flexophore-based similarity relationships generated using automatically determined similarity limits is shown in
Figure S1D, highlighting that several TRPV1 antagonists share structural similarity with agonists or experimentally determined inactive compounds.
The top five scoring BM and PR structures, ranked by activity scores are highlighted in
Table 1, for both antagonists and agonists. BM and PR structures were labeled by frequency of apparition in the dataset, in descending order. BM-75 (3-[(decahydronaphthalen-1-yl)methyl]-1,1′-bi(cyclohexane)) and BM-230 (1-[(3,4-dicyclohexylcyclopentyl)methyl]-decahydronaphthalene) are two similar antagonist-specific Bemis–Murcko skeletons with high activity scores, BM-230 having a cyclopentane scaffold instead of cyclohexane, which also has one additional substitution forming an uncondensed tricyclic substructure instead of the bicyclohexane scaffold. In the case of agonists, BM-145 ({2-[10-(3-cyclohexylpropyl)-hexadecahydro-1H-indeno[5,4-e]azulen-3b-yl]ethyl}cyclohexane) and BM-106 ({2-[10-(5-cyclohexylpentyl)-hexadecahydro-1H-indeno[5,4-e]azulen-3b-yl]ethyl}cyclohexane) have highly similar structural skeletons and activity scores, the only difference between the structures being the number of linker atoms (three for BM-145 and five for BM-106). Additionally, structures of BM-264 ((11-cyclopropylundecyl)cyclohexane), BM-176 ((10-cyclopropyldecyl)cyclohexane) and BM-205 ((3-cyclopropylpropyl)cyclohexane) are comprised of two cyclic substructures (cyclohexane and cyclopropane) linked together by an aliphatic chain, the activity score increasing with the number of linker atoms (3 for BM-205, 10 for BM-176 and 11 for BM-264).
Interestingly, the top five scoring plain rings for antagonists are nitrogen heterocycles. Among these fragments, PR-126 (pteridine), PR-127 (pyrido[3,2-d]pyrimidine) and PR-87 (pyrido[2,3-d]pyrimidine) are variations of the same scaffold. Furthermore, azocane (PR-100) had a higher activity score than 2,3,4,5-tetrahydro-1H-2-benzazepine (PR-136), the latter being specific to TRPV1 antagonist CPZ and other related derivatives. The PR with the highest score for agonists (PR-126, 1,2-dihydroisoquinolin-1-one) share structural similarities with the highly ranking antagonist-specific PR-128 (1,2-dihydroquinoxalin-2-one). Moreover, two out of the top five scoring agonist-specific plain rings, PR-57 ((2S,10R,11R)-12,14-dioxatetracyclo[8.7.0.0²,⁶.0¹¹,¹⁵]heptadeca-3,8-dien-5-one) and PR-45 ((1S,2R,10S,11R)-12,14,18-trioxapentacyclo[11.4.1.0¹,¹⁰.0²,⁶.0¹¹,¹⁵]octadeca-3,8-dien-5-one), are also highly similar.
The distributions of activity scores for antagonists, agonists and inactive compounds are shown in
Figure S2. As observed, antagonists had overall higher AAS values than agonists, since antagonists represented a significantly larger population among the biologically active compounds.
The predictive power of the established average activity score was assessed by generating ROC curves and calculating ROC AUC values. The ROC AUC value for antagonist activity scores was 0.963 (
Figure S3A), while the same parameter was 0.986 for predicting agonists (
Figure S3B), denoting high predictive accuracies in both cases.
3.3. Binary Classification
ROC curves were generated based on activity classes and molecular descriptors in order to build classification models using cutoff values. We chose to include a minimum of three and a maximum of eight molecular descriptors as independent variables. ROC AUC values were calculated for all descriptors to assess the discriminatory power of each variable. The eight descriptors were chosen based on four criteria: satisfactory ROC AUC values, statistically significant differences between values of active and inactive molecules, correlation coefficients between each pair of descriptors lower than 0.75 and ease of describing the respective molecular property.
The selected molecular descriptors and their classification performance parameters are presented in
Table 2 and
Table 3. The individual ROC curves that were used for choosing the descriptor cutoff values are shown in
Figure S4.
We chose to further discuss some of the selected molecular descriptors for AMG-517 (N-[4-[6-[4-(trifluoromethyl)phenyl]pyrimidin-4-yl]oxy-1,3-benzothiazol-2-yl]acetamide), a potent selective TRPV1 antagonist. Judging by the chosen features, TRPV1 antagonists have secondary nitrogen atoms in their molecules, between 0 and 8 halogen atoms, between 0 and 4 hexa-atomic heterocycles, and 1–15 hydrogen bond acceptors. AMG-517 (
Figure S5) has in its structure one secondary nitrogen (substituted amide), three halogen atoms (fluorine), one hexa-atomic heterocycle (pyrimidine), and nine hydrogen bond acceptors (three fluorine atoms, four nitrogens, two oxygens). Moreover, AMG-517 respects seven out of the eight criteria for descriptor threshold values, the only unsatisfied feature being the minimum sum of atom-type E-State for secondary nitrogens. Three of the established criteria require the presence of at least two halogen atoms, at least one hexa-atomic heterocycle, and a minimum five hydrogen bond acceptors.
In the case of TRPV1 agonists, it can be noted that active molecules have 0–3 hydroxyl groups in their molecules, logP values between 0.86 and 10.93, hybridization rations between 0 and 0.95, 0–3 halogen atoms, 0–5 nitrogen atoms, average molecular weight between 5.25 and 9.00, 0–22 atoms in the longest aliphatic chain, and between 3 and 21 atoms in the largest pi system. The established criteria for agonists classification were logP values above 3.023, hybridization ratios higher than 0.383, at least one hydroxyl group, at least 3 atoms in the longest aliphatic chain, the absence of halogen atoms, less than 10 atoms in the largest pi system, less than 3 nitrogen atoms and an average molecular weight lower than 7.25. For instance, the well-known TRPV1 agonist capsaicin ((E)-N-[(4-hydroxy-3-methoxyphenyl)methyl]-8-methylnon-6-enamide) respected all the threshold criteria, having a logP value of 3.983, a hybridization ratio of 0.5, one hydroxyl group, nine atoms in the longest aliphatic chain, no halogen atoms, eight atoms in the largest pi system, one nitrogen atom, and an average molecular weight of 6.228 (
Figure S5).
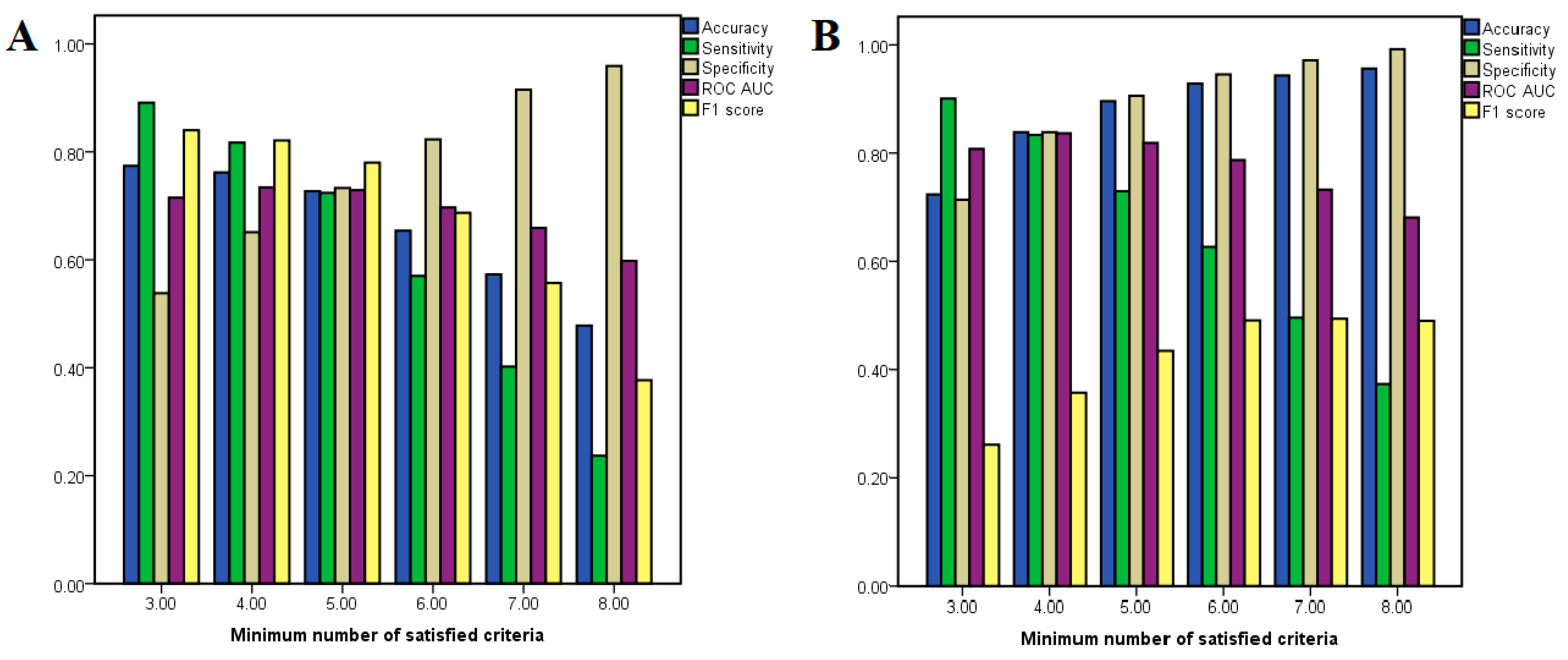
Notably, neither AMG-517 nor capsaicin met any of the required criteria for being classified as the opposite type of active compound, and only one molecular descriptor was included in classification models of both agonists and antagonists (number of halogen atoms). The optimal number of required criteria for considering a molecule either an antagonist or an agonist in our classification problem was established by calculating the performance metrics (such as accuracy and ROC AUC) after varying the minimum number of satisfied molecular descriptor thresholds from three to eight. The performance metrics for each classification condition are shown in
Figure 3. In the case of antagonists, it can be noted that the model accuracy decreased when the minimum number of required criteria is increased, while in the case of agonists the accuracy varies in proportion to the number of required criteria.
For classifying antagonists, the accuracy varied between 47.8 and 77.4%, while for agonist classification models the same parameter ranged between 72.5 and 95.8%. The classification model for predicting antagonists with the most optimal balance between sensitivity (0.724) and specificity (0.733) had a minimum number of five satisfied criteria, the accuracy of the model being 72.7%, showing an ROC AUC value of 0.729. On the other hand, the most balanced model for classifying agonists had a minimum required criteria of four satisfied descriptor thresholds, the model’s accuracy being 83.9%, showing sensitivity and specificity values of 0.834 and 0.839, respectively and an ROC AUC of 0.837. Although the model that used a minimum number of five criteria had better accuracy (89.7%), the ROC AUC value was lower (0.819) and this classification model showed a higher preference for true negatives over true positives.
3.4. Molecular Docking Simulations
Molecular docking studies were carried out to estimate the binding affinities of potential repurposing candidates and to investigate the predicted binding modes. Molecular docking results were the third and final independent variable in the proposed repurposing predictive model. Two crystal structures were used in this study: activated TRPV1 bound to agonist RTX and TRPV1 in a closed state bound to antagonist CPZ. Both qualitative and quantitative validations of the docking procedure were performed. First of all, the accuracy of binding mode predictions was assessed by docking the co-crystallized ligands into the binding site and superposing the predicted conformation with the experimentally determined ligand pose. The RMSD values calculated after superposition were 1.1277 Å for CPZ and 1.2564 Å for RTX, showing low deviations from original conformation and satisfying accuracy for pose prediction (
Figure S6). Binding energies for the positive controls were −9.13 kcal/mol for CPZ and −11.55 kcal/mol for RTX, respectively.
A second validation of the docking protocol was performed by assessing the capability of the two TRPV1 conformations to discriminate against active and inactive ligands by analyzing the predicted binding energies or ligand efficiencies. Therefore, a selection of TRPV1 agonists (n = 194), antagonists (n = 222), inactive molecules (n = 488) and decoys (n = 500) were docked against the binding sites of active (PDB ID 7MZC) and inactive (PDB ID 5IS0) conformations of TRPV1. Molecular docking simulations on TRPV1 in closed conformation yielded binding energies ranging from −13.04 to −5.93 (−9.56 ± 1.096) kcal/mol for antagonists, −11.48 to −5.80 (−8.27 ± 1.260) kcal/mol for agonists, −11.05 to −3.39 (−8.12 ± 1.213) kcal/mol for inactive ligands and from −11.13 to −4.78 (−7.54 ± 1.171) for decoys. The differences in binding energies between antagonists and inactive molecules, and between antagonists and decoys, were statistically different (p < 0.05, Student’s independent t-test).
After docking on the open state receptor conformation, binding energies between −12.21 and −5.61 (−8.51 ± 1.412) kcal/mol were obtained for agonists, −12.15 and −5.62 (−9.20 ± 0.934) kcal/mol for antagonists, −11.48 and −3.92 (−8.13 ± 1.187) kcal/mol for inactive compounds and between −11.53 and −4.77 (−7.52 ± 1.088) kcal/mol for decoys. Although statistically significant differences were observed between the binding energies of agonists and inactive molecules and between the docking scores of agonists and decoys (p < 0.05, independent t-test), the differences were not strong enough to discriminate well between agonists and experimentally determined inactives. Due to this inconvenience, a derived parameter was calculated in order to solve the issue of comparable docking scores between agonists and inactive ligands. Thus, the ligand efficiency-dependent lipophilicity index (LELP) was calculated for all the docked ligands, which is expressed as logP divided by ligand efficiency. Mean LELP values were obtained as follows: 13.44 for antagonists, 18.90 for agonists, 9.40 for inactive molecules and 9.61 for decoys. Using LELP values, statistically significant differences were observed between agonists and inactive ligands and between agonists and decoys, respectively (p < 0.05).
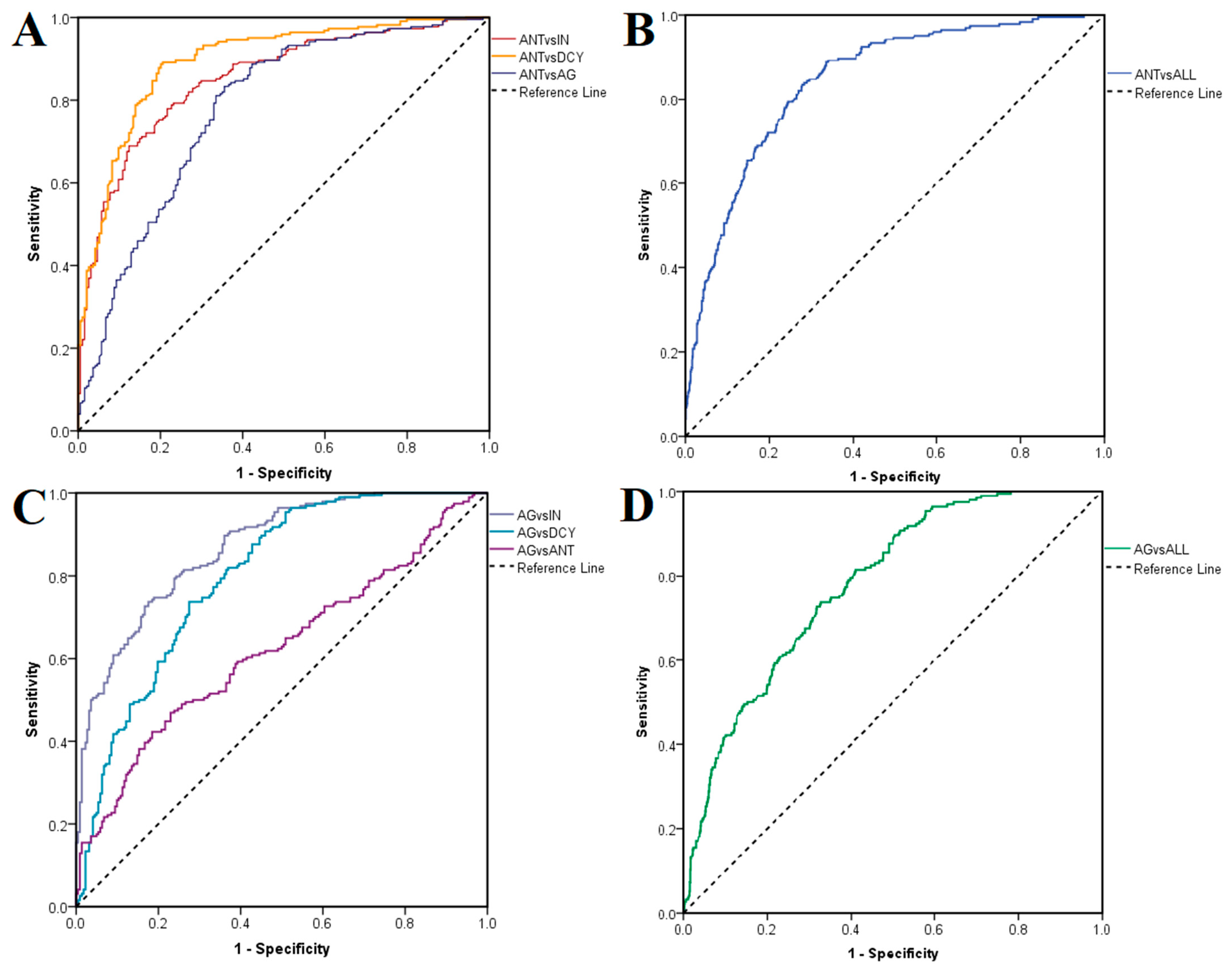
ROC curves were generated to assess the suitability of the docking procedure for discriminating between active and inactive ligands. In the first case, antagonists were labeled as positives and non-antagonists (agonists, inactive compounds and decoys) as negatives after docking on the closed conformation of TRPV1. ROC AUC values of 0.852 were obtained after testing antagonists against inactive ligands, 0.894 against decoys, 0.779 against agonists (
Figure 4A) and 0.844 against all non-antagonists (
Figure 4B). The same rationale was applied after docking on the open conformation, when agonists were treated as positives and non-agonists as negatives, LELP being used in this case instead of binding energy as a classifier. ROC AUC values were 0.868, 0.799 and 0.623 when testing agonists against inactive compounds, decoys and antagonists (
Figure 4C), and 0.780 when testing against all non-agonists, respectively (
Figure 4D). Thus, the best performance was observed when inactive ligands were treated as negatives. Notably, the molecular docking experiment showed greater accuracies in predicting true antagonists than true agonists.
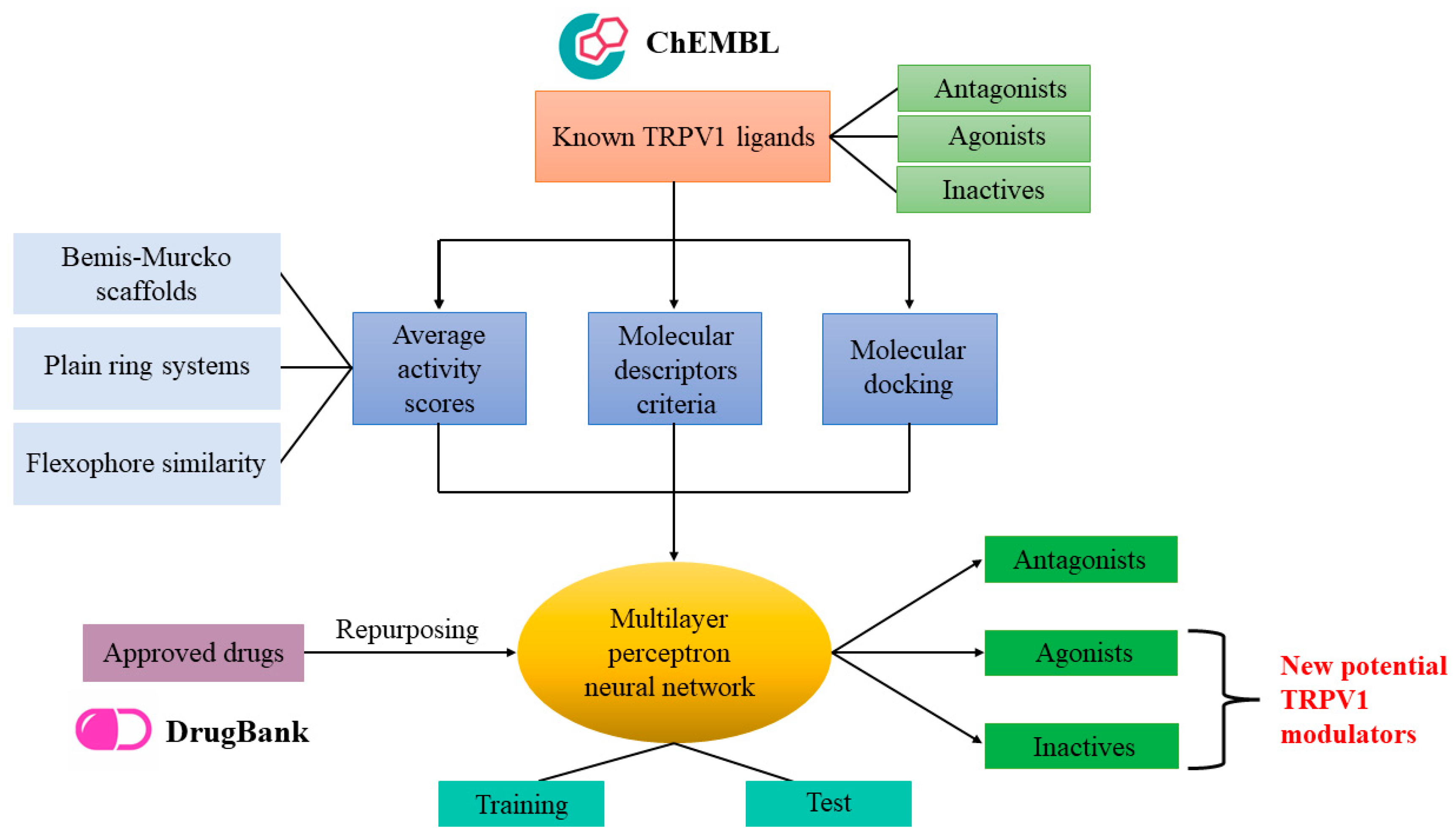
3.5. Integrated Predictive Model Based on Neural Networks
After establishing activity scores, the number of satisfied descriptor criteria, and binding affinities and efficacies for antagonists, agonists and inactive molecules, these data were integrated into one global predictive model in order to increase the predictive accuracy by adding weights to each of the aforementioned parameters. Since antagonist, agonist and inactive datasets are rather unbalanced, we generated the machine learning model using only the compounds that were selected for molecular docking, thus creating a more balanced training dataset. The machine learning algorithm that we selected for this task was the multilayer perceptron neural network since it also allows the prediction of multiple classes. The architecture with the most optimal parameters had the following characteristics: six input nodes (average activity scores and satisfied descriptor criteria for both antagonists and agonists, binding energies for antagonists, LELP values for agonists), one hidden layer with four neurons (which, in fact, represents the geometric mean between the number of input and output nodes) activated with tanh function, and the output layer with three nodes corresponding to the probabilities for each of the three classes, generated with the softmax function. The most optimal values for the hyperparameters were 0.5 for initial learning rate, 0.7 for momentum and 25 for maximum number of epochs.
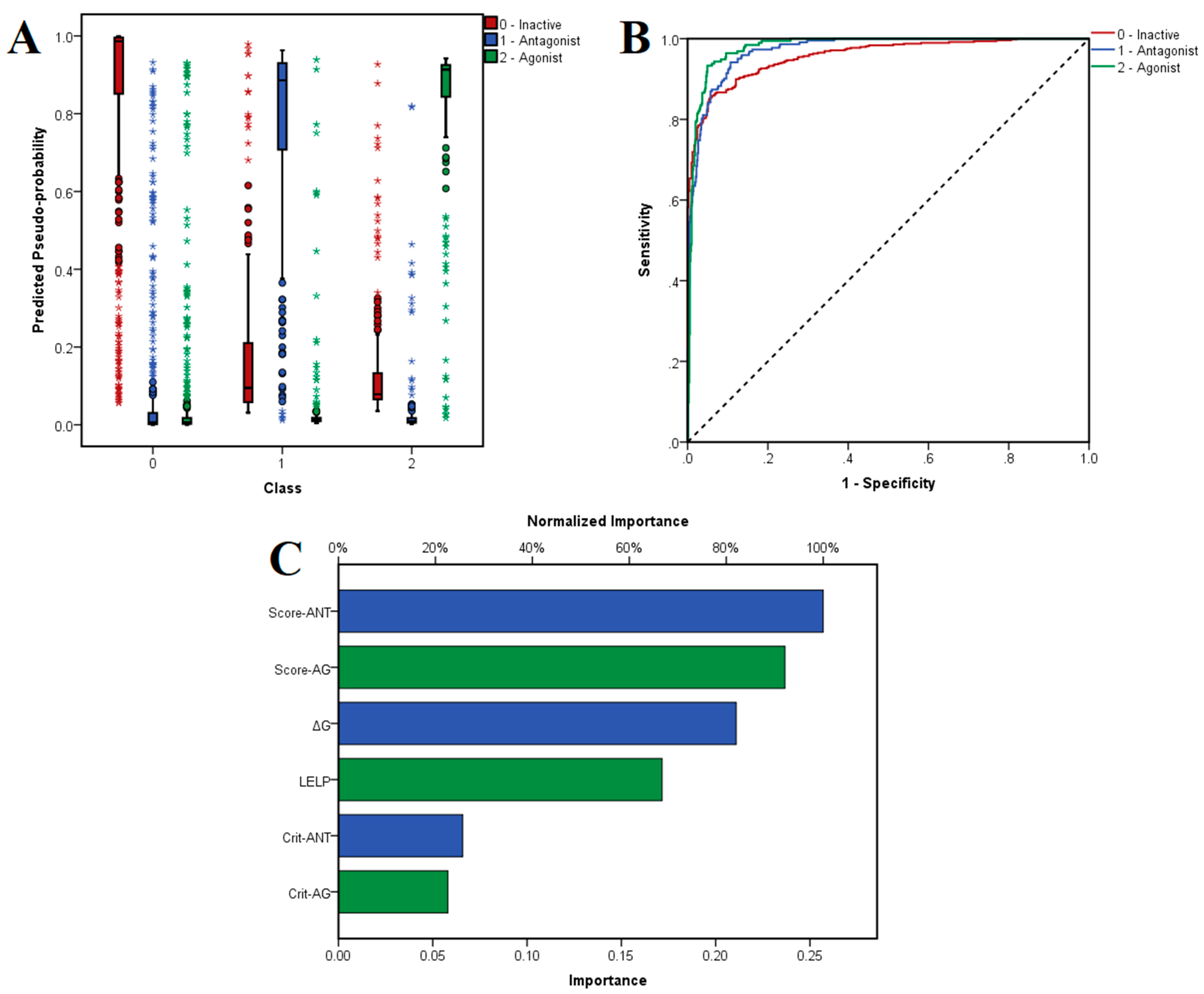
The chosen neural network correctly predicted 86.3% of inactive molecules, 86.2% of antagonists and 89.4% of antagonists in the training set, with an overall 86.9% prediction accuracy. Moreover, the trained model did not suffer from overfitting, since the algorithm correctly predicted 90.7% of the ligands from the test set: 89.7% of inactive compounds, 90.0% of antagonists and 94.3% of agonists. The distribution of the predicted pseudo-probabilities is shown in
Figure 5A. The overall prediction accuracy was 88.8% for predicting inactive compounds, 92.04% for antagonists and 93.70% for agonists, the model showing better accuracies for correctly predicting agonists and antagonists (
Table 4). ROC AUC values were 0.957 for classifying inactive ligands, 0.972 for antagonists and 0.980 for agonists (
Figure 5B). The generated classification model showed higher values for specificity over sensitivity, and thus the algorithm identifies true negatives relatively more accurately than true positives.
The independent variables with the highest importance in predicting the three classes were average activity scores for antagonists and agonists, followed by binding energy and LELP, while the numbers of satisfied molecular descriptor criteria for antagonists and agonists had the lowest weights (
Figure 5C).
3.6. Prediction of Potential TRPV1 Modulators through Drug Repurposing
The 1981 approved drugs retrieved from the DrugBank database were subjected to the virtual screening protocol to identify potentially new TRPV1 modulators. Firstly, graph mining techniques were used with DataWarrior software to append appropriate activity scores for the repurposing molecules. Then, scaffold analysis and flexophore similarity search were performed to retrieve Bemis–Murcko, plain rings and cluster similarity scores for approved drugs that had similar structural features with experimentally determined TRPV1 antagonists, agonists and inactive molecules. After performing the analysis, average activity scores varied between 0 and 6.23 (1.42 ± 1.151) for predicting antagonists and between 0 and 4.40 (0.41 ± 0.549) for predicting agonists. Only five compounds had higher activity scores than the mean score of antagonists (5.79) and only one drug showed a higher activity score than the mean value for agonists (3.01).
Among the screened compounds, 686 (34.6%) have in their structure Bemis–Murcko skeletons present among TRPV1 antagonists, and 725 (36.6%) molecules have BM skeletons observed within agonists. A total of 1348 (68%) compounds contain plain rings contained by structures of antagonists, and 1406 (80%) drugs contain plain rings commonly found within agonists, which denotes that agonists contain aromatic or non-aromatic rings that are not highly specific to TRPV1 agonistic activity. After performing the similarity search based on flexophore descriptors, we found that 333 (16.8%) and 146 (7.4%) approved drugs shared structural similarities higher than 0.8 with antagonists and agonists, respectively.
Furthermore, 1D and 2D molecular descriptors were generated for marketed drugs to establish the number of relevant molecular features that are shared with known TRPV1 ligands. We previously showed that a compound should satisfy at least five antagonist-specific molecular descriptor criteria to be classified as an antagonist and at least four agonist-specific criteria to be considered an agonist, with a good balance between sensitivity and specificity. After applying the thresholds for the 16 chosen molecular descriptors, 315 (15.9%) molecules satisfied the condition of being classified as antagonists, and 1121 (56.6%) compounds met the criteria for being considered potential agonists. These results hinted toward the fact that the minimum required number of descriptor criteria for classifying agonists is too permissive. On another note, we observed while training the neural networks that the machine learning algorithm performed more accurately when we used the total number of the satisfied descriptor criteria instead of the binary categorical values (1 for active antagonist or agonist, 0 for inactive) as classifiers. The last two observations led to the treatment of the molecular descriptor-derived property as an ordinal independent variable, rather than dichotomous.
Molecular docking experiments were carried out to estimate binding energies after predicting the interaction with a closed-state conformation of TRPV1, and the ligand efficiency-dependent lipophilicity index after simulating interactions with the agonist-bound conformation of the ion channel. Among the screened drugs, 79 (4%) molecules showed lower binding energies than the mean value for TRPV1 antagonists (−9.56 kcal/mol) after docking into the antagonist-specific binding site. After simulating the interactions with the agonist-specific conformation of the binding site, 276 (13.9%) compounds had lower energies than the mean value for agonists (−8.51 kcal/mol), and 147 (7.4%) showed LELP values higher than the mean for agonists (18.90).
The distribution of the established input variables for class prediction is depicted in
Figure S7. As observed, only the number of satisfied descriptor criteria for predicting agonists, predicted binding energies and calculated LELP followed normal distributions among the screened drugs.
The independent variables that were determined after performing the prerequisite screening were fed into the validated neural network. The distribution of the estimated probabilities corresponding to each of the predicted classes yielded after applying the integrated machine learning model is illustrated in
Figure 6A. The implemented model predicted 1112 (56.1%) compounds as inactive molecules, 258 (13%) as potential antagonists and 572 (28.9%) as potential agonists, if a 50% probability threshold was applied for being considered a positive. Moreover, 116 (5.9%) drugs from the predicted antagonist class and 210 (10.6%) from the predicted agonist class showed probabilities over 90%.
The top 10 (0.5%) predicted antagonists ranked by probabilities and their established pharmacological activity are shown in
Table 5, while the top 10 predicted agonists are shown in
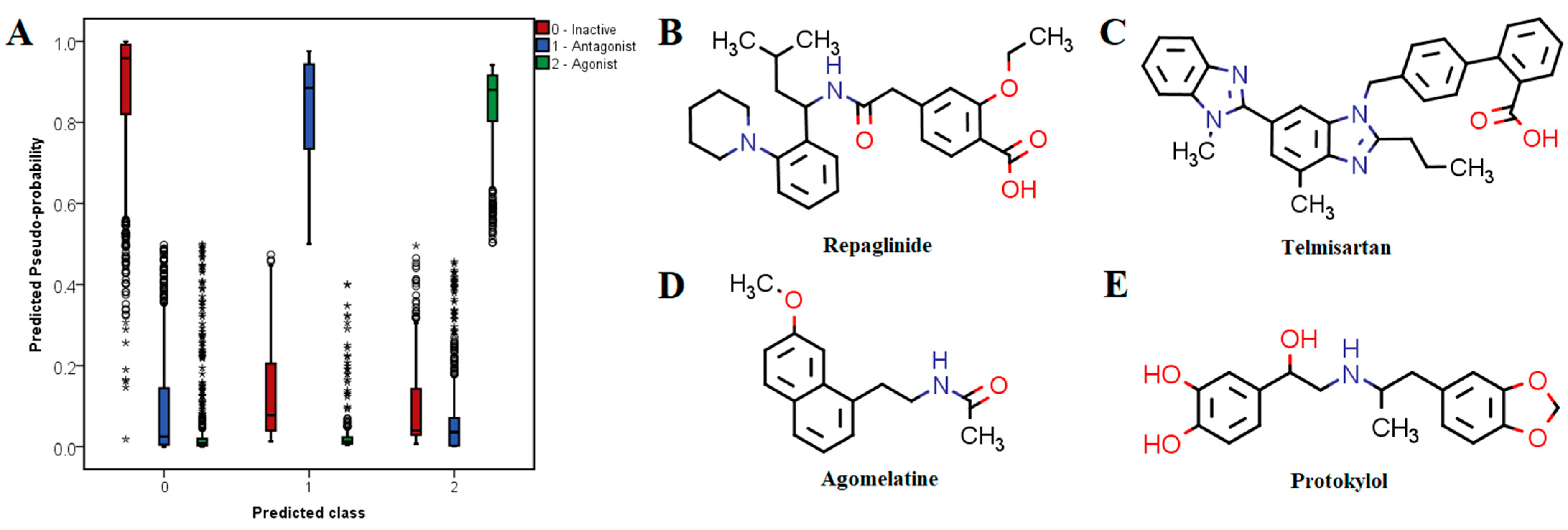
Table 6. Approved drugs with high probabilities of being active antagonists are very structurally and pharmacologically diverse. For instance, the top 10 predicted antagonists have entirely different therapeutic indications and had calculated probabilities over 95%. The top three potential antagonists were repaglinide (antihyperglycemic agent, ATP-dependent potassium channel blocker), telmisartan (antihypertensive, angiotensin II receptor blocker) and tafenoquine (antimalarial agent).
Some of the drugs with high predicted probabilities for acting as TRPV1 agonists were adrenergic receptor modulators, either sympathomimetics (e.g., protokylol, ephedrine, etilephrine and formoterol) or β-blockers (e.g., bisoprolol, esmolol, practolol, celiprolol, sotalol and labetalol), indicating that these molecules share common features with TRPV1 agonists. However, timolol, nadolol, carteolol and prazosin were predicted as potential antagonists. Lidocaine and other local anesthetics (e.g., butacaine, prilocaine, oxetacaine and procaine) were also identified as potential agonists. Calcitriol (active form of vitamin D) showed the highest probability of exerting TRPV1 agonist activity. Moreover, vitamin A and its derivatives (alitretinoin, isotretinoin) were identified among the top 10 potential agonists. Most of these compounds have high lipophilicity, which is a common property for TRPV1 agonists as seen from molecular descriptor analysis and LELP values.
The most promising candidates for repurposing as TRPV1 modulators with pharmacotherapeutic utility in pain relief were chosen based on three criteria: high probability of being active, favorable interactions with relevant residues within the binding site of TRPV1 and acceptable safety profiles. Therefore, three potential antagonists (repaglinide, telmisartan and agomelatine) and one potential agonist (protokylol) were proposed as repositioning candidates and were discussed in further detail (
Figure 6B–E).
Repaglinide, an antidiabetic drug acting as a blocker of ATP-dependent potassium channels [
54], showed the highest probability of blocking TRPV1 and had an average activity score of 5.42, its structure being characterized by a BM-24 skeleton (2-(4-cyclohexylbutyl)-1,1′-bi(cyclohexane)). Moreover, repaglinide contains in its molecule two phenyl rings (PR-1) and a piperidine scaffold (PR-5), both being present in structures of TRPV1 antagonists. Repaglinide also has a flexophore similarity of 87% with TRPV1 antagonist CHEMBL1779679. Furthermore, repaglinide satisfied three out of eight proposed molecular descriptor criteria, having in its molecule one secondary nitrogen, one hexa-atomic ring containing heteroatoms and six hydrogen bond acceptors. Telmisartan, an antihypertensive agent that blocks angiotensin II receptors [
55] showed an average activity score of 4.29, having also the second highest predicted probability of blocking TRPV1. Its structure contained no antagonist-specific BM scaffold, but the benzodiazole (PR-21) and benzene (PR-1) rings had high contributions to the overall score. Moreover, telmisartan had a flexophore similarity of 80.5% with TRPV1 antagonist CHEMBL3961718. Telmisartan satisfied only two molecular descriptor criteria, the measure of electronegative atom count relative to molecular size and molecular complexity. Another interesting potential TRPV1 antagonist was agomelatine, a melatonin naphthalene analog used to treat depressive disorders [
56], which had an estimated probability of binding to the channel of 80.25%. This atypical antidepressant had an average activity score of 4.00, its structure being derived from the decahydronaphthalene BM skeleton (BM-90) or naphthalene scaffold (PR-16), which are present among potent TRPV1 antagonists. Agomelatine had 83.1% flexophore similarity with TRPV1 antagonist CHEMBL400371. In fact, the discovered flexophore similarity pair is characterized by strikingly similar structures, since the two molecules share a large structural fragment ((7-methoxynaphthalen-1-yl)ethyl), while the acetamide moiety from agomelatine is replaced with (trifluoromethoxy)benzamide in the TRPV1 antagonist. Although agomelatine contains a secondary nitrogen, its structure did not satisfy any molecular descriptor criteria for antagonists.
Bronchodilator protokylol [
57] showed the second highest estimated probability for acting as a TRPV1 agonist. Protokylol had an average activity score of 2.76, its molecule containing the 5-(5-cyclohexylpentyl)-octahydro-1H-indene BM skeleton (BM-126), represented by the benzene (PR-1) and benzodioxole (PR-44) plain rings, these structural features being also present among TRPV1 agonists. A flexophore similarity match of 87.8% was observed between protokylol and nylidrin. Nylidrin (buphenine) is a β2-adrenoreceptor agonist acting as a vasodilator (withdrawn for lack of effectiveness) [
58], which has 15.85 µm potency for TRPV1 agonist activity (pEC
50 = 4.8 M), the result being declared inconclusive in the ChEMBL database. Protokylol met six out of eight molecular descriptor criteria for acting as a TRPV1 agonist, the only violated thresholds being for XlogP and hybridization ratio.
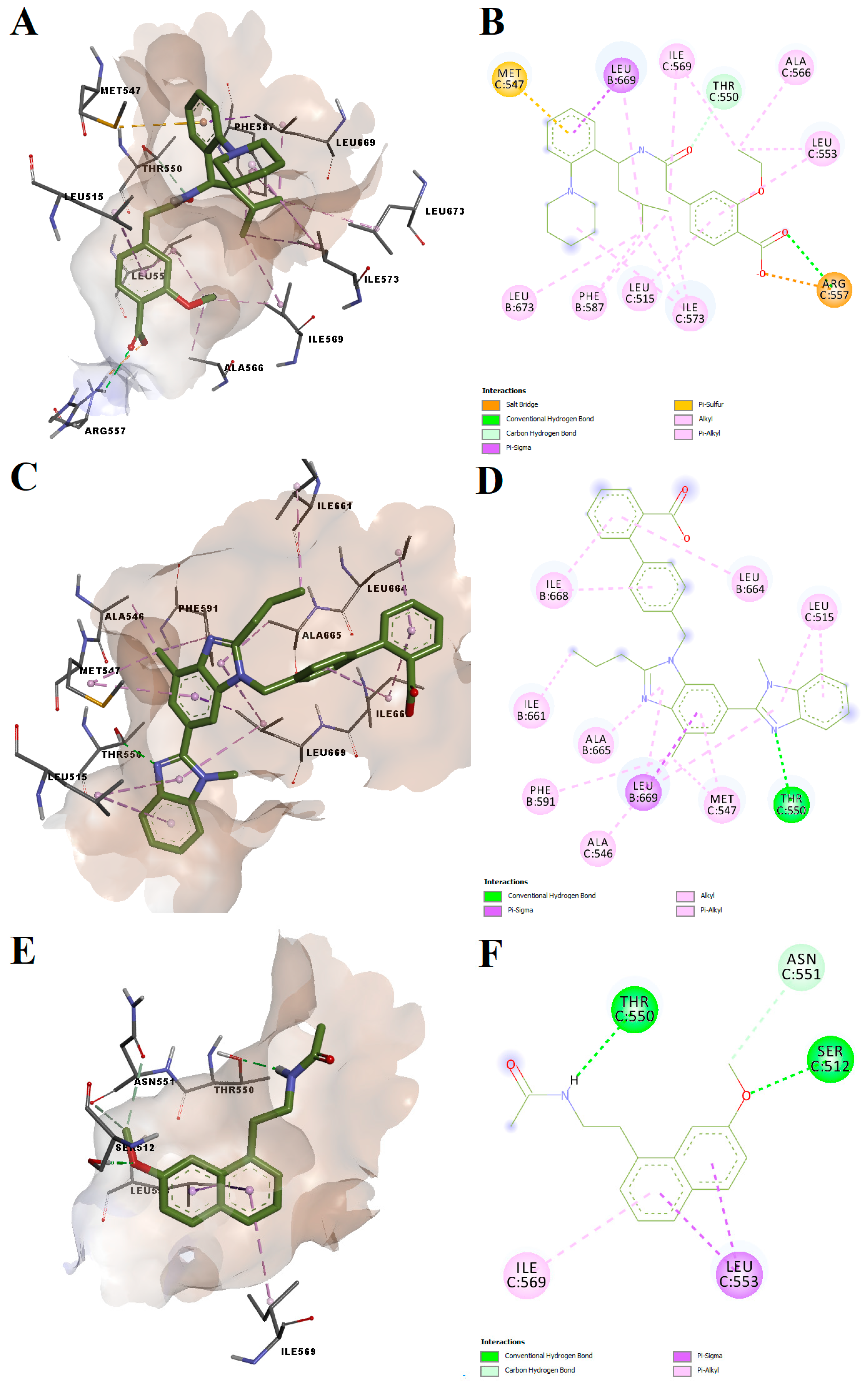
Repaglinide, telmisartan and agomelatine had binding energies after docking into the antagonist-specific conformation of the vanilloid pocket of −7.78, −9.20 and −7.69 kcal/mol, respectively. The predicted conformations of the three potential antagonists were not highly torsioned, while repaglinide and agomelatine adopted spatial orientations similar to antagonist CPZ (
Figure 7). All three ligands formed hydrogen bonds or carbon–hydrogen bonds with Thr550, located in the loop between TM4 and 5, which was shown to be a key residue for TRPV1 modulation [
32,
33]. Moreover, agomelatine forms a hydrogen bond with Ser512 from the loop connecting TM2 and TM3, while repaglinide forms a hydrogen bond and a salt bridge with Arg557, a residue that is relevant for CPZ antagonist activity. For instance, CPZ forms hydrogen bonds with Thr550, Ser512 and Arg557. Multiple non-polar interactions were also observed between the docked ligands and residues within the vanilloid binding pocket, the docked ligands showing high potential for blocking the TRPV1 channel by acting on the CPZ binding site. On the other hand, the structure of telmisartan lacks a vanillyl head analogous scaffold and therefore did not bind as deep into the vanilloid pocket as other ligands.
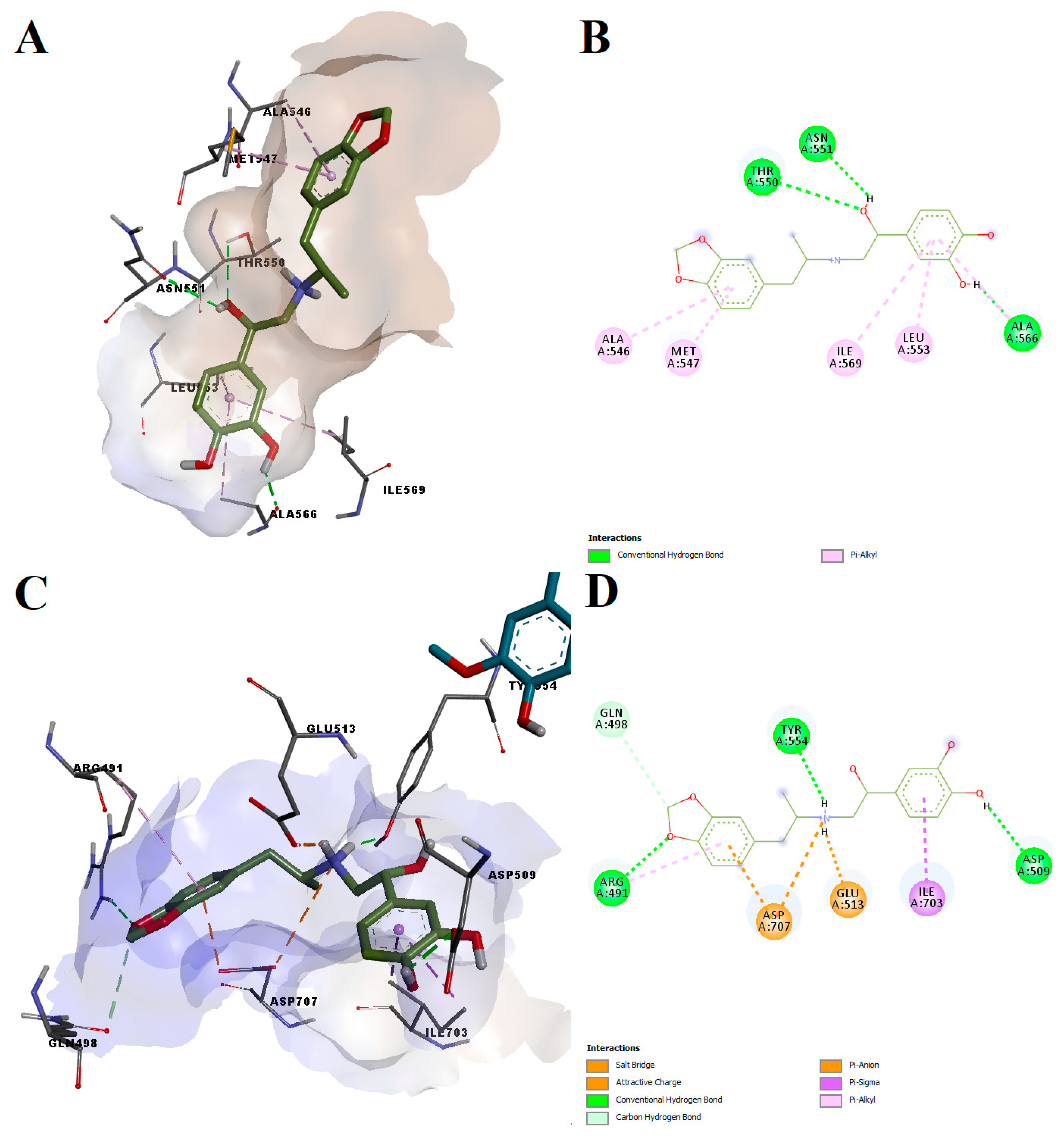
Protokylol showed a predicted binding energy of −8.27 kcal/mol after simulating the interaction with the agonist-bound conformation of the TRPV1 active site, the calculated LELP being 5.52. Similar to the other analyzed ligands, protokylol fit very well within the binding cavity (
Figure 8A), showing similar orientations with known TRPV1 ligands. Protokylol formed three hydrogen bonds with Thr550, Asn551 and Ala566 and multiple non-polar pi-alkyl interactions with other residues (
Figure 8B). Interestingly, after analyzing the third predicted conformation, we found that protokylol could potentially bind to the phosphoinositides-specific binding site (−7.52 kcal/mol,
Figure 8C,D). This binding site overlaps with the vanilloid binding pocket and can inhibit capsaicin-induced activation of TRPV1 [
59]. Protokylol formed a salt bridge and a pi-anion interaction with Asp707 and a pi-sigma interaction with Ile703, residues that were shown to be essential for TRPV1 modulation by phosphoinositides [
59]. Additionally, the protonated secondary amine moiety also interacted with Glu513 through attractive charges. Moreover, three hydrogen bonds and one carbon–hydrogen bond were formed with other residues.













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



















