
Effects of Isosakuranetin on Pharmacokinetic Changes of Tofacitinib in Rats with N-Dimethylnitrosamine-Induced Liver Cirrhosis
 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Animals
2.3. Induction of Liver Cirrhosis
2.4. Preliminary Study
2.5. Rat Plasma Protein Binding of Tofacitinib
2.6. Intravenous and Oral Administration of Tofacitinib
2.7. Tissue Distribution of Tofacitinib
2.8. Measurement of Vmax, Km and CLint
2.9. Immunoblot Analysis
2.10. HPLC Analysis
2.11. Pharmacokinetic Analysis
2.12. Statistical Analysis
3. Results
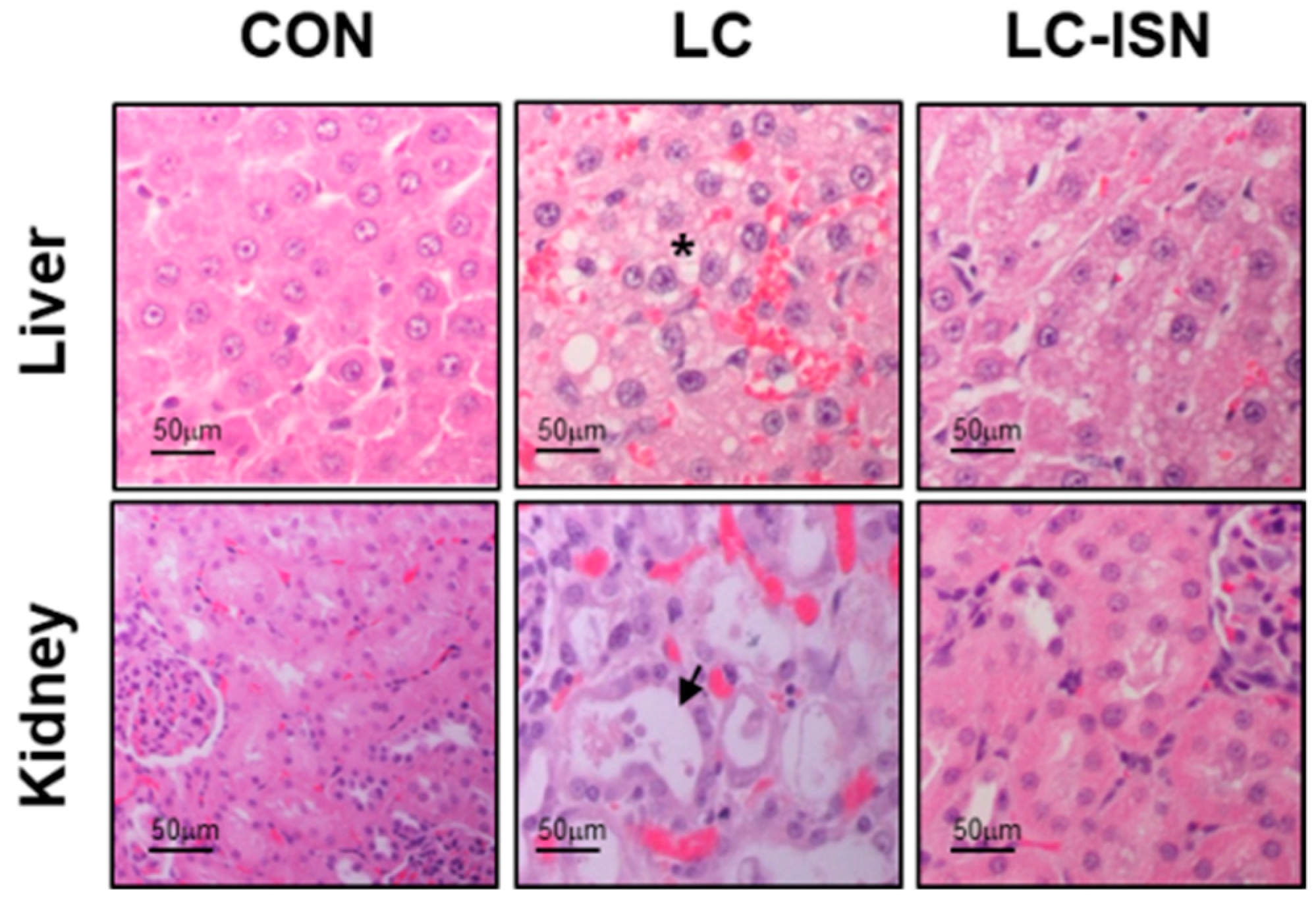
3.1. Preliminary Study
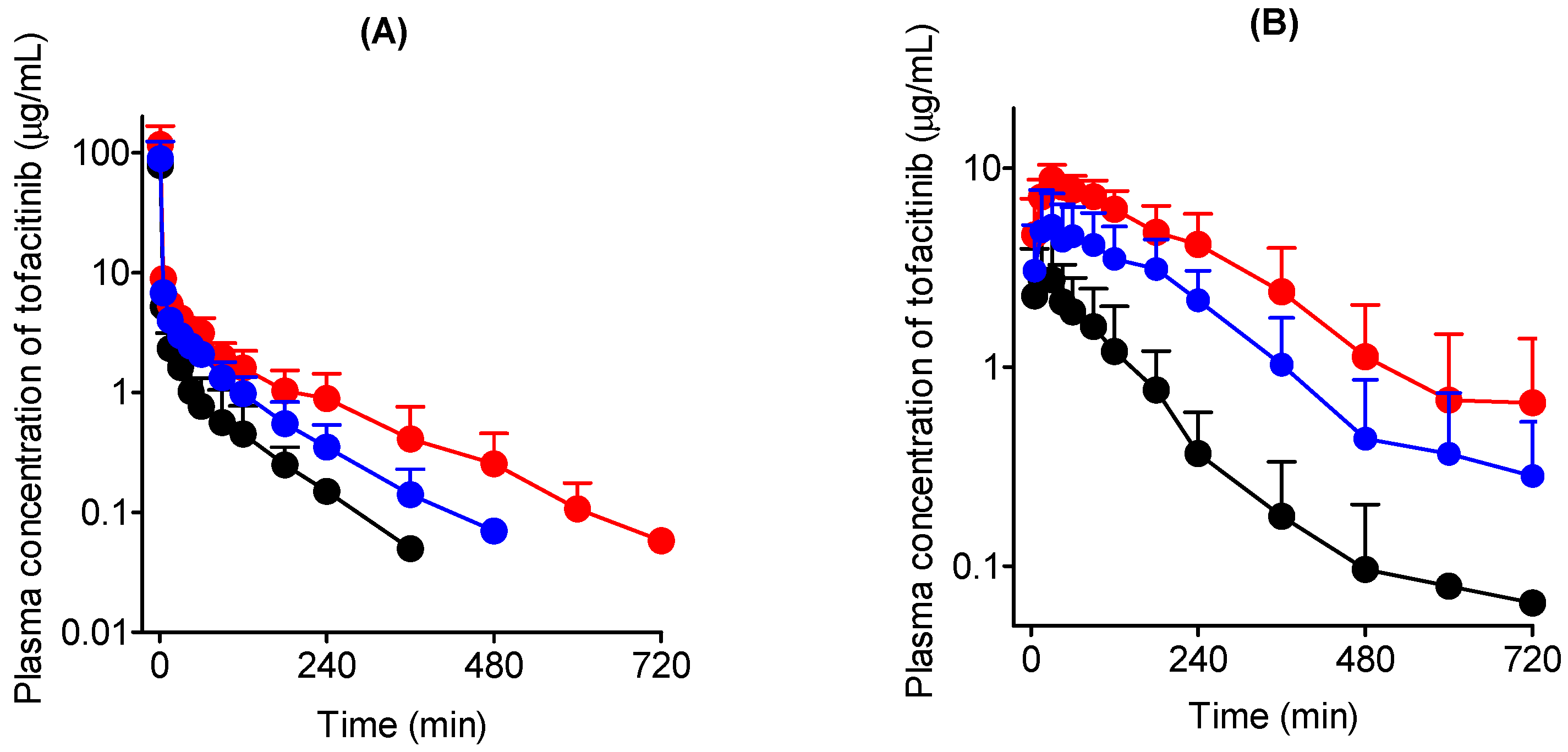
3.2. Intravenous Administration of Tofacitinib in Rats
3.3. Oral Administration of Tofacitinib in Rats
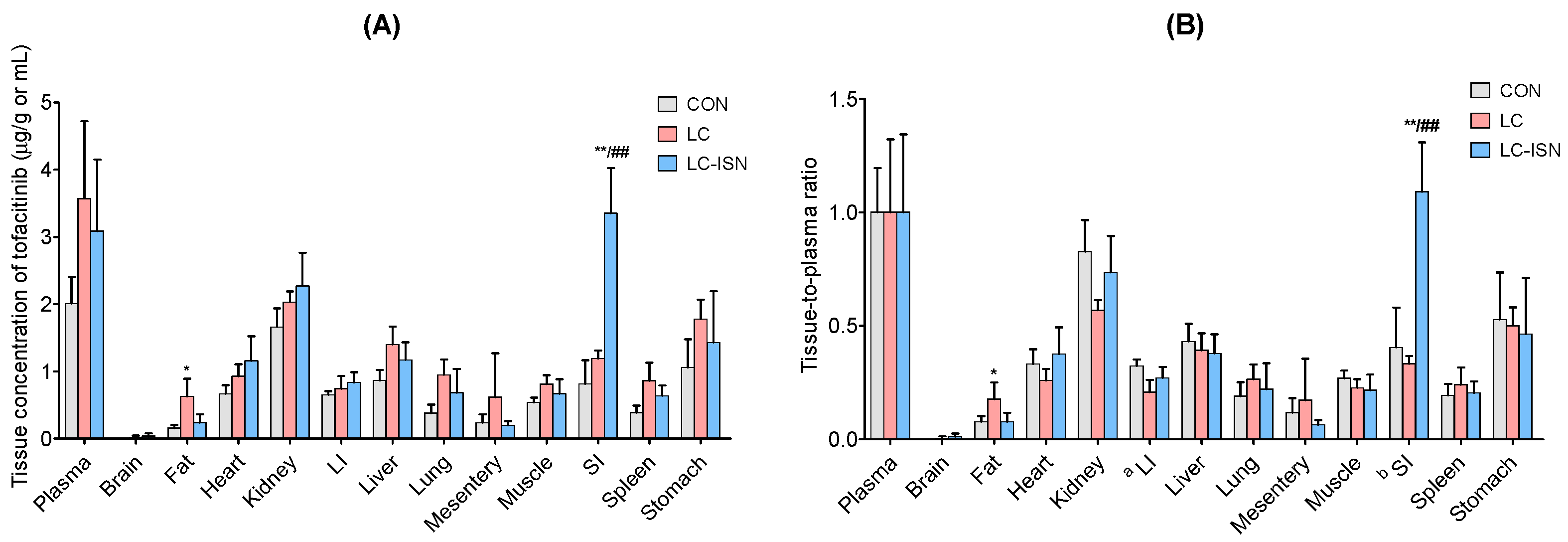
3.4. Tissue Distribution of Tofacitinib
3.5. In Vitro Metabolism of Tofacitinib
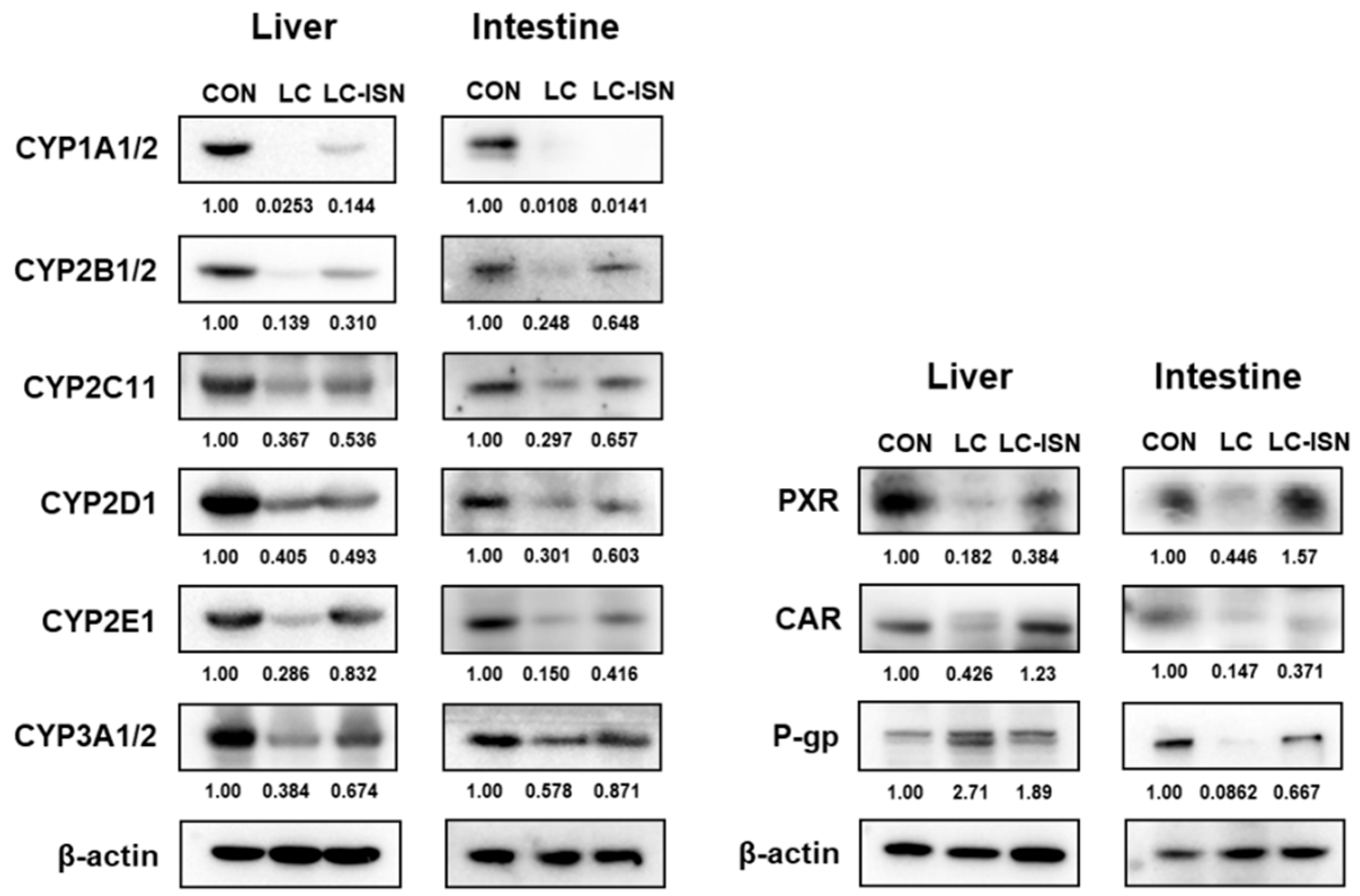
3.6. Expression of CYP Isoforms and Other Involved Proteins in the Hepatic and Intestinal Microsomes
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Claxton, L.; Taylor, M.; Soonasra, A.; Bourret, J.A.; Gerber, R.A. An economic evaluation of tofacitinib treatment in rheumatoid arthritis after methotrexate or after 1 or 2 TNF inhibitors from a U.S. payer perspective. J. Manag. Care Spec. Pharm. 2018, 24, 1010–1017. [Google Scholar] [CrossRef] [PubMed]
- Palmroth, M.; Kuuliala, K.; Peltomaa, R.; Virtanen, A.; Kuuliala, A.; Kurttila, A.; Kinnunen, A.; Leirisalo-Repo, M.; Silvennoinen, O.; Isomäki, P. Tofacitinib Suppresses Several JAK-STAT Pathways in Rheumatoid Arthritis In Vivo and Baseline Signaling Profile Associates With Treatment Response. Front. Immunol. 2021, 12, 738481. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT signaling pathway: From bench to clinic. Signal Transduct. Target. Ther. 2021, 6, 402. [Google Scholar] [CrossRef] [PubMed]
- Sandborn, W.J.; Peyrin-Biroulet, L.; Sharara, A.I.; Su, C.; Modesto, I.; Mundayat, R.; Gunay, L.M.; Salese, L.; Sands, B.E. Efficacy and safety of tofacitinib in ulcerative colitis based on prior tumor necrosis factor inhibitor failure status. Clin. Gastroenterol. Hepatol. 2022, 20, 591–601. [Google Scholar] [CrossRef]
- Fukuda, T.; Naganuma, M.; Kanai, T. Current new challenges in the management of ulcerative colitis. Intest. Res. 2019, 17, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Bachelez, H.; Van de Kerkhof, P.C.; Strohal, R.; Kubanov, A.; Valenzuela, F.; Lee, J.H.; Gupta, P. Tofacitinib versus etanercept or placebo in moderate-to-severe chronic plaque psoriasis: A phase 3 randomised non-inferiority trial. Lancet 2015, 386, 552–561. [Google Scholar] [CrossRef]
- Papp, K.A.; Menter, M.A.; Abe, M.; Elewski, B.; Feldman, S.R.; Gottlieb, A.B.; Langley, R.; Luger, T.; Thaci, D.; Buonanno, M.; et al. OPT Pivotal 1 and OPT Pivotal 2 investigators. Tofacitinib, an oral Janus kinase inhibitor, for the treatment of chronic plaque psoriasis: Results from two randomized, placebo-controlled, phase III trials. Br. J. Dermatol. 2015, 173, 949–961. [Google Scholar] [CrossRef]
- Hogan, S.; Wang, S.; Ibrahim, O.; Piliang, M.; Bergfeld, W. Long-term treatment with tofacitinib in severe alopecia areata: An update. J. Clin. Aesthet. Dermatol. 2019, 12, 12–14. [Google Scholar]
- Levy, L.L.; Urban, J.; King, B.A. Treatment of recalcitrant atopic dematitis with the oral Janus kinase inhibitor tofacitinib citrate. J. Am. Acad. Dermatol. 2015, 73, 395–399. [Google Scholar] [CrossRef]
- Mohanakrishnan, R.; Beier, S.; Deodhar, A. Tofacitinib for the treatment of active ankylosing spondylitis in adults. Expert Rev. Clin. Immunol. 2022, 18, 273–280. [Google Scholar] [CrossRef]
- Dowty, M.E.; Lin, J.; Ryder, T.F.; Wang, W.; Walker, G.S.; Vaz, A.; Prakash, C. The pharmacokinetics, metabolism, and clearance mechanisms of tofacitinib, a janus kinase inhibitor, in humans. Drug Metab. Dispos. 2014, 42, 759–773. [Google Scholar] [CrossRef] [PubMed]
- Bannwarth, B.; Kostine, M.; Poursac, N. A pharmacokinetic and clinical assessment of tofacitinib for the treatment of rheumatoid arthritis. Expert Opin. Drug Metab. Toxicol. 2013, 9, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Kim, S.H. Dose-dependent pharmacokinetics of tofacitinib in rats: Influence of hepatic and intestinal first-pass metabolism. Pharmaceutics 2019, 11, 318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radovanović-Dinić, B.; Tešić-Rajković, S.; Zivkovic, V.; Grgov, S. Clinical connection between rheumatoid arthritis and liver damage. Rheumatol. Int. 2018, 38, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Meng, Y.; Han, Z.; Ye, F.; Wei, L.; Zong, C. Mesenchymal stem cell therapy for liver disease: Full of chances and challenges. Cell biosci. 2020, 10, 123. [Google Scholar] [CrossRef] [PubMed]
- Pinter, M.; Trauner, M.; Peck-Radosavljevic, M.; Sieghart, W. Cancer and liver cirrhosis: Implications on prognosis and management. ESMO Open. 2016, 1, e000042. [Google Scholar] [CrossRef] [Green Version]
- Peterson, J.J.; Dwyer, J.T.; Beecher, G.R.; Bhagwat, S.A.; Gebhardt, S.E.; Haytowitz, D.B.; Holden, J.M. Flavanones in oranges, tangerines (mandarins), tangors, and tangelos: A compilation and review of the data from the analytical literature. J. Food Compos. Anal. 2006, 19, S66–S73. [Google Scholar] [CrossRef]
- Straub, I.; Krügel, U.; Mohr, F.; Teichert, J.; Rizun, O.; Konrad, M.; Oberwinkler, J.; Schaefer, M. Flavanones that selectively inhibit TRPM3 attenuate thermal nociception in vivo. Mol. Pharmacol. 2013, 84, 736–750. [Google Scholar] [CrossRef] [Green Version]
- Jia, S.; Zhang, Y.; Yu, J. Antinociceptive effects of isosakuranetin in a rat model of peripheral neuropathy. Pharmacology 2017, 100, 201–207. [Google Scholar] [CrossRef]
- Lee, U.; Oh, E. Pharmacokinetic changes of drugs in a rat model of liver cirrhosis induced by dimethylnitrosamine, alone and in combination with diabetes mellitus induced by streptozotocin. Biopharm. Drug Dispos. 2015, 36, 1–14. [Google Scholar] [CrossRef]
- Bae, S.K.; Lee, S.J.; Kim, T.; Kim, J.W.; Lee, I.; Kim, S.G.; Lee, M.G. Pharmacokinetics and therapeutic effects of oltipraz after consecutive or intermittent oral administration in rats with liver cirrhosis induced by dimethylnitrosamine. J. Pharm. Sci. 2006, 95, 985–997. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Yoon, S.; Moon, J.O. The flavonoid naringenin inhibits dimethylnitrosamine-induced liver damage in rats. Biol. Pharm. Bull. 2004, 27, 72–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chooi, K.F.; Kuppan Rajendran, D.B.; Phang, S.S.; Toh, H.H. The dimethylnitrosamine induced liver fibrosis model in the rat. J.Vis. Exp. 2016, 112, e54208. [Google Scholar] [CrossRef] [PubMed]
- Ullah, H.; Khan, A.; Baig, M.W.; Ullah, N.; Ahmed, N.; Tipu, M.K.; Ali, H.; Khan, S. Poncirin attenuates CCl4-induced liver injury through inhibition of oxidative stress and inflammatory cytokines in mice. BMC complement. Med. Ther. 2020, 20, 115. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Lee, J.S.; Lee, M.G. Stability, blood partition and plasma protein binding of ipriflavone, an isoflavone derivative. Biopharm. Drug Dispos. 1999, 20, 355–360. [Google Scholar] [CrossRef]
- Svein, Ø.; Theodor, W.G. Comparison of equilibrium time in dialysis experiments using spiked plasma or spiked buffer. J. Pharm. Sci. 1982, 71, 127–128. [Google Scholar]
- Shin, W.G.; Lee, M.G.; Lee, M.H.; Kim, N.D. Factors influencing the protein binding of vancomycin. Biopharm. Drug Dispos. 1991, 12, 637–646. [Google Scholar] [CrossRef]
- Kim, J.E.; Park, M.Y.; Kim, S.H. Simple determination and quantification of tofacitinib, a JAK inhibitor, in rat plasma, urine and tissue homogenates by HPLC and its application to a pharmacokinetic study. J. Pharm. Investig. 2020, 50, 603–612. [Google Scholar] [CrossRef]
- Park, H.J.; Bae, S.H.; Kim, S.H. Dose-independent pharmacokinetics of loganin in rats: Effect of intestinal first-pass metabolism on bioavailability. J. Pharm. Investig. 2021, 51, 767–776. [Google Scholar] [CrossRef]
- Gwak, E.H.; Yoo, H.Y.; Kim, S.H. Effects of diabetes mellitus on the disposition of tofacitinib, a Janus kinase inhibitor, in rats. Biomol. Ther. 2020, 28, 361–369. [Google Scholar] [CrossRef]
- Bae, S.H.; Chang, S.Y.; Kim, S.H. Slower elimination of tofacitinib in acute renal failure rat Models: Contribution of hepatic metabolism and renal excretion. Pharmaceutics 2020, 12, 714. [Google Scholar] [CrossRef] [PubMed]
- Duggleby, R.G. Analysis of enzyme progress curves by nonlinear regression. Methods Enzymol. 1995, 249, 61–90. [Google Scholar] [PubMed]
- Bae, S.H.; Park, J.H.; Choi, H.G.; Kim, H.; Kim, S.H. Imidazole antifungal drugs inhibit the cell proliferation and invasion of human breast cancer cells. Biomol. Ther (Seoul). 2018, 26, 494–502. [Google Scholar] [CrossRef]
- Gibaldi, M.; Perrier, D. Pharmacokinetics, 2nd ed.; Marcel-Dekker: New York, NY, USA, 1982. [Google Scholar]
- Chiou, W.L. Critical evaluation of the potential error in pharmacokinetic studies of using the linear trapezoidal rule method for the calculation of the area under the plasma level-time curve. J. Pharmacokinet. Biopharm. 1978, 6, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Goeting, N.L.; Fleming, J.S.; Gallagher, P.; Walmsely, B.H.; Karran, S.J. Alterations in liver blood flow and reticuloendothelial function in progressive cirrhosis in the rat. J. Nucl. Med. 1986, 27, 1751–1754. [Google Scholar] [PubMed]
- Bastien, M.C.; Leblond, F.; Pichette, V.; Villeneuve, J.P. Differential alteration of cytochrome P450 isoenzymes in two experimental models of cirrhosis. Can. J. Physiol. Pharmacol. 2000, 78, 912–919. [Google Scholar] [CrossRef] [PubMed]
- Burk, O.; Koch, I.; Raucy, J.; Hustert, E.; Eichelbaum, M.; Brockmöller, J.; Zanger, U.M.; Wojnowski, L. The induction of cytochrome P450 3A5 (CYP3A5) in the human liver and intestine is mediated by the xenobiotic sensors pregnane X receptor (PXR) and constitutively activated receptor (CAR). J. Biol. Chem. 2004, 279, 38379–38385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanada, K.; Nakai, K.; Tanaka, H.; Suzuki, F.; Kumada, H.; Ohno, Y.; Ozawa, S.; Ogata, H. Effect of nuclear receptor downregulation on hepatic expression of cytochrome P450 and transporters in chronic hepatitis C in association with fibrosis development. Drug Metab. Pharmacokinet. 2012, 27, 301–306. [Google Scholar] [CrossRef] [Green Version]
- Daujat-Chavanieu, M.; Gerbal-Chaloin, S. Regulation of CAR and PXR expression in health and disease. Cells 2020, 9, 2395. [Google Scholar] [CrossRef]
- Hussa, D.A. 2013 new drug update: What do new approvals hold for the elderly? Consult. Pharm. 2014, 29, 224–238. [Google Scholar] [CrossRef]
- Grover, A.; Benet, L.Z. Effects of drug transporters on volume of distribution. AAPS J. 2009, 11, 250–261. [Google Scholar] [CrossRef] [Green Version]
- Jeong, H.J.; Lee, S.H.; Kang, H.E. Changes in digoxin pharmacokinetics associated with hepatic P-glycoprotein upregulation in rats with non-alcoholic fatty liver disease. Fundam. Clin. Pharmacol. 2021, 35, 1100–1108. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Miao, M.X.; Sun, B.B.; Wang, Z.J.; Tang, X.G.; Chen, Y.; Zhao, K.J.; Liu, X.D.; Liu, L. Acute liver failure enhances oral plasma exposure of zidovudine in rats by downregulation of hepatic UGT2B7 and intestinal P-gp. Acta. Pharmacol. Sin. 2017, 38, 1554–1565. [Google Scholar] [CrossRef] [PubMed]
- Aguirre Valadez, J.M.; Rivera-Espinosa, L.; Méndez-Guerrero, O.; Chávez-Pacheco, J.L.; García Juárez, I.; Torre, A. Intestinal permeability in a patient with liver cirrhosis. Ther. Clin. Risk Manag. 2016, 12, 1729–1748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Cho, Y.K.; Jung, Y.S.; Kim, Y.C.; Lee, M.G. Effects of Escherichia coli lipopolysaccharide on telithromycin pharmacokinetics in rats: Inhibition of metabolism via CYP3A. Antimicrob. Agents. Chemother. 2008, 52, 1046–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, L.; Zhou, X.; Huang, N.; Li, H.; Li, T.; Yao, K.; Tian, Y.; Hu, C.A.; Yin, Y. Functions of pregnane X receptor in self-detoxification. Amino acids. 2017, 49, 1999–2007. [Google Scholar] [CrossRef]
- Kobayashi, K.; Kuze, J.; Abe, S.; Takehara, S.; Minegishi, G.; Igarashi, K.; Kitajima, S.; Kanno, J.; Yamamoto, T.; Oshimura, M.; et al. CYP3A4 induction in the liver and intestine of pregnane X receptor/CYP3A-Humanized Mice: Approaches by mass spectrometry imaging and portal blood analysis. Mol. Pharmacol. 2019, 96, 600–608. [Google Scholar] [CrossRef]
- Hernández-Aquino, E.; Muriel, P. Beneficial effects of naringenin in liver diseases: Molecular mechanisms. World J. Gastroenterol. 2018, 24, 1679–1707. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | CON (n = 3) | LC (n = 3) | LC-ISN (n = 3) |
|---|---|---|---|
| Body weight (g) | |||
| Initial | 109 ± 1.15 | 105 ± 3.06 | 105 ± 2.65 |
| Final | 383 ± 14.9 | 188 ± 1.15 ** | 288 ± 67.8 |
| Plasma | |||
| Total protein (g/dL) | 6.85 ± 0.288 | 4.09 ± 0.731 ** | 6.17 ± 0.466 ## |
| Albumin (g/dL) | 3.74 ± 0.407 | 2.09 ± 0.484 ** | 3.24 ± 0.205 # |
| Urea nitrogen (mg/dL) | 11.7 ± 1.14 | 19.1 ± 6.58 | 13.0 ± 2.00 |
| GOT (IU/L) | 39.3 ± 12.3 | 111 ± 19.0 ** | 69.5 ± 19.5 |
| GPT (IU/L) | 20.0 ± 2.03 | 47.3 ± 11.3 * | 36.0 ± 6.22 |
| ALP (K-AU) | 50.8 ± 6.68 | 282 ± 89.6 ** | 118 ± 17.5 # |
| Total cholesterol (mg/dL) | 77.2 ± 6.22 | 142 ± 22.6 ** | 81.1 ± 11.5 ## |
| SCR (mg/dL) | 0.570 ± 0.0942 | 0.597 ± 0.0869 | 0.667 ± 0.0415 |
| CLCR (mL/min/kg) | 3.65 ± 0.823 | 1.71 ± 0.778 * | 2.43 ± 0.391 |
| Liver weight (% of body weight) | 3.25 ± 0.263 | 2.23 ± 0.490 * | 2.71 ± 0.257 |
| Kidney weight (% of body weight) | 0.734 ± 0.0279 | 1.08 ± 0.0379 ** | 0.856 ± 0.124 # |
| Plasma protein binding (%) | 25.1 ± 2.75 | 18.0 ± 1.18 ** | 19.7 ± 1.21 * |
| Parameters | CON (n = 5) | LC (n = 6) | LC-ISN (n = 7) |
|---|---|---|---|
| Body weight (g) | 430 ± 20.0 | 249 ± 67.6 *** | 309 ± 30.6 *** |
| Terminal half-life (min) | 35.4 ± 19.2 | 113 ± 43.7 ** | 58.2 ± 16.8 # |
| AUC (μg∙min/mL) | 440 ± 211 | 1135 ± 391 ** | 737 ± 107 # |
| CL (mL/min/kg) | 24.1 ± 6.27 | 11.0 ± 2.06 *** | 13.8 ± 1.73 *** |
| CLR (mL/min/kg) | 3.73 ± 1.09 | 4.65 ± 2.74 | 4.10 ± 1.64 |
| CLNR (mL/min/kg) | 21.1 ± 4.57 | 6.35 ± 1.27 *** | 9.68 ± 2.40 *** |
| Vss (mL/kg) | 429 ± 253 | 798 ± 177 | 800 ± 326 |
| Ae0–24 h (% of dose) | 13.9 ± 2.28 | 40.2 ± 18.9 * | 30.1 ± 12.3 |
| GI24 h (% of dose) | 0.164 ± 0.102 | 2.07 ± 2.12 | 0.166 ± 0.135 # |
| Parameters | CON (n = 6) | LC (n = 7) | LC-ISN (n = 7) |
|---|---|---|---|
| Body weight (g) | 386 ± 28.2 | 222 ± 36.6 *** | 255 ± 38.6 *** |
| AUC (μg∙min/mL) | 384 ± 122 | 2031 ± 500 *** | 1240 ± 128 ***,### |
| Cmax (μg/mL) | 3.53 ± 1.46 | 9.10 ± 1.26 *** | 6.08 ± 1.99 *,## |
| Tmax (min) | 50.0 ± 44.2 | 55.7 ± 36.4 | 85.7 ± 79.7 |
| CLR (mL/min/kg) | 6.98 ± 2.99 | 3.62 ± 1.78 | 5.71 ± 2.47 |
| Ae0–24 h (% of dose) | 11.8 ± 4.08 | 35.8 ± 16.9 * | 35.9 ± 16.8 * |
| GI24 h (% of dose) | 0.106 ± 0.101 | 1.73 ± 2.04 | 0.340 ± 0.288 |
| F (%) | 43.6 | 89.5 | 84.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bae, S.H.; Choi, H.G.; Park, S.Y.; Chang, S.-Y.; Kim, H.; Kim, S.H. Effects of Isosakuranetin on Pharmacokinetic Changes of Tofacitinib in Rats with N-Dimethylnitrosamine-Induced Liver Cirrhosis. Pharmaceutics 2022, 14, 2684. https://doi.org/10.3390/pharmaceutics14122684
Bae SH, Choi HG, Park SY, Chang S-Y, Kim H, Kim SH. Effects of Isosakuranetin on Pharmacokinetic Changes of Tofacitinib in Rats with N-Dimethylnitrosamine-Induced Liver Cirrhosis. Pharmaceutics. 2022; 14(12):2684. https://doi.org/10.3390/pharmaceutics14122684
Chicago/Turabian StyleBae, Sung Hun, Hyeon Gyeom Choi, So Yeon Park, Sun-Young Chang, Hyoungsu Kim, and So Hee Kim. 2022. "Effects of Isosakuranetin on Pharmacokinetic Changes of Tofacitinib in Rats with N-Dimethylnitrosamine-Induced Liver Cirrhosis" Pharmaceutics 14, no. 12: 2684. https://doi.org/10.3390/pharmaceutics14122684