
Identification of a Quinone Derivative as a YAP/TEAD Activity Modulator from a Repurposing Library

 , , ,
, , ,  , ,
, ,  , , ,
, , ,  and
and
Abstract
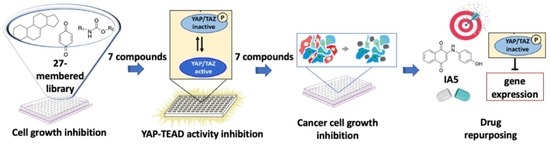
:
1. Introduction
2. Materials and Methods
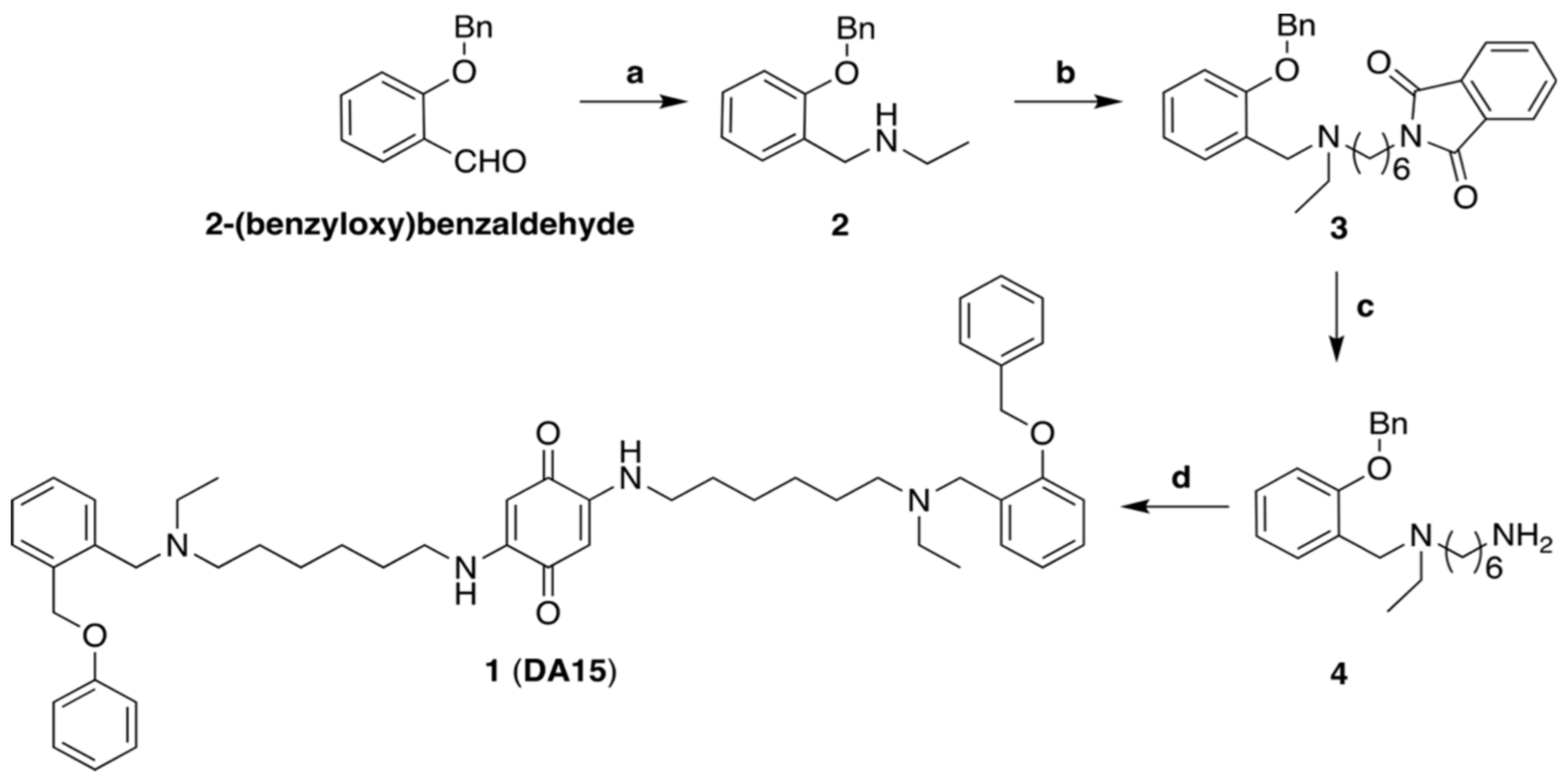
2.1. Chemistry
2.2. Synthesis of N-(2-(Benzyloxy)benzyl)ethanamine (2)
2.3. Synthesis of 2-(6-((2-(Benzyloxy)benzyl)(ethyl)amino)hexyl)isoindoline-1,3-dione (3)
2.4. Synthesis of N1-(2-(Benzyloxy)benzyl)-N1-ethylhexane-1,6-diamine (4)
2.5. Synthesis of 2-((6-((2-(Benzyloxy)benzyl)(ethyl)amino)hexyl)amino)-5-((6-(ethyl(2-(phenoxymethyl) benzyl)amino)hexyl)amino)cyclohexa-2,5-diene-1,4-dione (1)
2.6. Physicochemical Descriptors
2.7. Characterization of the Chemical-Space: Principal Component Analysis (PCA)
2.8. Hierarchical Clustering Analysis
2.9. Cell Culture
2.10. Cell Viability Assay
2.11. Dual Luciferase Reporter Assay
2.12. Immunoblotting
2.13. Immunofluorescence Staining
2.14. Real Time PCR (qPCR)
2.15. Statistical Analysis
3. Results
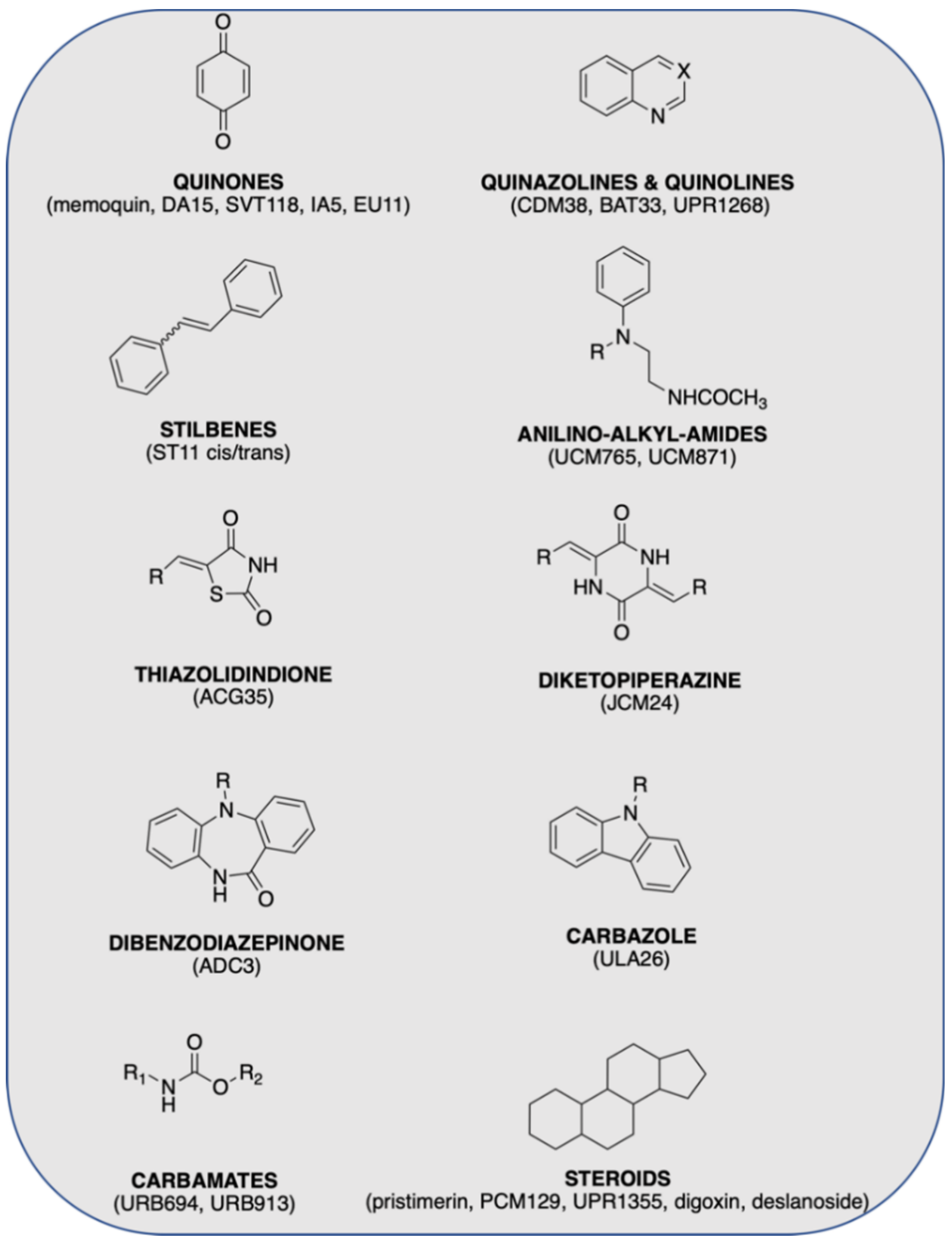
3.1. Chemical Library Collection
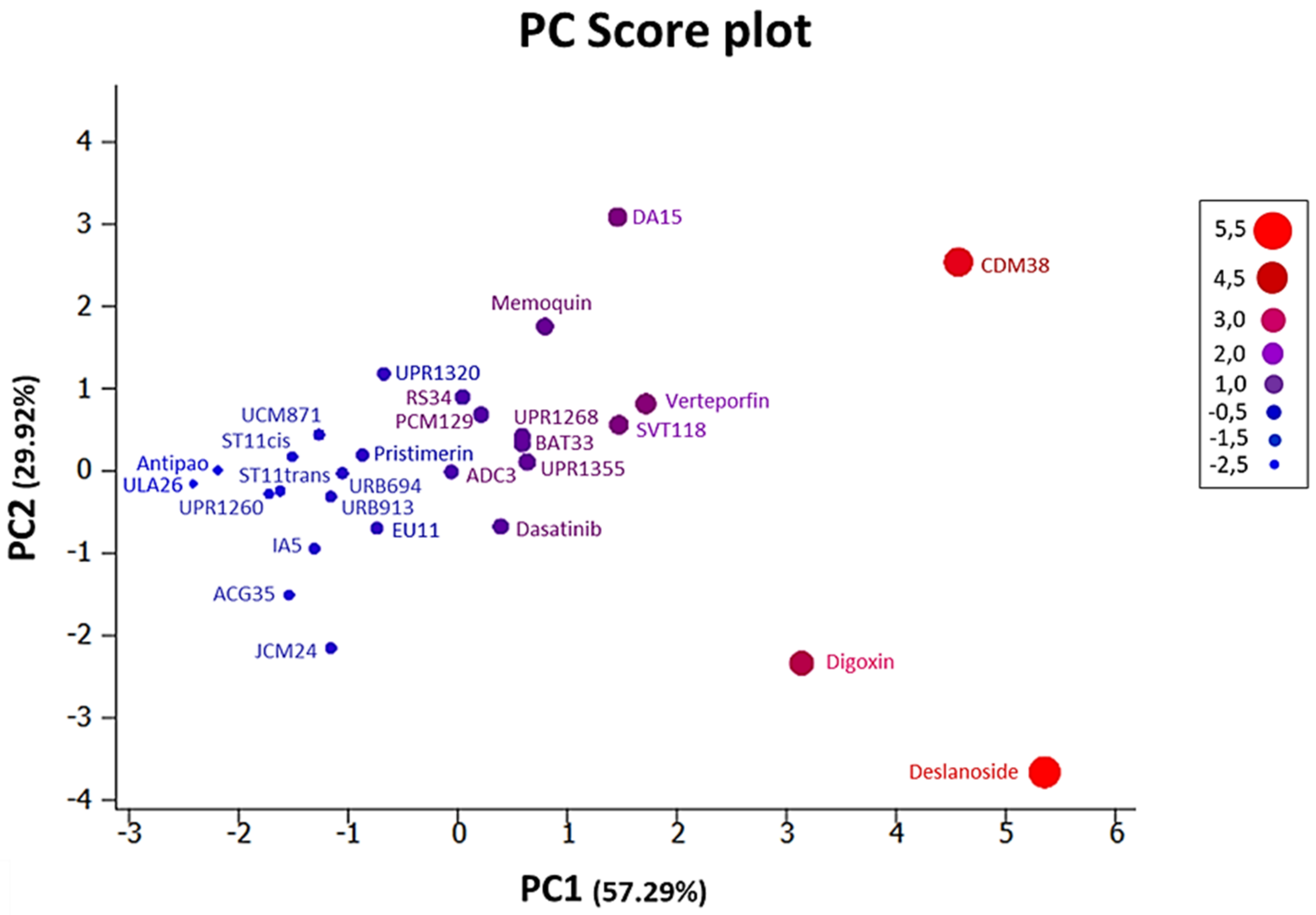
3.2. Chemoinformatic Analysis: Physicochemical Descriptors and PCA
3.3. Chemistry
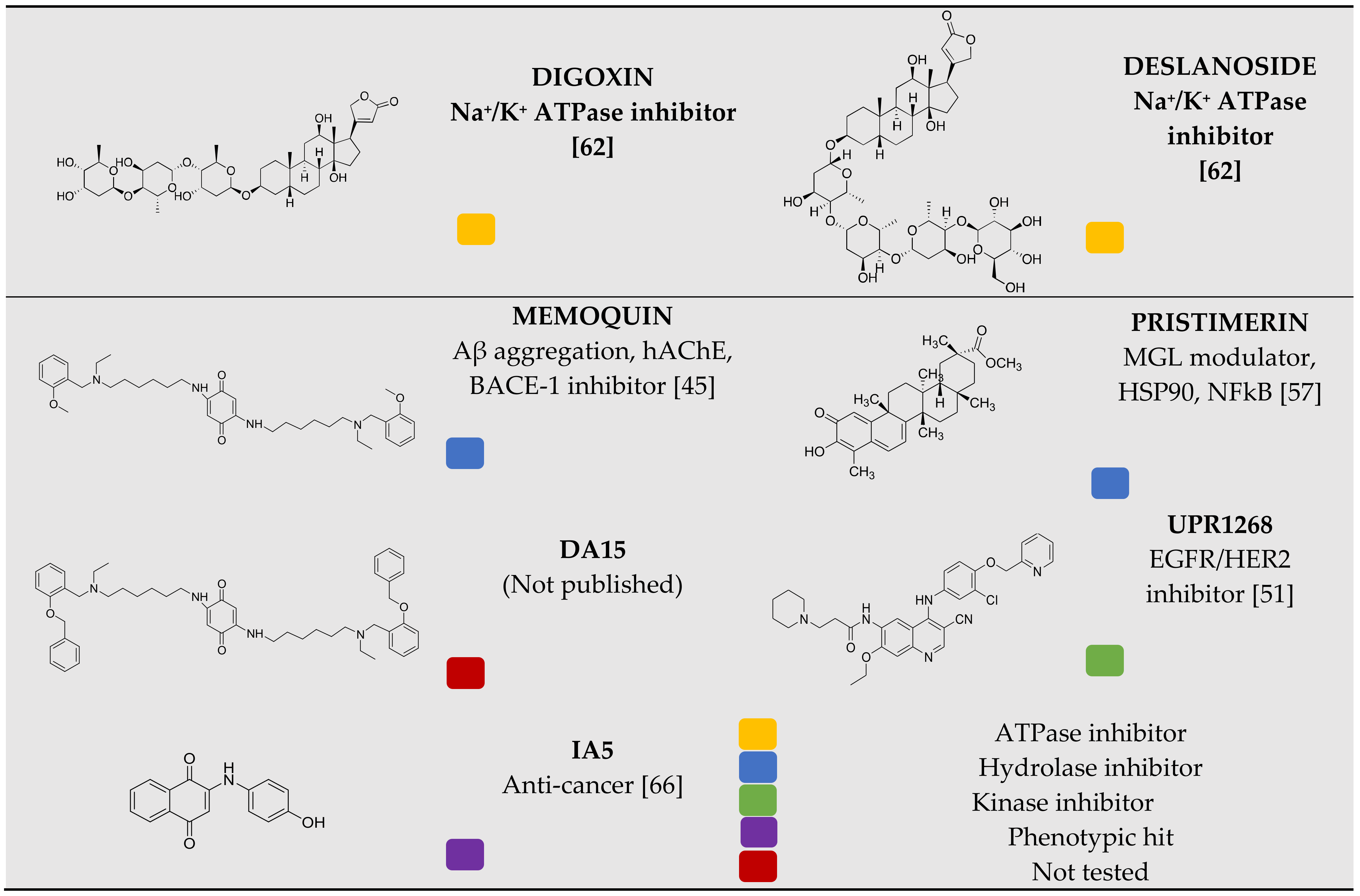
3.4. Identification of Compounds with the Potential to Inhibit Cell Growth
3.5. Inhibition of the Activity of the YAP–TEAD Complex
3.6. Cancer Cell Growth Inhibition and YAP Protein Levels and Phosphorylation Changes
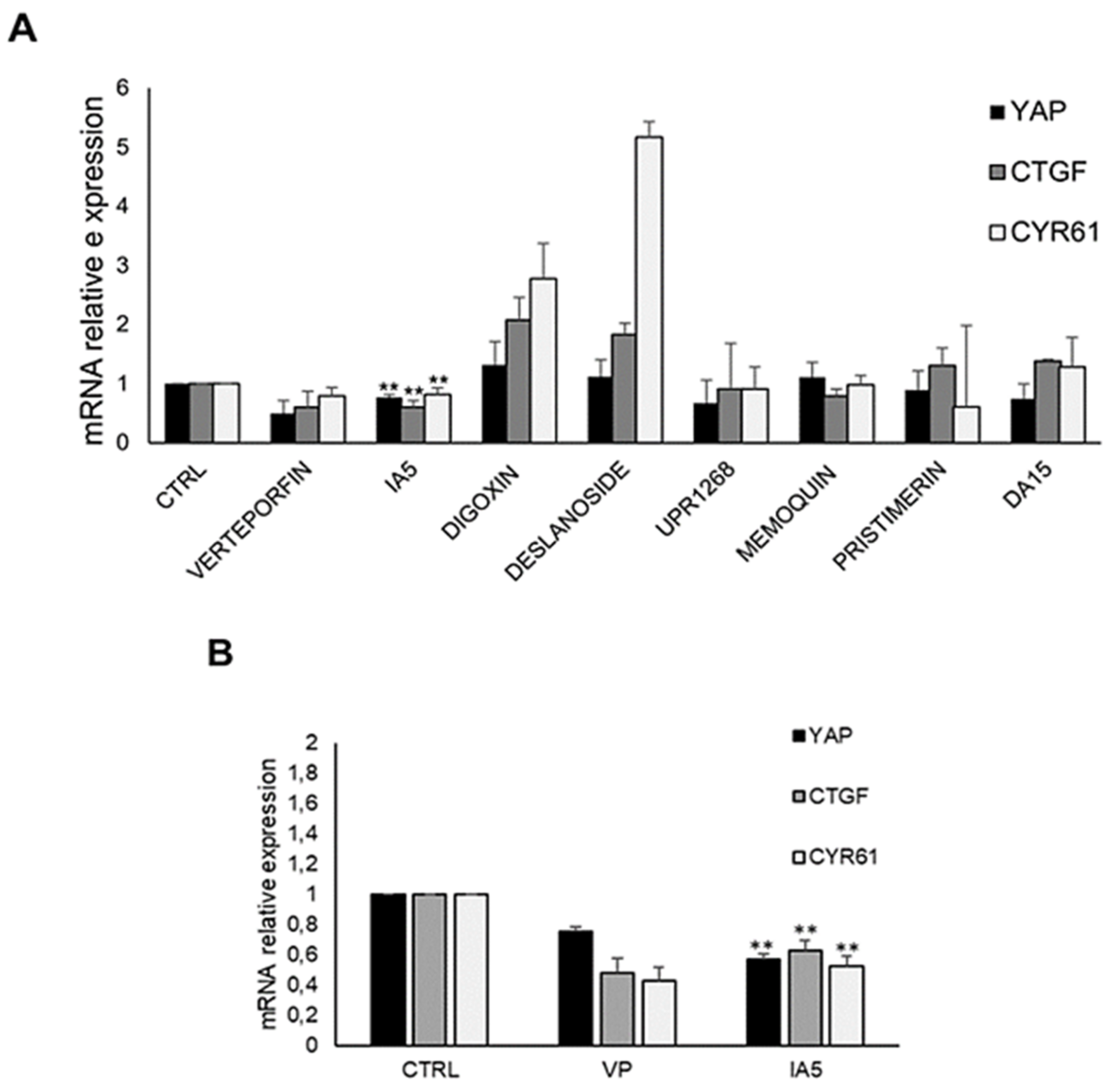
3.7. IA5 Reduces YAP-TEAD Target Genes Expression
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maugeri-Saccà, M.; Barba, M.; Pizzuti, L.; Vici, P.; Di Lauro, L.; Dattilo, R.; Vitale, I.; Bartucci, M.; Mottolese, M.; De Maria, R. The Hippo transducers TAZ and YAP in breast cancer: Oncogenic activities and clinical implications. Expert Rev. Mol. Med. 2015, 17, e14. [Google Scholar] [CrossRef] [PubMed]
- Bartucci, M.; Dattilo, R.; Moriconi, C.; Pagliuca, A.; Mottolese, M.; Federici, G.; Di Benedetto, A.; Todaro, M.; Stassi, G.; Sperati, F.; et al. TAZ is required for metastatic activity and chemoresistance of breast cancer stem cells. Oncogene 2014, 34, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Shi, S.; Guo, Z.; Zhang, X.; Han, S.; Yang, A.; Wen, W.; Zhu, Q. Overexpression of YAP and TAZ is an independent predictor of prognosis in colorectal cancer and related to the proliferation and metastasis of colon cancer cells. PLoS ONE 2013, 8, e65539. [Google Scholar] [CrossRef] [PubMed]
- An Integrative Analysis of the Tumorigenic Role of TAZ in Human Non–Small Cell Lung Cancer|Clinical Cancer Research, (n.d.). Available online: https://clincancerres.aacrjournals.org/content/20/17/4660.long (accessed on 1 April 2020).
- Santucci, M.; Vignudelli, T.; Ferrari, S.; Mor, M.; Scalvini, L.; Bolognesi, M.L.; Uliassi, E.; Costi, M.P. The Hippo Pathway and YAP/TAZ–TEAD Protein–Protein Interaction as Targets for Regenerative Medicine and Cancer Treatment. J. Med. Chem. 2015, 58, 4857–4873. [Google Scholar] [CrossRef]
- Lamar, J.M.; Xiao, Y.; Norton, E.; Jiang, Z.-G.; Gerhard, G.M.; Kooner, S.; Warren, J.S.A.; Hynes, R.O. SRC tyrosine kinase activates the YAP/TAZ axis and thereby drives tumor growth and metastasis. J. Biol. Chem. 2019, 294, 2302–2317. [Google Scholar] [CrossRef] [Green Version]
- Tremblay, A.M.; Camargo, F.D. Hippo signaling in mammalian stem cells. Semin. Cell Dev. Biol. 2012, 23, 818–826. [Google Scholar] [CrossRef]
- Yu, F.-X.; Guan, K.-L. The Hippo pathway: Regulators and regulations. Genes Dev. 2013, 27, 355–371. [Google Scholar] [CrossRef] [Green Version]
- Sayedyahossein, S.; Li, Z.; Hedman, A.C.; Morgan, C.; Sacks, D.B. IQGAP1 Binds to Yes-associated Protein (YAP) and Modulates Its Transcriptional Activity. J. Biol. Chem. 2016, 291, 19261–19273. [Google Scholar] [CrossRef] [Green Version]
- Taha, Z.; Van Rensburg, H.J.J.; Yang, X. The Hippo Pathway: Immunity and Cancer. Cancers 2018, 10, 94. [Google Scholar] [CrossRef] [Green Version]
- Holden, J.; Cunningham, C. Targeting the Hippo Pathway and Cancer through the TEAD Family of Transcription Factors. Cancers 2018, 10, 81. [Google Scholar] [CrossRef] [Green Version]
- Liu-Chittenden, Y.; Huang, B.; Shim, J.S.; Chen, Q.; Lee, S.-J.; Anders, R.A.; Liu, J.O.; Pan, D. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012, 26, 1300–1305. [Google Scholar] [CrossRef] [Green Version]
- Elisi, G.M.; Santucci, M.; D’Arca, D.; Lauriola, A.; Marverti, G.; Losi, L.; Scalvini, L.; Bolognesi, M.L.; Mor, M.; Costi, M.P. Repurposing of Drugs Targeting YAP-TEAD Functions. Cancers 2018, 10, 329. [Google Scholar] [CrossRef] [Green Version]
- Pobbati, A.V.; Hong, W. A combat with the YAP/TAZ-TEAD oncoproteins for cancer therapy. Theranostics 2020, 10, 3622–3635. [Google Scholar] [CrossRef]
- Stanger, B.Z. Quit your YAPing: A new target for cancer therapy. Genes Dev. 2012, 26, 1263–1267. [Google Scholar] [CrossRef] [Green Version]
- Dong, L.; Lin, F.; Wu, W.; Liu, Y.; Huang, W. Verteporfin inhibits YAP-induced bladder cancer cell growth and invasion via Hippo signaling pathway. Int. J. Med. Sci. 2018, 15, 645–652. [Google Scholar] [CrossRef] [Green Version]
- Eales, K.L.; Wilkinson, E.; Cruickshank, G.; Tucker, J.; Tennant, D.A. Verteporfin selectively kills hypoxic glioma cells through iron-binding and increased production of reactive oxygen species. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef]
- Nouri, K.; Azad, T.; Ling, M.; Van Rensburg, H.J.J.; Pipchuk, A.; Shen, H.; Hao, Y.; Zhang, J.; Yang, X. Identification of Celastrol as a Novel YAP-TEAD Inhibitor for Cancer Therapy by High Throughput Screening with Ultrasensitive YAP/TAZ–TEAD Biosensors. Cancers 2019, 11, 1596. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Lin, Z.; Zhou, Z.; Shen, H.C.; Yan, S.F.; Mayweg, A.V.; Xu, Z.; Qin, N.; Wong, J.C.; Zhang, Z.; et al. Structure-Based Design and Synthesis of Potent Cyclic Peptides Inhibiting the YAP-TEAD Protein-Protein Interaction. ACS Med. Chem. Lett. 2014, 5, 993–998. [Google Scholar] [CrossRef]
- Crook, Z.R.; Sevilla, G.P.; Friend, D.; Brusniak, M.-Y.; Bandaranayake, A.D.; Clarke, M.; Gewe, M.; Mhyre, A.J.; Baker, D.; Strong, R.K.; et al. Mammalian display screening of diverse cystine-dense peptides for difficult to drug targets. Nat. Commun. 2017, 8, 2244. [Google Scholar] [CrossRef]
- Jiao, S.; Wang, H.; Shi, Z.; Dong, A.; Zhang, W.; Song, X.; He, F.; Wang, Y.; Zhang, Z.; Wang, W.; et al. A Peptide Mimicking VGLL4 Function Acts as a YAP Antagonist Therapy against Gastric Cancer. Cancer Cell 2014, 25, 166–180. [Google Scholar] [CrossRef] [Green Version]
- Smith, S.A.; Sessions, R.B.; Shoemark, D.K.; Williams, C.; Ebrahimighaei, R.; McNeill, M.C.; Crump, M.P.; McKay, T.R.; Harris, G.; Newby, A.C.; et al. Antiproliferative and Antimigratory Effects of a Novel YAP–TEAD Interaction Inhibitor Identified Using in Silico Molecular Docking. J. Med. Chem. 2019, 62, 1291–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sudol, M.; Shields, D.C.; Farooq, A. Structures of YAP protein domains reveal promising targets for development of new cancer drugs. Semin. Cell Dev. Biol. 2012, 23, 827–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Li, P.-X.; Wu, J.; Gao, Y.-J.; Yin, M.-X.; Lin, Y.; Yang, M.; Chen, D.-P.; Sun, H.-P.; Liu, Z.-B.; et al. Involvement of the Hippo pathway in regeneration and fibrogenesis after ischaemic acute kidney injury: YAP is the key effector. Clin. Sci. 2016, 130, 349–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, W.; Wang, J.; Li, Y.; Tao, H.; Xiong, H.; Lian, F.; Gao, J.; Ma, H.; Lu, T.; Zhang, D.; et al. Discovery and biological evaluation of vinylsulfonamide derivatives as highly potent, covalent TEAD autopalmitoylation inhibitors. Eur. J. Med. Chem. 2019, 184, 111767. [Google Scholar] [CrossRef]
- Kaneda, A.; Seike, T.; Uemori, T.; Myojo, K.; Aida, K.; Danjo, T.; Nakajima, T.; Yamaguchi, D.; Hamada, T.; Tsuji, Y.; et al. Abstract 3086: Discovery of a first-in-class TEAD inhibitor which directly inhibits YAP/TAZ-TEAD protein-protein interaction and shows a potent anti-tumor effect in malignant pleural mesothelioma. In Experimental and Molecular Therapeutics; American Association for Cancer Research: Philadelphia, PA, USA, 2019. [Google Scholar] [CrossRef]
- Torchet, R.; Druart, K.; Ruano, L.C.; Moine-Franel, A.; Borges, H.; Doppelt-Azeroual, O.; Brancotte, B.; Mareuil, F.; Nilges, M.; Ménager, H.; et al. The iPPI-DB initiative: A community-centered database of protein–protein interaction modulators. Bioinformatics 2021, 37, 89–96. [Google Scholar] [CrossRef]
- Kouassi, K.A.R.; Ganiyou, A.; Benié, A.; Koné, M.G.-R.; Nobel, N.K.; Bohoussou, K.V.; Coulibaly, W.K. Identification of Potential C-kit Protein Kinase Inhibitors Associated with Human Liver Cancer: Atom-based 3D-QSAR Modeling, Pharmacophores-based Virtual Screening and Molecular Docking Studies. Am. J. Pharmacol. Sci. 2021, 9, 1–29. [Google Scholar] [CrossRef]
- González-Medina, M.; Prieto-Martínez, F.D.; Naveja, J.J.; Méndez-Lucio, O.; El-Elimat, T.; Pearce, C.J.; Oberlies, N.H.; Figueroa, M.; Medina-Franco, J.L. Chemoinformatic expedition of the chemical space of fungal products. Future Med. Chem. 2016, 8, 1399–1412. [Google Scholar] [CrossRef] [Green Version]
- De Souza, A.S.; Ferreira, L.L.G.; De Oliveira, A.S.; Andricopulo, A.D. Quantitative Structure–Activity Relationships for Structurally Diverse Chemotypes Having Anti-Trypanosoma cruzi Activity. Int. J. Mol. Sci. 2019, 20, 2801. [Google Scholar] [CrossRef] [Green Version]
- Rogers, D.; Hahn, M. Extended-Connectivity Fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef]
- Bajusz, D.; Rácz, A.; Héberger, K. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? J. Cheminform. 2015, 7, 20. [Google Scholar] [CrossRef] [Green Version]
- Shao, J.; Tanner, S.W.; Thompson, N.; Cheatham, T.E. Clustering Molecular Dynamics Trajectories: 1. Characterizing the Performance of Different Clustering Algorithms. J. Chem. Theory Comput. 2007, 3, 2312–2334. [Google Scholar] [CrossRef]
- Voicu, A.; Duteanu, N.; Voicu, M.; Vlad, D.; Dumitrascu, V. The rcdk and cluster R packages applied to drug candidate selection. J. Cheminform. 2020, 12, 3. [Google Scholar] [CrossRef] [Green Version]
- Rácz, A.; Bajusz, D.; Héberger, K. Life beyond the Tanimoto coefficient: Similarity measures for interaction fingerprints. J. Cheminform. 2018, 10, 48. [Google Scholar] [CrossRef] [Green Version]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [Green Version]
- Churi, C.R.; Shroff, R.; Wang, Y.; Rashid, A.; Kang, H.; Weatherly, J.; Zuo, M.; Zinner, R.; Hong, D.; Meric-Bernstam, F.; et al. Mutation Profiling in Cholangiocarcinoma: Prognostic and Therapeutic Implications. PLoS ONE 2014, 9, e115383. [Google Scholar] [CrossRef] [Green Version]
- Corvaisier, M.; Bauzone, M.; Corfiotti, F.; Renaud, F.; El Amrani, M.; Monté, D.; Truant, S.; Leteurtre, E.; Formstecher, P.; Van Seuningen, I.; et al. Regulation of cellular quiescence by YAP/TAZ and Cyclin E1 in colon cancer cells: Implication in chemoresistance and cancer relapse. Oncotarget 2016, 7, 56699–56712. [Google Scholar] [CrossRef] [Green Version]
- Keskin, O.; Gursoy, A.; Ma, B.; Nussinov, R. Principles of Protein−Protein Interactions: What are the Preferred Ways For Proteins To Interact? Chem. Rev. 2008, 108, 1225–1244. [Google Scholar] [CrossRef]
- Kuenemann, M.A.; Bourbon, L.M.L.; Labbé, C.M.; Villoutreix, B.; Sperandio, O. Which Three-Dimensional Characteristics Make Efficient Inhibitors of Protein–Protein Interactions? J. Chem. Inf. Model. 2014, 54, 3067–3079. [Google Scholar] [CrossRef]
- Benet, L.Z.; Hosey, C.M.; Ursu, O.; Oprea, T. BDDCS, the Rule of 5 and drugability. Adv. Drug Deliv. Rev. 2016, 101, 89–98. [Google Scholar] [CrossRef] [Green Version]
- Björklund, M. Be careful with your principal components. Evolution 2019, 73, 2151–2158. [Google Scholar] [CrossRef] [PubMed]
- Rodionova, O.; Kucheryavskiy, S.; Pomerantsev, A. Efficient tools for principal component analysis of complex data—A tutorial. Chemom. Intell. Lab. Syst. 2021, 213, 104304. [Google Scholar] [CrossRef]
- Cavalli, A.; Bolognesi, M.L.; Capsoni, S.; Andrisano, V.; Bartolini, M.; Margotti, E.; Cattaneo, A.; Recanatini, M.; Melchiorre, C. A Small Molecule Targeting the Multifactorial Nature of Alzheimer’s Disease. Angew. Chem. Int. Ed. 2007, 46, 3689–3692. [Google Scholar] [CrossRef] [PubMed]
- Bongarzone, S.; Tran, H.N.A.; Cavalli, A.; Roberti, M.; Carloni, P.; Legname, G.; Bolognesi, M.L. Parallel Synthesis, Evaluation, and Preliminary Structure−Activity Relationship of 2,5-Diamino-1,4-benzoquinones as a Novel Class of Bivalent Anti-Prion Compound. J. Med. Chem. 2010, 53, 8197–8201. [Google Scholar] [CrossRef]
- Kawahara, G.; Gasperini, M.J.; Myers, J.A.; Widrick, J.J.; Eran, A.; Serafini, P.R.; Alexander, M.S.; Pletcher, M.T.; Morris, C.A.; Kunkel, L.M. Dystrophic muscle improvement in zebrafish via increased heme oxygenase signaling. Hum. Mol. Genet. 2013, 23, 1869–1878. [Google Scholar] [CrossRef] [Green Version]
- Uliassi, E.; Fiorani, G.; Krauth-Siegel, R.L.; Bergamini, C.; Fato, R.; Bianchini, G.; Menéndez, J.C.; Molina, M.T.; López-Montero, E.; Falchi, F.; et al. Crassiflorone derivatives that inhibit Trypanosoma brucei glyceraldehyde-3-phosphate dehydrogenase (Tb GAPDH) and Trypanosoma cruzi trypanothione reductase (Tc TR) and display trypanocidal activity. Eur. J. Med. Chem. 2017, 141, 138–148. [Google Scholar] [CrossRef]
- Bolognesi, M.L.; Marucci, G.; Angeli, P.; Buccioni, M.; Minarini, A.; Rosini, M.; Tumiatti, V.; Melchiorre, C. Analogues of Prazosin That Bear a Benextramine-Related Polyamine Backbone Exhibit Different Antagonism toward α1-Adrenoreceptor Subtypes. J. Med. Chem. 2000, 44, 362–371. [Google Scholar] [CrossRef]
- Bolognesi, M.L.; Budriesi, R.; Chiarini, A.; Poggesi, E.; Leonardi, A.; Melchiorre, C. Design, Synthesis, and Biological Activity of Prazosin-Related Antagonists. Role of the Piperazine and Furan Units of Prazosin on the Selectivity for α1-Adrenoreceptor Subtypes. J. Med. Chem. 1998, 41, 4844–4853. [Google Scholar] [CrossRef]
- Carmi, C.; Galvani, E.; Vacondio, F.; Rivara, S.; Lodola, A.; Russo, S.; Aiello, S.; Bordi, F.; Costantino, G.; Cavazzoni, A.; et al. Irreversible Inhibition of Epidermal Growth Factor Receptor Activity by 3-Aminopropanamides. J. Med. Chem. 2012, 55, 2251–2264. [Google Scholar] [CrossRef] [Green Version]
- Roberti, M.; Pizzirani, D.; Recanatini, M.; Simoni, D.; Grimaudo, S.; Di Cristina, A.; Abbadessa, V.; Gebbia, A.N.; Tolomeo, M. Identification of a Terphenyl Derivative that Blocks the Cell Cycle in the G0−G1 Phase and Induces Differentiation in Leukemia Cells. J. Med. Chem. 2006, 49, 3012–3018. [Google Scholar] [CrossRef]
- Gandini, A.; Bartolini, M.; Tedesco, D.; Martinez-Gonzalez, L.; Roca, C.; Campillo, N.E.; Zaldivar-Diez, J.; Perez, C.; Zuccheri, G.; Miti, A.; et al. Tau-Centric Multitarget Approach for Alzheimer’s Disease: Development of First-in-Class Dual Glycogen Synthase Kinase 3β and Tau-Aggregation Inhibitors. J. Med. Chem. 2018, 61, 7640–7656. [Google Scholar] [CrossRef]
- Bolognesi, M.L.; Tran, H.N.A.; Staderini, M.; Monaco, A.; López-Cobeñas, A.; Bongarzone, S.; Biarnés, X.; López-Alvarado, P.; Cabezas, N.; Caramelli, M.; et al. Discovery of a Class of Diketopiperazines as Antiprion Compounds. ChemMedChem 2010, 5, 1324–1334. [Google Scholar] [CrossRef]
- Bolognesi, M.L.; Cavalli, A.; Valgimigli, L.; Bartolini, M.; Rosini, M.; Andrisano, V.; Recanatini, M.; Melchiorre, C. Multi-Target-Directed Drug Design Strategy: From a Dual Binding Site Acetylcholinesterase Inhibitor to a Trifunctional Compound against Alzheimer’s Disease. J. Med. Chem. 2007, 50, 6446–6449. [Google Scholar] [CrossRef]
- Uliassi, E.; Pena-Altamira, E.; Morales, A.V.; Massenzio, F.; Petralla, S.; Rossi, M.; Roberti, M.; Gonzalez, L.M.; Martinez, A.; Monti, B.; et al. A Focused Library of Psychotropic Analogues with Neuroprotective and Neuroregenerative Potential. ACS Chem. Neurosci. 2018, 10, 279–294. [Google Scholar] [CrossRef]
- King, A.R.; Dotsey, E.Y.; Lodola, A.; Jung, K.-M.; Ghomian, A.; Qiu, Y.; Fu, J.; Mor, M.; Piomelli, D. Discovery of Potent and Reversible Monoacylglycerol Lipase Inhibitors. Chem. Biol. 2009, 16, 1045–1052. [Google Scholar] [CrossRef] [Green Version]
- Tarzia, G.; Duranti, A.; Gatti, G.; Piersanti, G.; Tontini, A.; Rivara, S.; Lodola, A.; Plazzi, P.V.; Mor, M.; Kathuria, S.; et al. Synthesis and Structure-Activity Relationships of FAAH Inhibitors: Cyclohexylcarbamic Acid Biphenyl Esters with Chemical Modulation at the Proximal Phenyl Ring. ChemMedChem 2006, 1, 130–139. [Google Scholar] [CrossRef] [Green Version]
- Duranti, A.; Tontini, A.; Antonietti, F.; Vacondio, F.; Fioni, A.; Silva, C.; Lodola, A.; Rivara, S.; Solorzano, C.; Piomelli, D.; et al. N-(2-Oxo-3-oxetanyl)carbamic Acid Esters as N-Acylethanolamine Acid Amidase Inhibitors: Synthesis and Structure–Activity and Structure–Property Relationships. J. Med. Chem. 2012, 55, 4824–4836. [Google Scholar] [CrossRef] [Green Version]
- Drakes, M.L.; Stiff, P.J. Regulation of Ovarian Cancer Prognosis by Immune Cells in the Tumor Microenvironment. Cancers 2018, 10, 302. [Google Scholar] [CrossRef] [Green Version]
- Hassan-Mohamed, I.; Giorgio, C.; Incerti, M.; Russo, S.; Pala, D.; Pasquale, E.B.; Zanotti, I.; Vicini, P.; Barocelli, E.; Rivara, S.; et al. UniPR129 is a competitive small molecule Eph-ephrin antagonist blocking in vitro angiogenesis at low micromolar concentrations: Eph antagonist and angiogenesis. Br. J. Pharmacol. 2014, 171, 5195–5208. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, A.; Allen, J.C.; Harigaya, S. Possible involvement of cardiac Na+, K+-adenosine triphosphatase in the mechanism of action of cardiac glycosides. J. Pharmacol. Exp. Ther. 1969, 168, 31–41. [Google Scholar]
- Rivara, S.; Lodola, A.; Mor, M.; Bedini, A.; Spadoni, G.; Lucini, V.; Pannacci, M.; Fraschini, F.; Scaglione, F.; Sanchez, R.O.; et al. N-(Substituted-anilinoethyl)amides: Design, Synthesis, and Pharmacological Characterization of a New Class of Melatonin Receptor Ligands. J. Med. Chem. 2007, 50, 6618–6626. [Google Scholar] [CrossRef]
- Rivara, S.; Pala, D.; Lodola, A.; Mor, M.; Lucini, V.; Dugnani, S.; Scaglione, F.; Bedini, A.; Lucarini, S.; Tarzia, G.; et al. MT1-Selective Melatonin Receptor Ligands: Synthesis, Pharmacological Evaluation, and Molecular Dynamics Investigation of N-{[(3-O-Substituted)anilino]alkyl}amides. ChemMedChem 2012, 7, 1954–1964. [Google Scholar] [CrossRef]
- Song, S.; Honjo, S.; Jin, J.; Chang, S.-S.; Scott, A.W.; Chen, Q.; Kalhor, N.; Correa, A.M.; Hofstetter, W.L.; Albarracin, C.T.; et al. The Hippo Coactivator YAP1 Mediates EGFR Overexpression and Confers Chemoresistance in Esophageal Cancer. Clin. Cancer Res. 2015, 21, 2580–2590. [Google Scholar] [CrossRef] [Green Version]
- Murphey, R.D.; Stern, H.M.; Straub, C.T.; Zon, L.I. A Chemical Genetic Screen for Cell Cycle Inhibitors in Zebrafish Embryos. Chem. Biol. Drug Des. 2006, 68, 213–219. [Google Scholar] [CrossRef]
- Yousef, B.; Hassan, H.M.; Guerram, M.; Hamdi, A.M.; Wang, B.; Zhang, L.-Y.; Jiang, Z.-Z. Pristimerin inhibits proliferation, migration and invasion, and induces apoptosis in HCT-116 colorectal cancer cells. Biomed. Pharmacother. 2016, 79, 112–119. [Google Scholar] [CrossRef]
- Galvani, E.; Giovannetti, E.; Saccani, F.; Cavazzoni, A.; Leon, L.G.; Dekker, H.; Alfieri, R.; Carmi, C.; Mor, M.; Ardizzoni, A.; et al. Molecular Mechanisms Underlying the Antitumor Activity of 3-Aminopropanamide Irreversible Inhibitors of the Epidermal Growth Factor Receptor in Non–Small Cell Lung Cancer. Neoplasia 2013, 15, 61–72, IN18. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Zhang, W.; Pan, Y.; Gao, Y.; Deng, L.; Li, F.; Li, F.; Ma, X.; Hou, S.; Xueyan, M.; et al. YAP Suppresses Lung Squamous Cell Carcinoma Progression via Deregulation of the DNp63–GPX2 Axis and ROS Accumulation. Cancer Res. 2017, 77, 5769–5781. [Google Scholar] [CrossRef] [Green Version]
- Solberg, N.; Krauss, S. Luciferase Assay to Study the Activity of a Cloned Promoter DNA Fragment. In Gene Regulation; Humana Press: Totowa, NJ, USA, 2013; Volume 977, pp. 65–78. [Google Scholar] [CrossRef]
- Li, Z.; Feng, J.; Gou, J.; Jia, J.; Yi, T.; Cui, T. Verteporfin, a suppressor of YAP–TEAD complex, presents promising antitumor properties on ovarian cancer. OncoTargets Ther. 2016, 9, 5371–5381. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.-W.; Liu, Y.-Z.; Pan, J.-X. Verteporfin induces apoptosis and eliminates cancer stem-like cells in uveal melanoma in the absence of light activation. Am. J. Cancer Res. 2016, 6, 2816–2830. [Google Scholar]
- Su, M.; Zhan, L.; Zhang, Y.; Zhang, J. Yes-activated protein promotes primary resistance of BRAF V600E mutant metastatic colorectal cancer cells to mitogen-activated protein kinase pathway inhibitors. J. Gastrointest. Oncol. 2021, 12, 953–963. [Google Scholar] [CrossRef]
- Ling, H.-H.; Kuo, C.-C.; Lin, B.-X.; Huang, Y.-H.; Lin, C.-W. Elevation of YAP promotes the epithelial-mesenchymal transition and tumor aggressiveness in colorectal cancer. Exp. Cell Res. 2017, 350, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Plouffe, S.W.; Lin, K.C.; Moore, J.L.; Tan, F.E.; Ma, S.; Ye, Z.; Qiu, Y.; Ren, B.; Guan, K.-L. The Hippo pathway effector proteins YAP and TAZ have both distinct and overlapping functions in the cell. J. Biol. Chem. 2018, 293, 11230–11240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Zhu, X.; Feng, W.; Yu, Y.; Jeong, K.; Guo, W.; Lu, Y.; Mills, G.B. Verteporfin inhibits YAP function through up-regulating 14-3-3σ sequestering YAP in the cytoplasm. Am. J. Cancer Res. 2015, 6, 27–37. [Google Scholar] [PubMed]
- Lei, Q.-Y.; Zhang, H.; Zhao, B.; Zha, Z.; Bai, F.; Pei, X.-H.; Zhao, S.; Xiong, Y.; Guan, K.-L. TAZ Promotes Cell Proliferation and Epithelial-Mesenchymal Transition and Is Inhibited by the Hippo Pathway. Mol. Cell. Biol. 2008, 28, 2426–2436. [Google Scholar] [CrossRef] [Green Version]
- Bolton, J.L.; Dunlap, T. Formation and Biological Targets of Quinones: Cytotoxic versus Cytoprotective Effects. Chem. Res. Toxicol. 2016, 30, 13–37. [Google Scholar] [CrossRef]
- Wang, W.; Lin, P.; Zhang, Z.; Zhong, Y.; Kong, D. Digoxin Enhances the Anticancer Effect on Non-Small Cell Lung Cancer While Reducing the Cardiotoxicity of Adriamycin. Front. Pharmacol. 2020, 11, 186. [Google Scholar] [CrossRef] [Green Version]
- Shao, D.; Zhai, P.; Del Re, D.P.; Sciarretta, S.; Yabuta, N.; Nojima, H.; Lim, D.-S.; Pan, D.; Sadoshima, J. A functional interaction between Hippo-YAP signalling and FoxO1 mediates the oxidative stress response. Nat. Commun. 2014, 5, 3315. [Google Scholar] [CrossRef] [Green Version]
- Sperandio, O.; Reynès, C.H.; Camproux, A.-C.; Villoutreix, B. Rationalizing the chemical space of protein–protein interaction inhibitors. Drug Discov. Today 2010, 15, 220–229. [Google Scholar] [CrossRef]
- Sieveking, I.; Thomas, P.; Estévez, J.C.; Quiñones, N.; Cuéllar, M.A.; Villena, J.; Espinosa-Bustos, C.; Fierro, A.; Tapia, R.A.; Maya, J.D.; et al. 2-Phenylaminonaphthoquinones and related compounds: Synthesis, trypanocidal and cytotoxic activities. Bioorg. Med. Chem. 2014, 22, 4609–4620. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 (µM)-Cancer Cell Lines (48 h) | |||||
|---|---|---|---|---|---|
| Compound | A2780 | A2780/CP | HCA46 | HT29 | HCT116 |
| VERTEPORFIN | 12.38 ± 2.15 | 12.3 ± 0.76 | 5.55 ± 1.2 | 3.1 ± 0.32 | 14.7 ± 2.79 |
| MEMOQUIN | 9.53 ± 0.81 | 14.03 ± 1.05 | 14.59 ± 0.7 | 9.82 ± 0.8 | 2.89 ± 0.08 |
| IA5 | 3.8 ± 1.06 | 5.35 ± 0.95 | 19.79 ± 4.0 | 2.68 ± 0.72 | 4.17 ± 1.94 |
| UPR1268 | 6.17 ± 0.71 | 4.5 ± 1.7 | 2.39 ± 0.3 | 4.81 ± 0.25 | 6.79 ± 0.39 |
| DA15 | 1.71 ± 0.12 | 2.04 ± 0.66 | >2.5 | 1.78 ± 0.46 | 1.59 ± 0.45 |
| PRISTIMERIN | 0.44 ± 0.03 | 0.19 ± 0.01 | 0.94 ± 0.4 | 0.8 ± 0.2 | 0.64 ± 0.15 |
| DIGOXIN | 0.06 ± 0.01 | 0.04 ± 0.0 | 0.38 ± 0.15 | 0.04 ± 0.01 | 0.08 ± 0.0 |
| DESLANOSIDE | 0.1 ± 0.03 | 0.05 ± 0.0 | 0.19 ± 0.03 | 0.07 ± 0.0 | 0.12 ± 0.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lauriola, A.; Uliassi, E.; Santucci, M.; Bolognesi, M.L.; Mor, M.; Scalvini, L.; Elisi, G.M.; Gozzi, G.; Tagliazucchi, L.; Marverti, G.; et al. Identification of a Quinone Derivative as a YAP/TEAD Activity Modulator from a Repurposing Library. Pharmaceutics 2022, 14, 391. https://doi.org/10.3390/pharmaceutics14020391
Lauriola A, Uliassi E, Santucci M, Bolognesi ML, Mor M, Scalvini L, Elisi GM, Gozzi G, Tagliazucchi L, Marverti G, et al. Identification of a Quinone Derivative as a YAP/TEAD Activity Modulator from a Repurposing Library. Pharmaceutics. 2022; 14(2):391. https://doi.org/10.3390/pharmaceutics14020391
Chicago/Turabian StyleLauriola, Angela, Elisa Uliassi, Matteo Santucci, Maria Laura Bolognesi, Marco Mor, Laura Scalvini, Gian Marco Elisi, Gaia Gozzi, Lorenzo Tagliazucchi, Gaetano Marverti, and et al. 2022. "Identification of a Quinone Derivative as a YAP/TEAD Activity Modulator from a Repurposing Library" Pharmaceutics 14, no. 2: 391. https://doi.org/10.3390/pharmaceutics14020391
APA StyleLauriola, A., Uliassi, E., Santucci, M., Bolognesi, M. L., Mor, M., Scalvini, L., Elisi, G. M., Gozzi, G., Tagliazucchi, L., Marverti, G., Ferrari, S., Losi, L., D’Arca, D., & Costi, M. P. (2022). Identification of a Quinone Derivative as a YAP/TEAD Activity Modulator from a Repurposing Library. Pharmaceutics, 14(2), 391. https://doi.org/10.3390/pharmaceutics14020391