
Dually Active Polycation/miRNA Nanoparticles for the Treatment of Fibrosis in Alcohol-Associated Liver Disease

 , , , , and
, , , , and 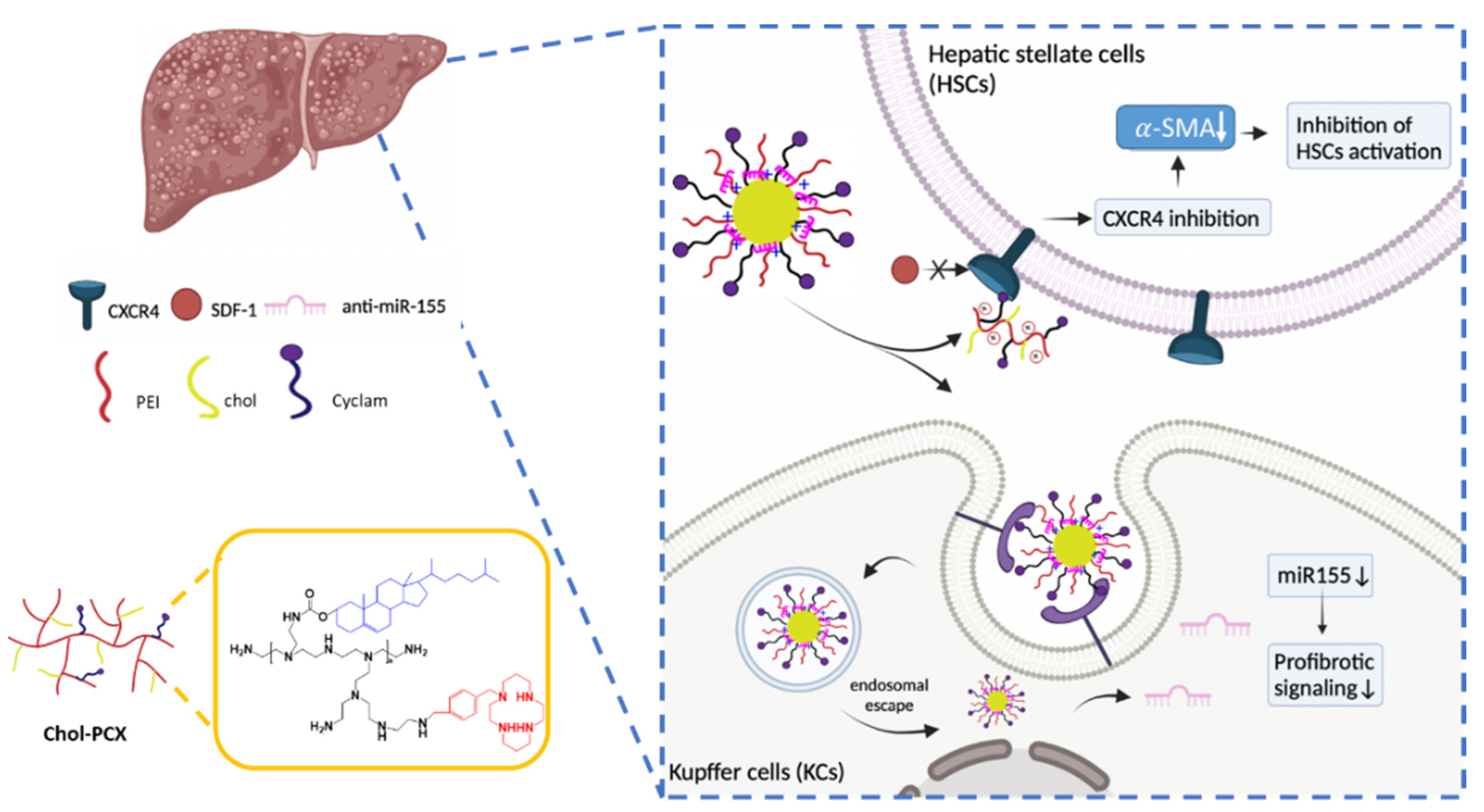{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis and Characterization of Chol-PCX
2.3. Preparation and Characterization of PCX/miRNA Nanoparticles
2.4. Cell Viability and Intracellular Trafficking
2.5. In Vitro miRNA Transfection
2.6. CXCR4 Antagonism Assay
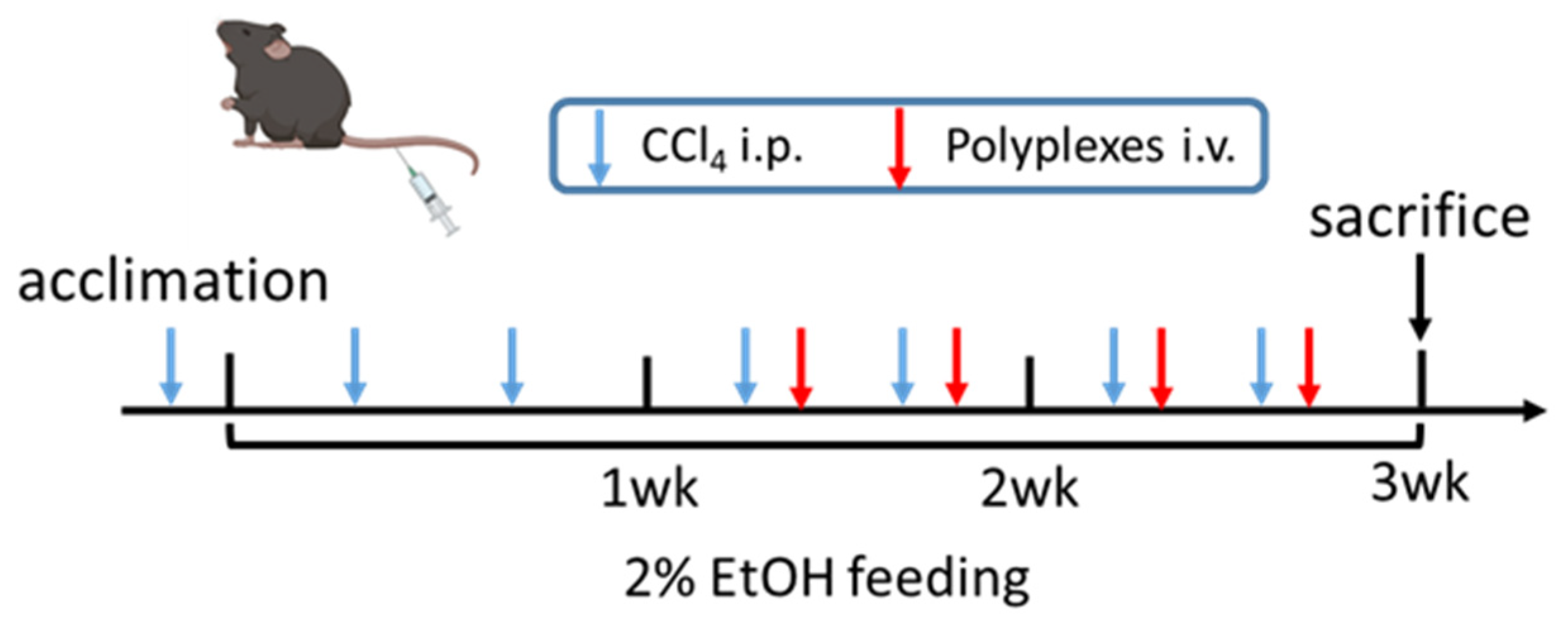
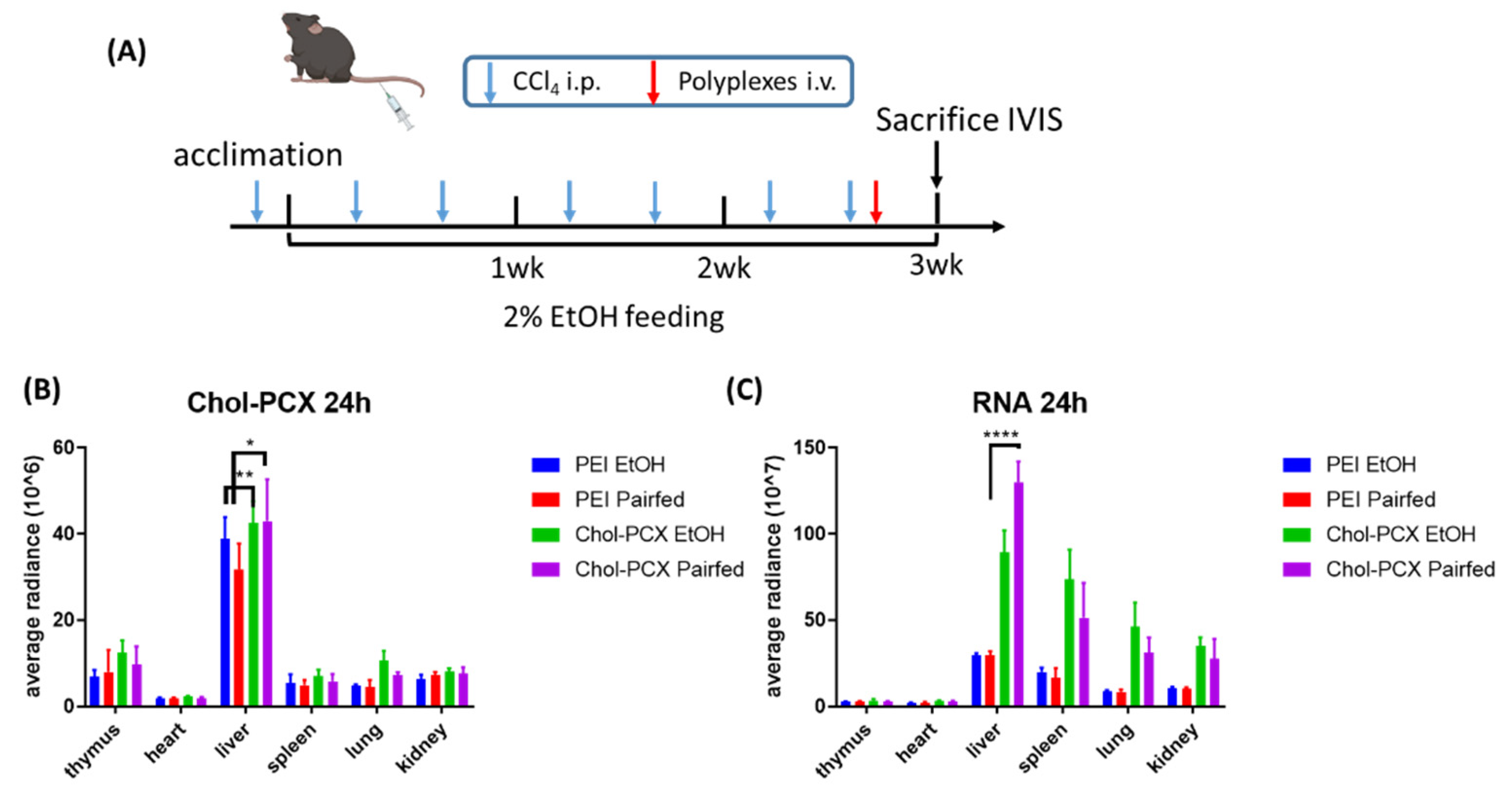
2.7. In Vivo Anti-miR-155 Therapy in AALD Fibrosis
2.8. Biodistribution
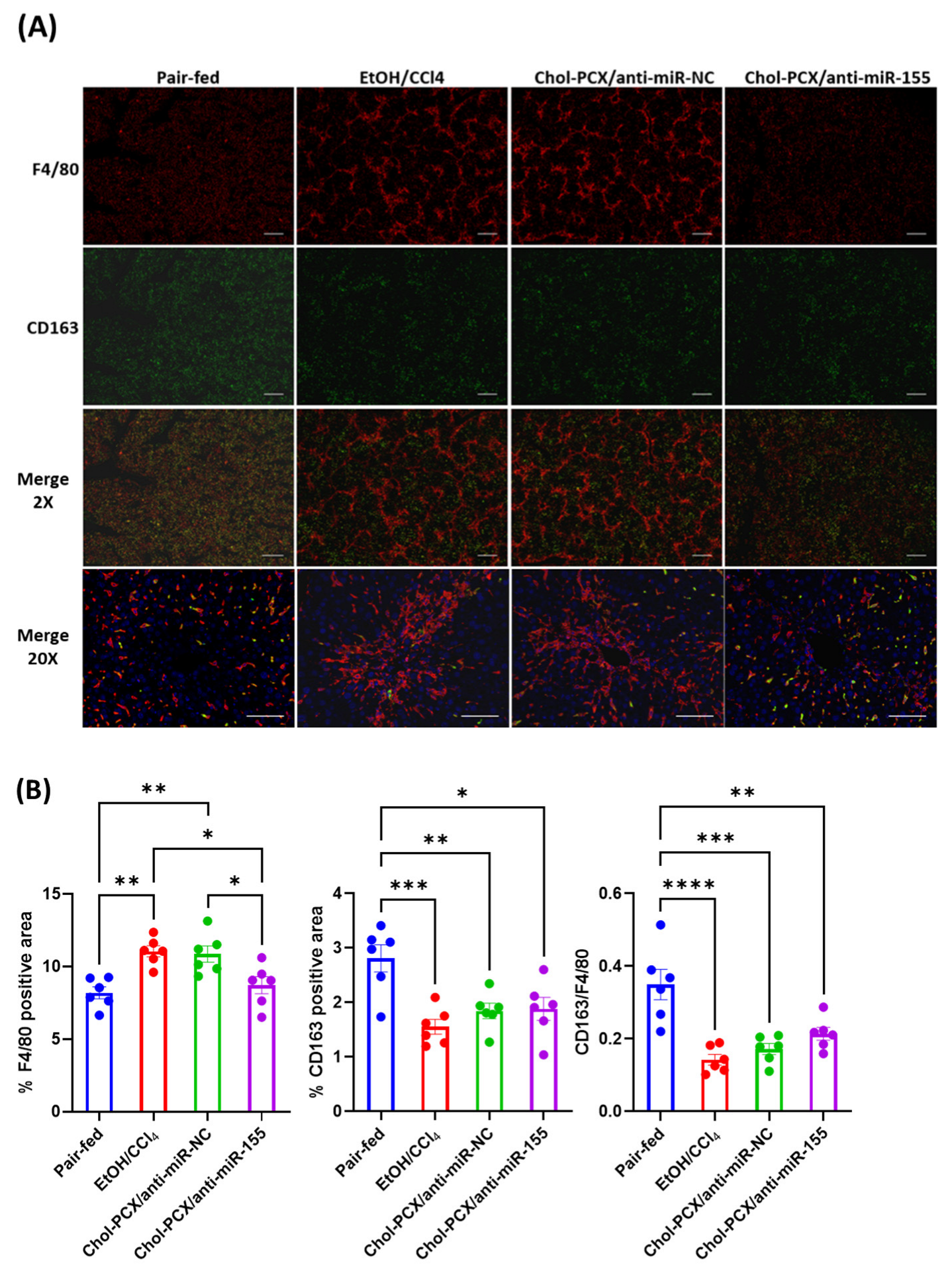
2.9. Histology and Immunohistochemistry
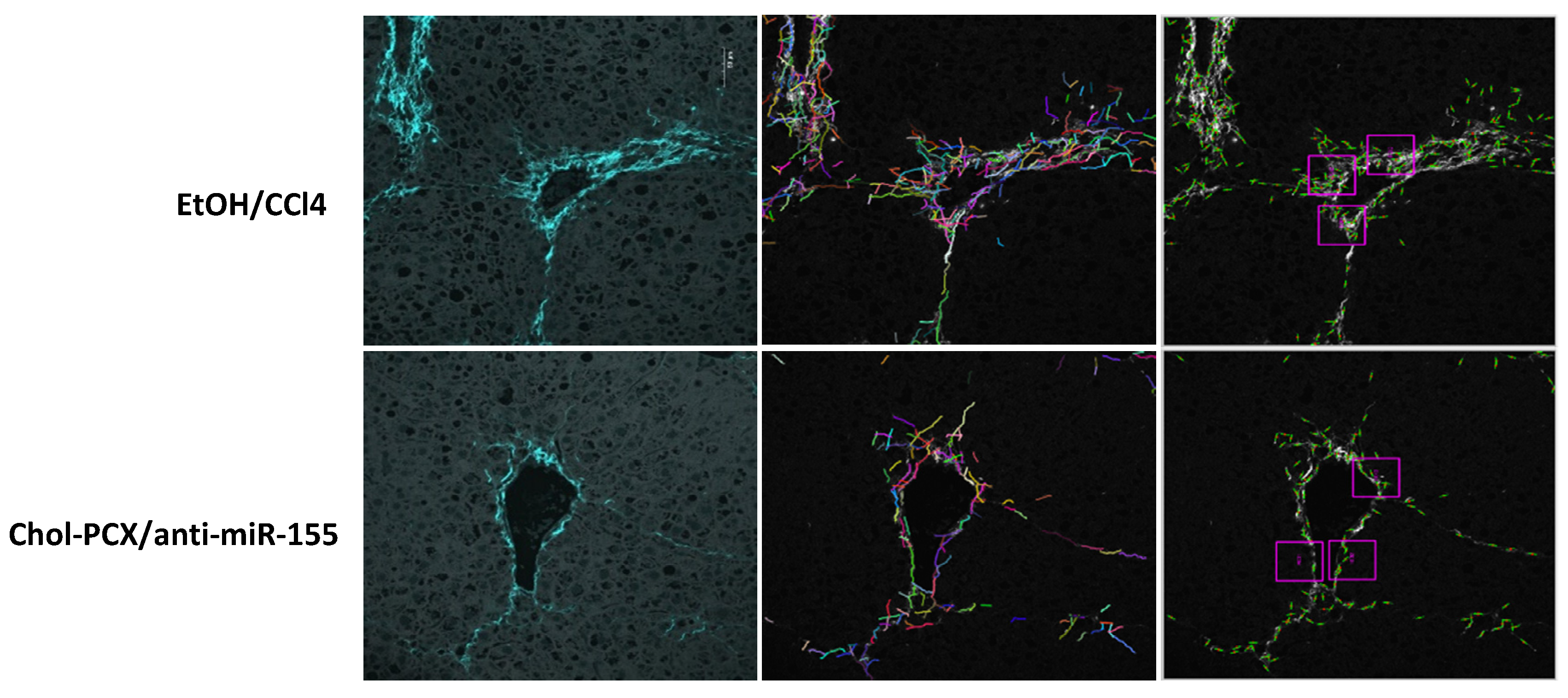
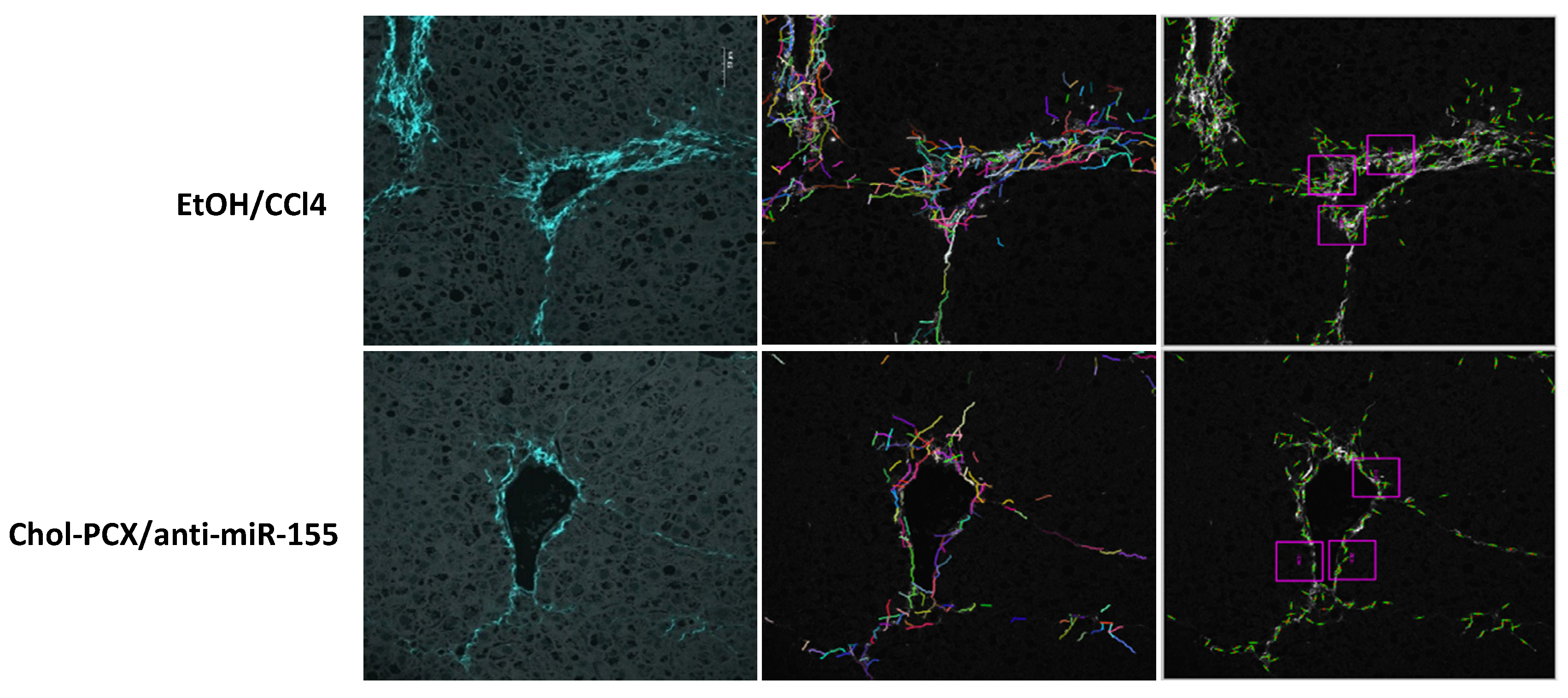
2.10. Second Harmonic Generation (SHG) Imaging
2.11. Statistical Analysis
3. Results
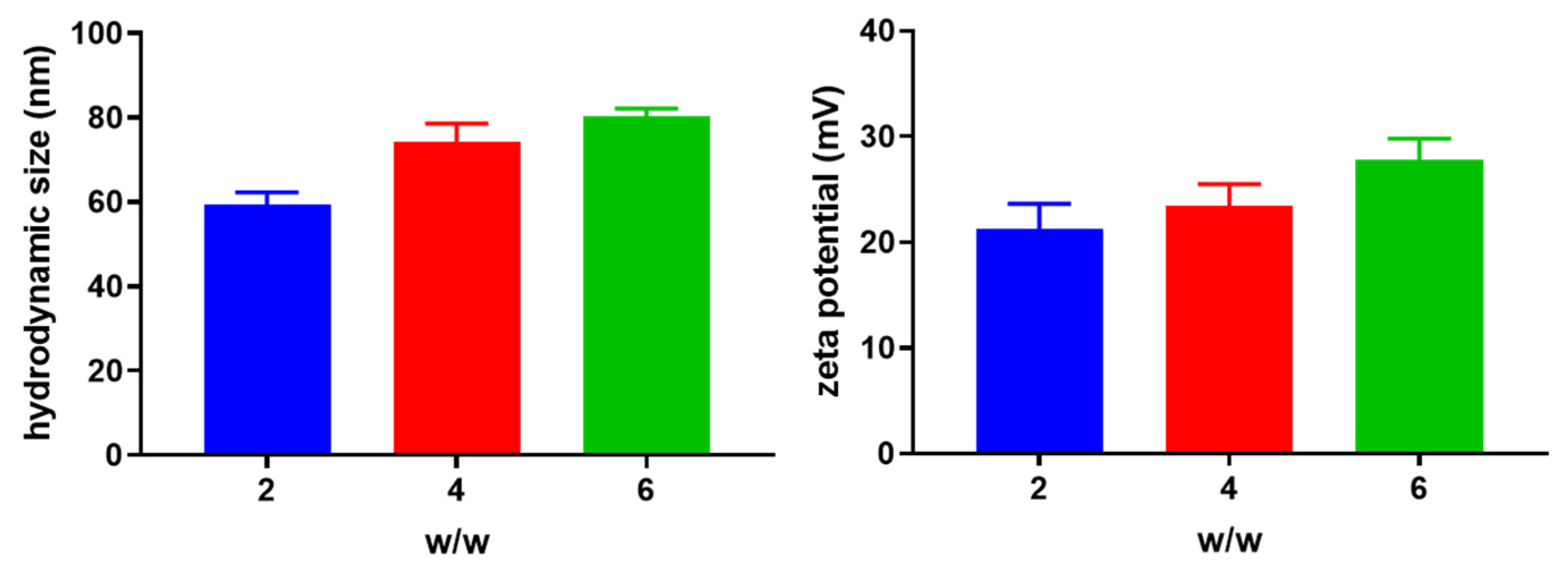
3.1. Preparation and Characterization of miRNA Nanoparticles
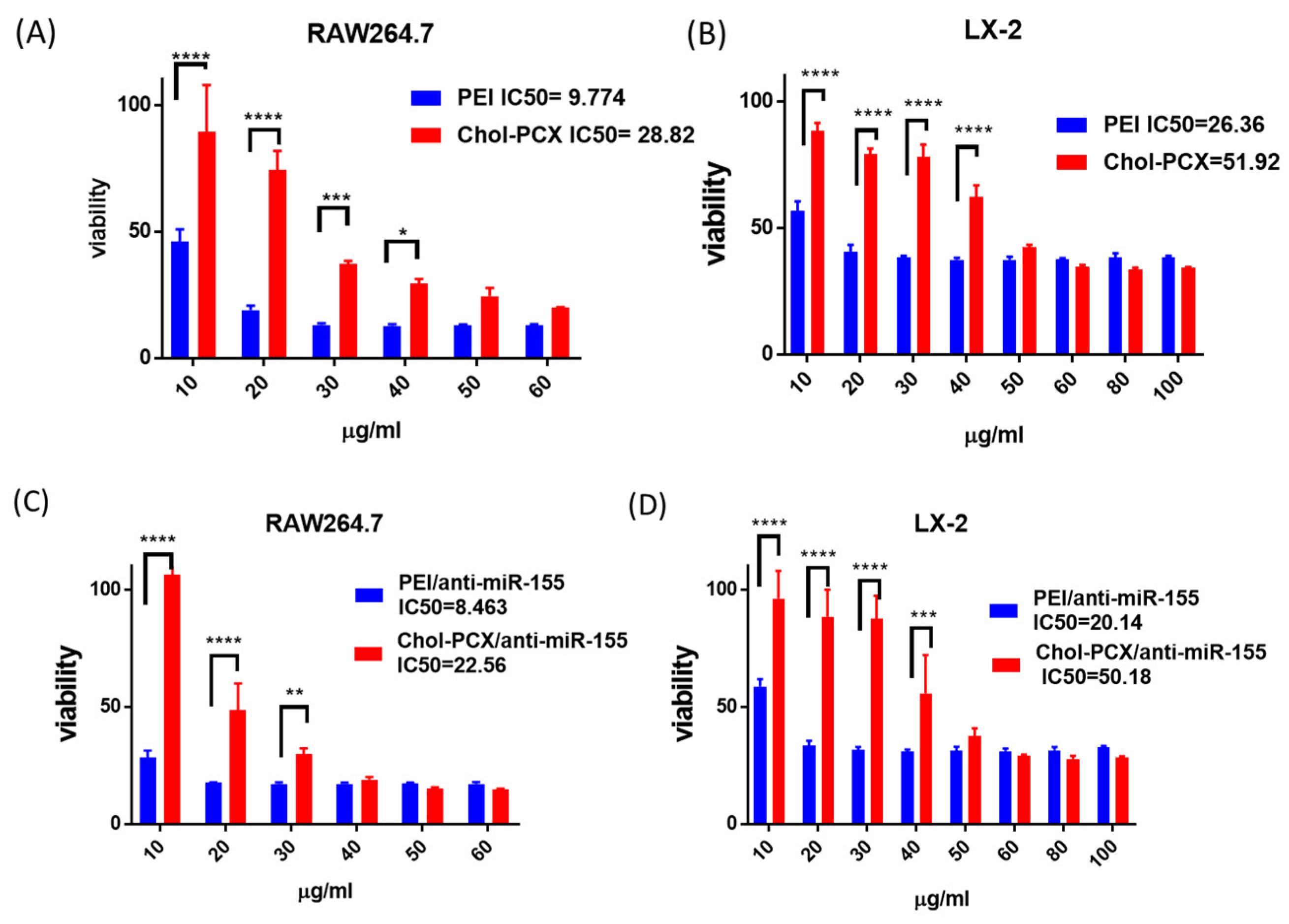
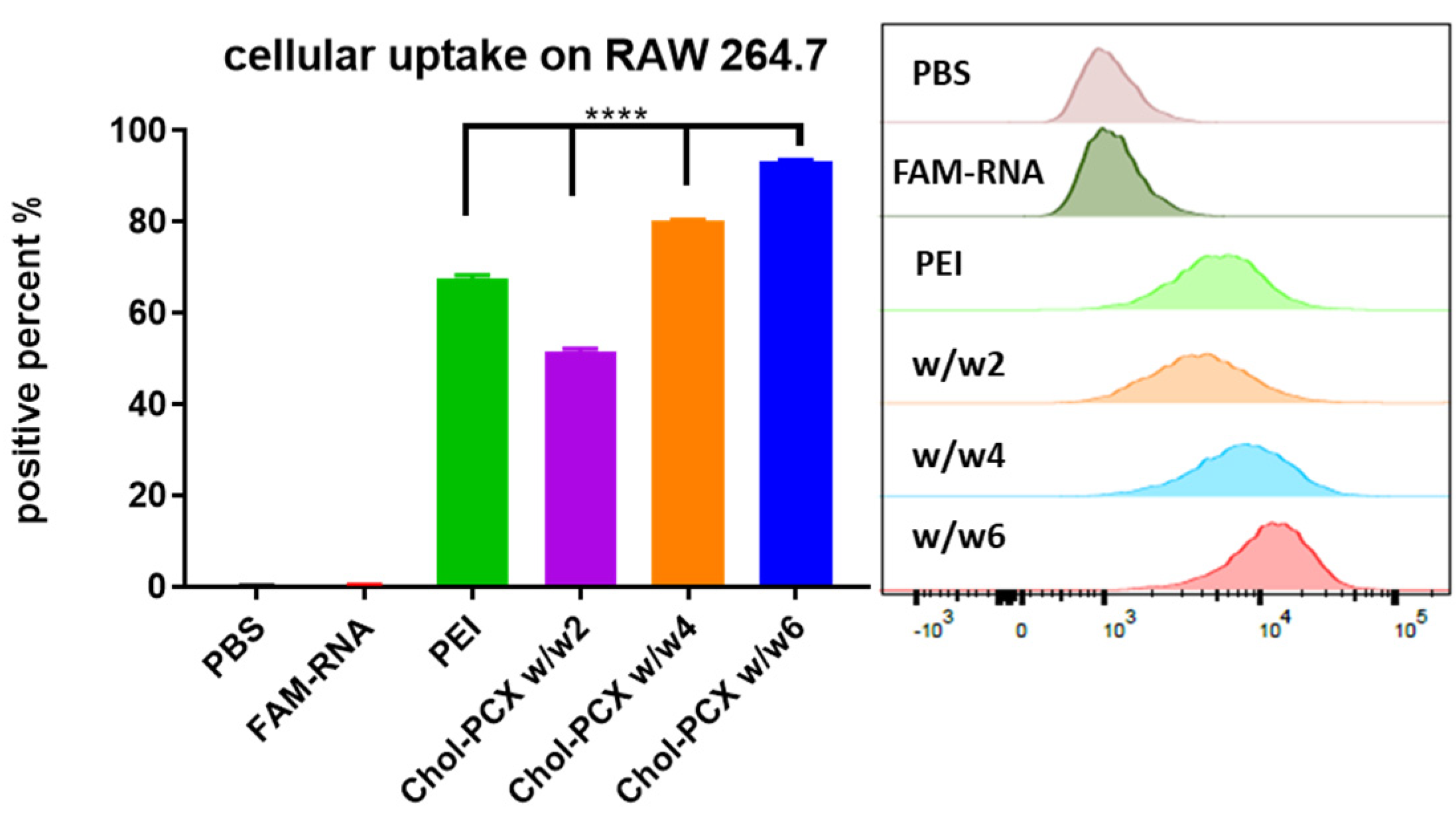
3.2. Cytotoxicity and Intracellular Trafficking
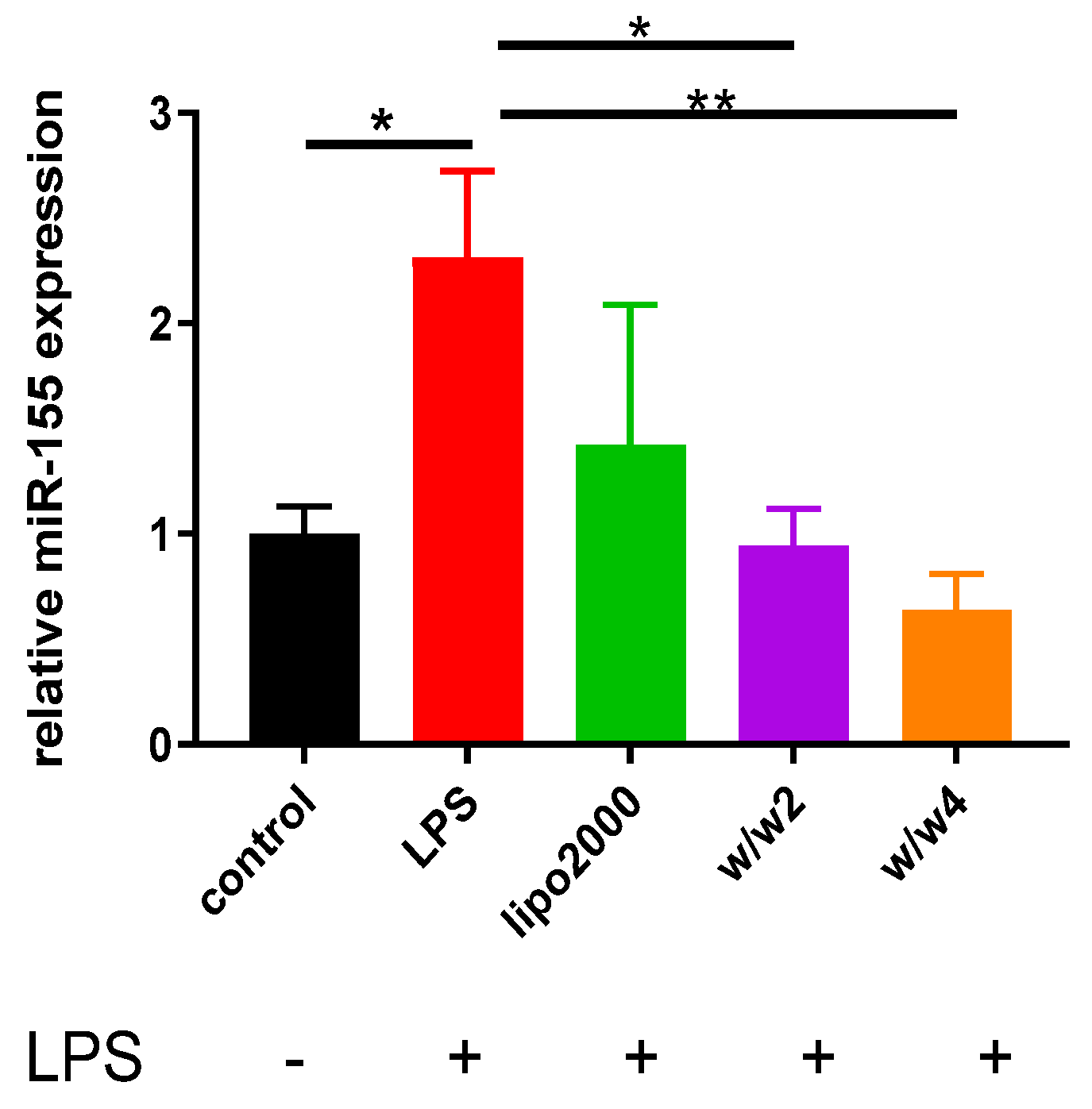
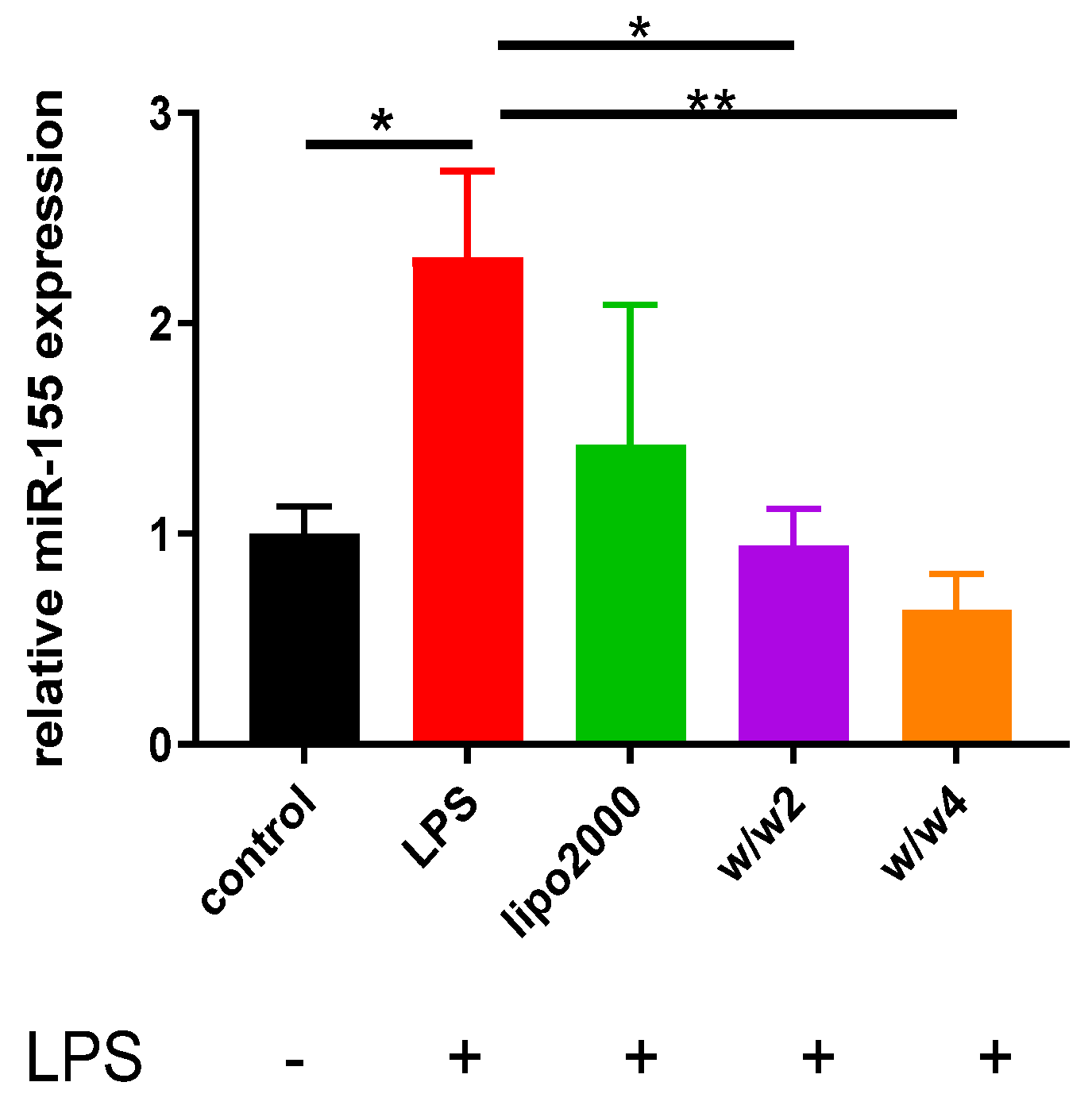
3.3. Chol-PCX/anti-miR-155 Nanoparticles Downregulate miR-155 Expression in Macrophages
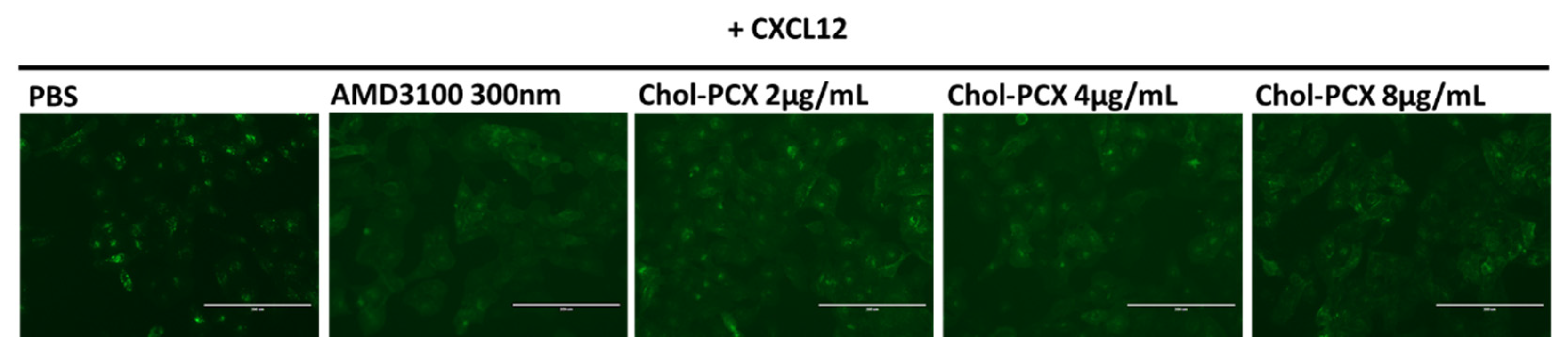
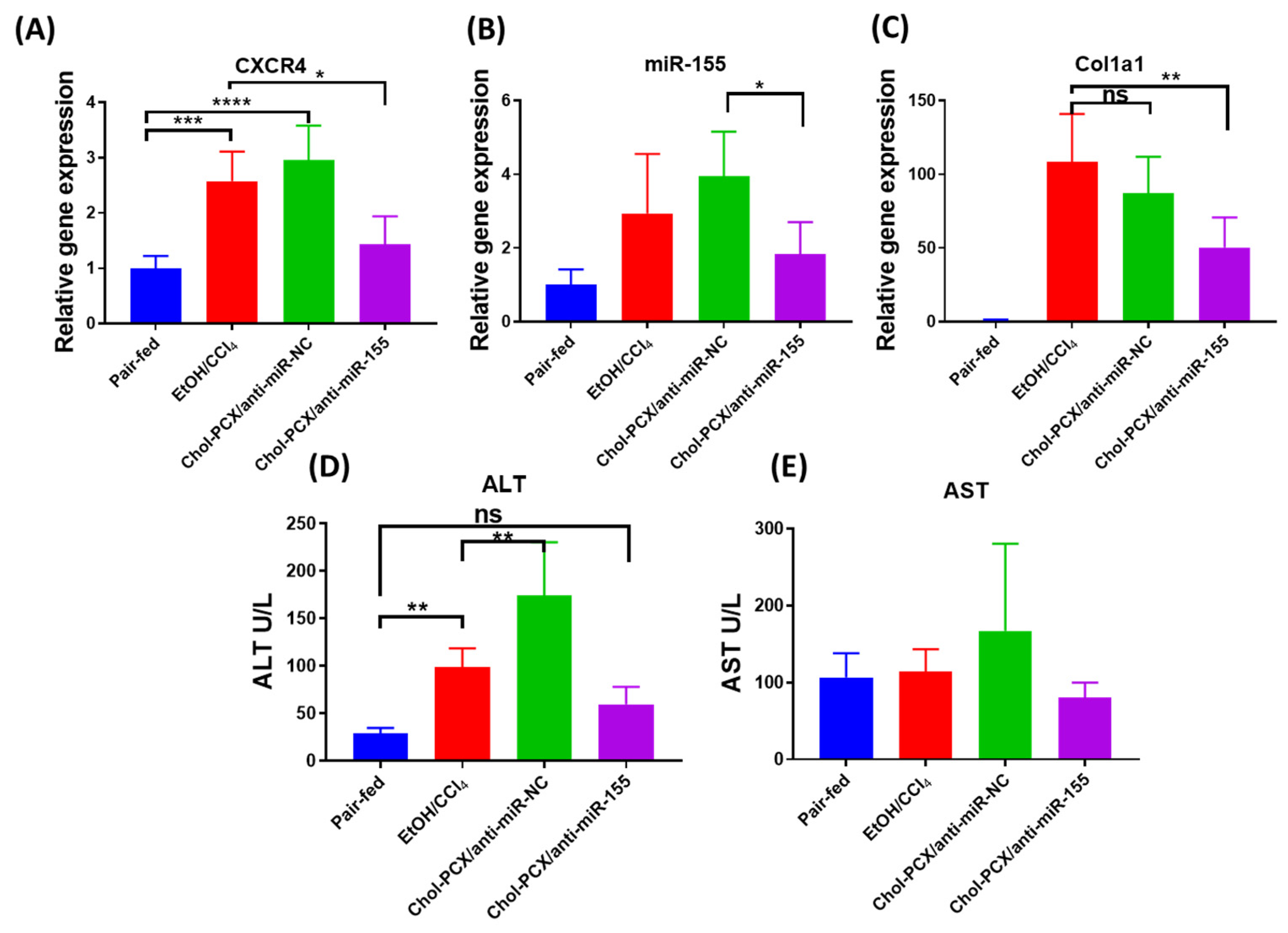
3.4. Inhibition of CXCR4 by Chol-PCX In Vitro
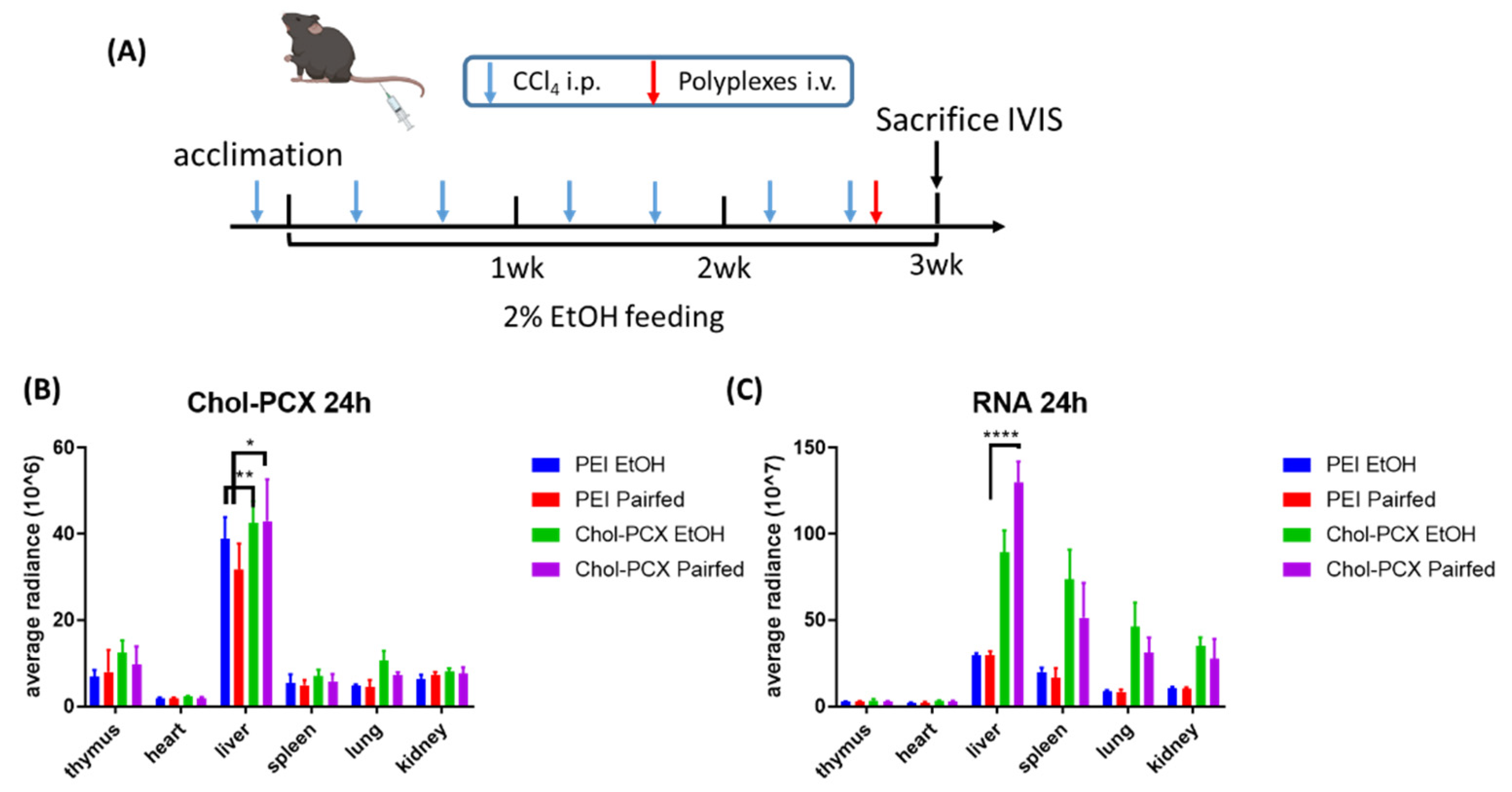
3.5. Chol-PCX Nanoparticles Biodistribution in AALD Fibrosis Model
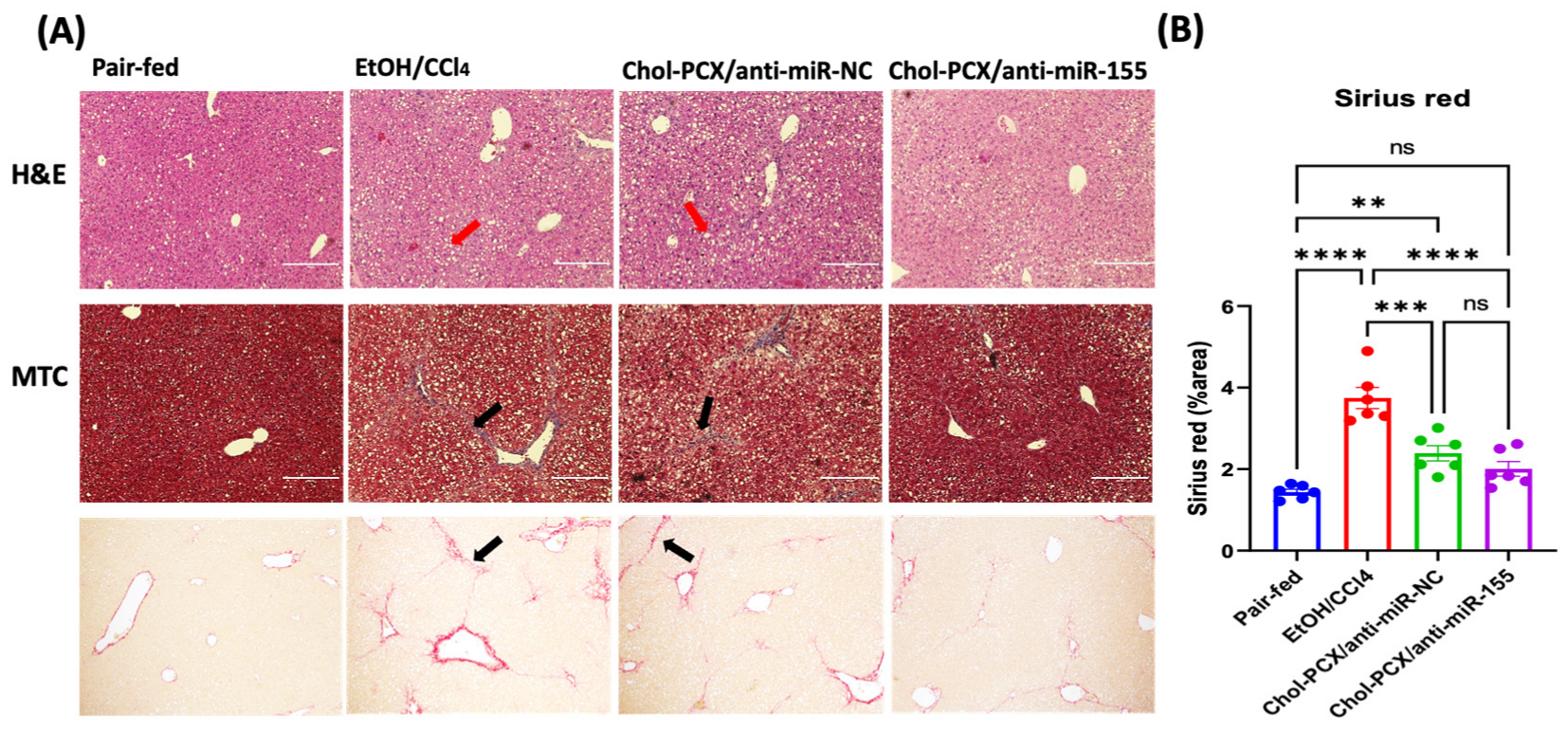
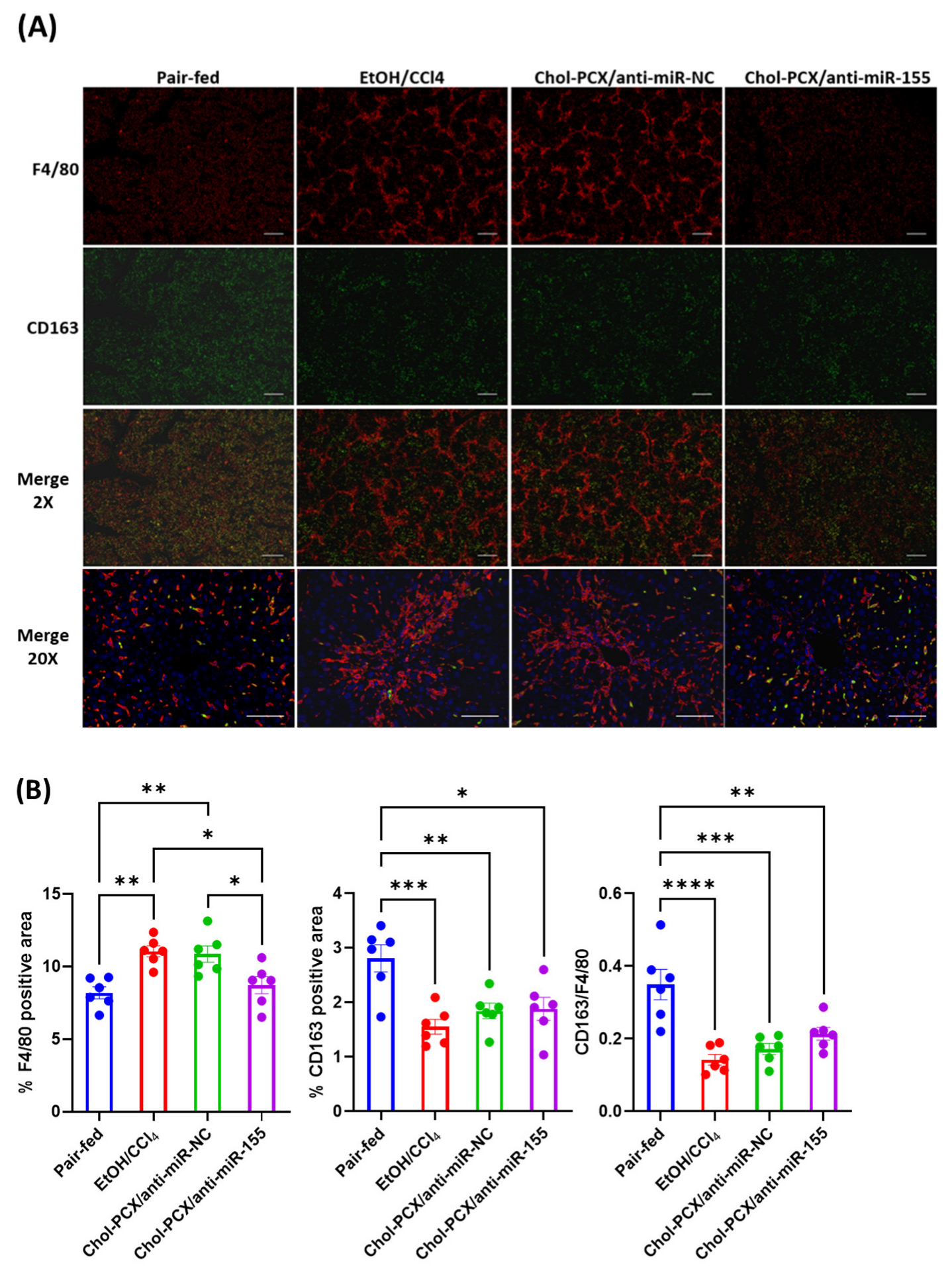
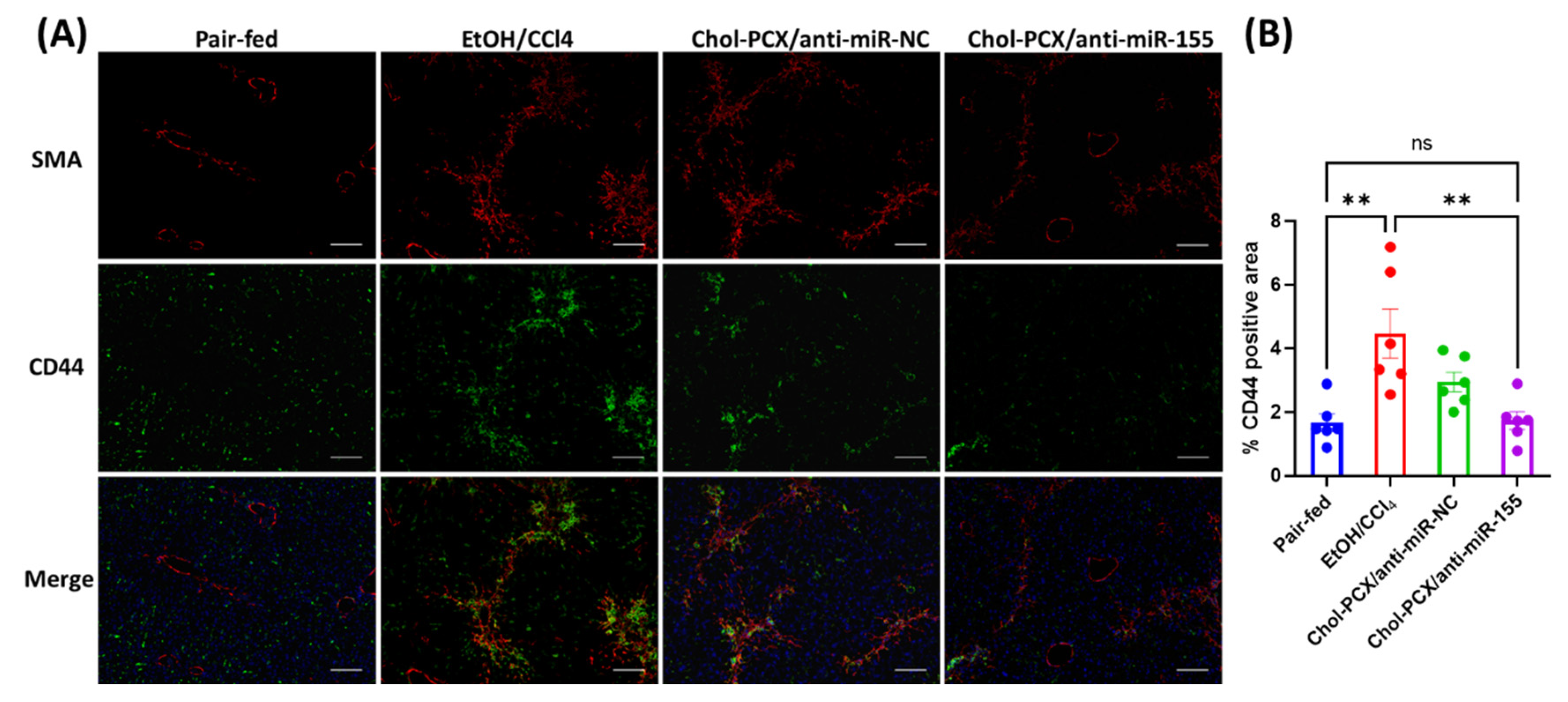
3.6. Chol-PCX/anti-miR-155 Treatment Ameliorates AALD Fibrosis
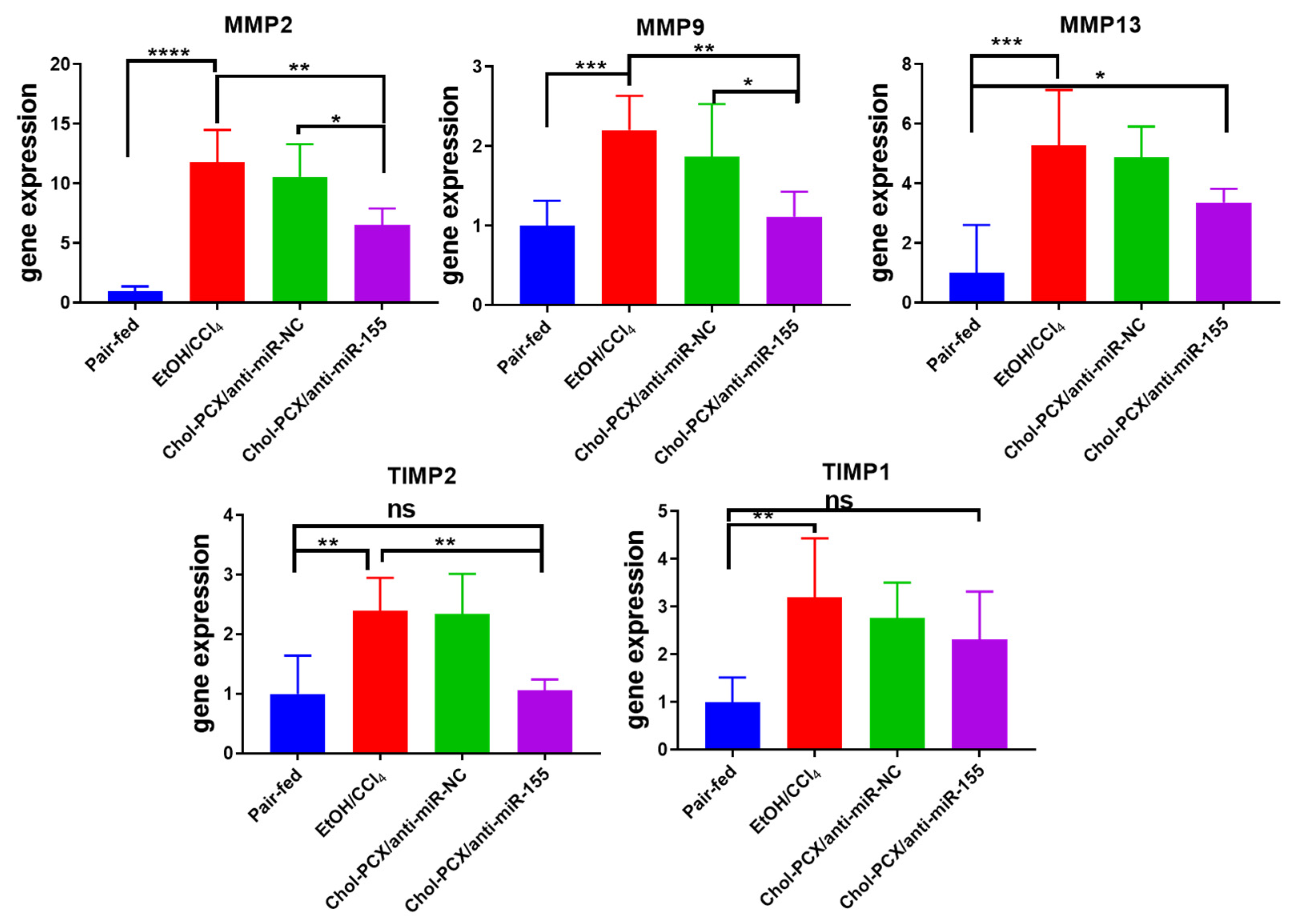
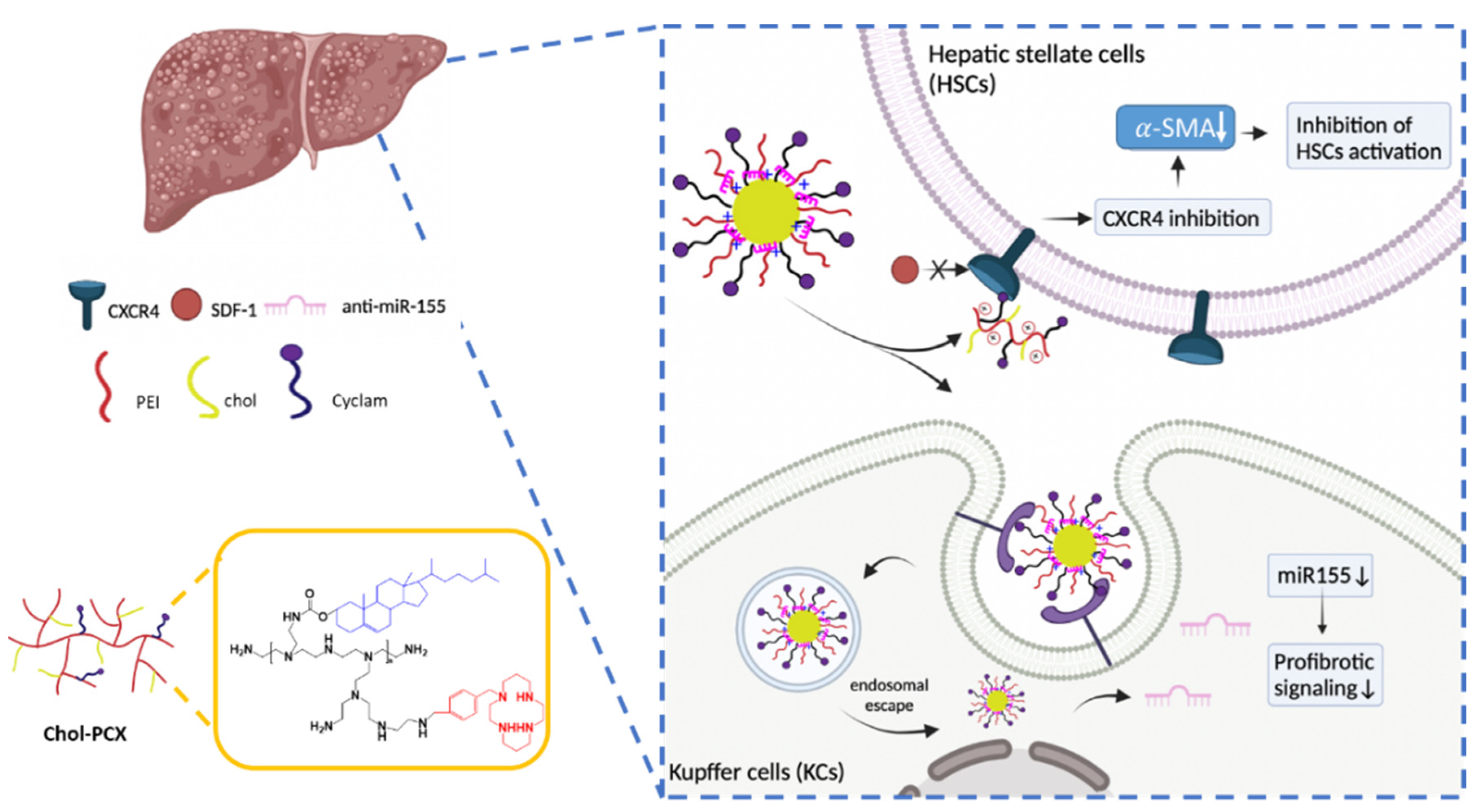
3.7. Analysis of the Therapeutic Mechanism of Action of the Nanoparticles in AALD Fibrosis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Bajaj, J.S. Alcohol, liver disease and the gut microbiota. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Rehm, J.; Shield, K.D. Global burden of alcohol use disorders and alcohol liver disease. Biomedicines 2019, 7, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, B.; Bataller, R. Alcoholic liver disease: Pathogenesis and new therapeutic targets. Gastroenterology 2011, 141, 1572–1585. [Google Scholar] [CrossRef] [Green Version]
- Teschke, R. Alcoholic liver disease: Alcohol metabolism, cascade of molecular mechanisms, cellular targets, and clinical aspects. Biomedicines 2018, 6, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shim, Y.-R.; Jeong, W.-I. Recent advances of sterile inflammation and inter-organ cross-talk in alcoholic liver disease. Exp. Mol. Med. 2020, 52, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Marrone, G.; Shah, V.H.; Gracia-Sancho, J. Sinusoidal communication in liver fibrosis and regeneration. J. Hepatol. 2016, 65, 608–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, C.; Tacke, F. Hepatic macrophages in homeostasis and liver diseases: From pathogenesis to novel therapeutic strategies. Cell. Mol. Immunol. 2016, 13, 316–327. [Google Scholar] [CrossRef] [Green Version]
- Anselmo, A.C.; Gupta, V.; Zern, B.J.; Pan, D.; Zakrewsky, M.; Muzykantov, V.; Mitragotri, S. Delivering nanoparticles to lungs while avoiding liver and spleen through adsorption on red blood cells. ACS Nano 2013, 7, 11129–11137. [Google Scholar] [CrossRef] [Green Version]
- Yoo, J.-W.; Chambers, E.; Mitragotri, S. Factors that control the circulation time of nanoparticles in blood: Challenges, solutions and future prospects. Curr. Pharm. Des. 2010, 16, 2298–2307. [Google Scholar] [CrossRef]
- Sung, Y.-C.; Liu, Y.-C.; Chao, P.-H.; Chang, C.-C.; Jin, P.-R.; Lin, T.-T.; Lin, J.-A.; Cheng, H.-T.; Wang, J.; Lai, C.P. Combined delivery of sorafenib and a MEK inhibitor using CXCR4-targeted nanoparticles reduces hepatic fibrosis and prevents tumor development. Theranostics 2018, 8, 894. [Google Scholar] [CrossRef]
- Lin, T.-T.; Gao, D.-Y.; Liu, Y.-C.; Sung, Y.-C.; Wan, D.; Liu, J.-Y.; Chiang, T.; Wang, L.; Chen, Y. Development and characterization of sorafenib-loaded PLGA nanoparticles for the systemic treatment of liver fibrosis. J. Control. Release 2016, 221, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Paunovska, K.; Da Silva Sanchez, A.J.; Sago, C.D.; Gan, Z.; Lokugamage, M.P.; Islam, F.Z.; Kalathoor, S.; Krupczak, B.R.; Dahlman, J.E. Nanoparticles containing oxidized cholesterol deliver mRNA to the liver microenvironment at clinically relevant doses. Adv. Mater. 2019, 31, 1807748. [Google Scholar] [CrossRef] [PubMed]
- Maugeri, M.; Nawaz, M.; Papadimitriou, A.; Angerfors, A.; Camponeschi, A.; Na, M.; Hölttä, M.; Skantze, P.; Johansson, S.; Sundqvist, M. Linkage between endosomal escape of LNP-mRNA and loading into EVs for transport to other cells. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, L.R.; Wan, Q.; Wen, D.; Zhang, Y.; Yu, J.; Kang, J.K.; Zhu, C.; McKinnon, E.L.; Gu, Z.; Qiang, L. Targeted delivery of notch inhibitor attenuates obesity-induced glucose intolerance and liver fibrosis. ACS Nano 2020, 14, 6878–6886. [Google Scholar] [CrossRef] [PubMed]
- Torres, J.-L.; Novo-Veleiro, I.; Manzanedo, L.; Alvela-Suárez, L.; Macías, R.; Laso, F.-J.; Marcos, M. Role of microRNAs in alcohol-induced liver disorders and non-alcoholic fatty liver disease. World J. Gastroenterol. 2018, 24, 4104. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Rana, T.M. Therapeutic targeting of microRNAs: Current status and future challenges. Nat. Rev. Drug Discov. 2014, 13, 622–638. [Google Scholar] [CrossRef]
- Burnett, J.C.; Rossi, J.J. RNA-based therapeutics: Current progress and future prospects. Chem. Biol. 2012, 19, 60–71. [Google Scholar] [CrossRef] [Green Version]
- Bala, S.; Szabo, G. MicroRNA signature in alcoholic liver disease. Int. J. Hepatol. 2012, 2012. [Google Scholar] [CrossRef] [Green Version]
- Bala, S.; Csak, T.; Saha, B.; Zatsiorsky, J.; Kodys, K.; Catalano, D.; Satishchandran, A.; Szabo, G. The pro-inflammatory effects of miR-155 promote liver fibrosis and alcohol-induced steatohepatitis. J. Hepatol. 2016, 64, 1378–1387. [Google Scholar] [CrossRef] [Green Version]
- Szabo, G.; Bala, S. Alcoholic liver disease and the gut-liver axis. World J. Gastroenterol. WJG 2010, 16, 1321. [Google Scholar] [CrossRef]
- Seth, D.; Haber, P.S.; Syn, W.K.; Diehl, A.M.; Day, C.P. Pathogenesis of alcohol-induced liver disease: Classical concepts and recent advances. J. Gastroenterol. Hepatol. 2011, 26, 1089–1105. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Huang, Y.; Reiberger, T.; Duyverman, A.M.; Huang, P.; Samuel, R.; Hiddingh, L.; Roberge, S.; Koppel, C.; Lauwers, G.Y. Differential effects of sorafenib on liver versus tumor fibrosis mediated by stromal-derived factor 1 alpha/C-X-C receptor type 4 axis and myeloid differentiation antigen–positive myeloid cell infiltration in mice. Hepatology 2014, 59, 1435–1447. [Google Scholar] [CrossRef] [PubMed]
- Hong, F.; Tuyama, A.; Lee, T.F.; Loke, J.; Agarwal, R.; Cheng, X.; Garg, A.; Fiel, M.I.; Schwartz, M.; Walewski, J. Hepatic stellate cells express functional CXCR4: Role in stromal cell–derived factor-1α–mediated stellate cell activation. Hepatology 2009, 49, 2055–2067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Hang, Y.; Wang, Y.; Sleightholm, R.; Prajapati, D.R.; Bader, J.; Yu, A.; Tang, W.; Jaramillo, L.; Li, J.; et al. Stromal Modulation and Treatment of Metastatic Pancreatic Cancer with Local Intraperitoneal Triple miRNA/siRNA Nanotherapy. ACS Nano 2020, 14, 255–271. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Wang, Y.; Li, J.; Hang, Y.; Jaramillo, L.; Wehrkamp, C.J.; Phillippi, M.A.; Mohr, A.M.; Chen, Y.; Talmon, G.A.; et al. Cholangiocarcinoma therapy with nanoparticles that combine downregulation of MicroRNA-210 with inhibition of cancer cell invasiveness. Theranostics 2018, 8, 4305–4320. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xie, Y.; Williams, J.; Hang, Y.; Richter, L.; Becker, M.; Amador, C.; Oupicky, D.; Hyde, R.K. Use of polymeric CXCR4 inhibitors as siRNA delivery vehicles for the treatment of acute myeloid leukemia. Cancer Gene Ther. 2019, 27, 45–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerlach, L.O.; Skerlj, R.T.; Bridger, G.J.; Schwartz, T.W. Molecular interactions of cyclam and bicyclam non-peptide antagonists with the CXCR4 chemokine receptor. J. Biol. Chem. 2001, 276, 14153–14160. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Yu, F.; Zhang, F.; Chen, G.; Wang, K.; Sun, M.; Li, J.; Oupický, D. Cyclam-modified PEI for combined VEGF siRNA silencing and CXCR4 inhibition to treat metastatic breast cancer. Biomacromolecules 2018, 19, 392–401. [Google Scholar] [CrossRef]
- Xu, L.; Hui, A.; Albanis, E.; Arthur, M.; O’byrne, S.; Blaner, W.; Mukherjee, P.; Friedman, S.; Eng, F. Human hepatic stellate cell lines, LX-1 and LX-2: New tools for analysis of hepatic fibrosis. Gut 2005, 54, 142–151. [Google Scholar] [CrossRef] [Green Version]
- Chiang, D.J.; Roychowdhury, S.; Bush, K.; McMullen, M.R.; Pisano, S.; Niese, K.; Olman, M.A.; Pritchard, M.T.; Nagy, L.E. Adenosine 2A receptor antagonist prevented and reversed liver fibrosis in a mouse model of ethanol-exacerbated liver fibrosis. PLoS ONE 2013, 8, e69114. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Magee, D.; Treanor, D.; Crellin, D.; Shires, M.; Smith, K.; Mohee, K.; Quirke, P. Colour normalisation in digital histopathology images. In Proceedings of the Optical Tissue Image analysis in Microscopy, Histopathology and Endoscopy (MICCAI Workshop), Citeseer, London, UK, 24 September 2009; pp. 100–111. [Google Scholar]
- Ruifrok, A.C.; Johnston, D.A. Quantification of histochemical staining by color deconvolution. Anal. Quant. Cytol. Histol. 2001, 23, 291–299. [Google Scholar] [PubMed]
- Toth, Z.E.; Mezey, E. Simultaneous visualization of multiple antigens with tyramide signal amplification using antibodies from the same species. J. Histochem. Cytochem. 2007, 55, 545–554. [Google Scholar] [CrossRef] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bredfeldt, J.S.; Liu, Y.; Conklin, M.W.; Keely, P.J.; Mackie, T.R.; Eliceiri, K.W. Automated quantification of aligned collagen for human breast carcinoma prognosis. J. Pathol. Inform. 2014, 5, 28. [Google Scholar] [CrossRef] [PubMed]
- Bredfeldt, J.S.; Liu, Y.; Pehlke, C.A.; Conklin, M.W.; Szulczewski, J.M.; Inman, D.R.; Keely, P.J.; Nowak, R.D.; Mackie, T.R.; Eliceiri, K.W. Computational segmentation of collagen fibers from second-harmonic generation images of breast cancer. J. Biomed. Opt. 2014, 19, 16007. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Keikhosravi, A.; Mehta, G.S.; Drifka, C.R.; Eliceiri, K.W. Methods for quantifying fibrillar collagen alignment. In Fibrosis; Springer: Berlin/Heidelberg, Germany, 2017; pp. 429–451. [Google Scholar]
- Wu, P.; Luo, X.; Wu, H.; Yu, F.; Wang, K.; Sun, M.; Oupicky, D. Cholesterol Modification Enhances Antimetastatic Activity and siRNA Delivery Efficacy of Poly (ethylenimine)-Based CXCR4 Antagonists. Macromol. Biosci. 2018, 18, 1800234. [Google Scholar] [CrossRef] [PubMed]
- Taranejoo, S.; Liu, J.; Verma, P.; Hourigan, K. A review of the developments of characteristics of PEI derivatives for gene delivery applications. J. Appl. Polym. Sci. 2015, 132, 42096. [Google Scholar] [CrossRef]
- Cicchi, R.; Vogler, N.; Kapsokalyvas, D.; Dietzek, B.; Popp, J.; Pavone, F.S. From molecular structure to tissue architecture: Collagen organization probed by SHG microscopy. J. Biophotonics 2013, 6, 129–142. [Google Scholar] [CrossRef]
- Lackner, C.; Tiniakos, D. Fibrosis and alcohol-related liver disease. J. Hepatol. 2019, 70, 294–304. [Google Scholar] [CrossRef]
- Urashima, S.; Tsutsumi, M.; Ozaki, K.; Tsuchishima, M.; Shimanaka, K.; Ueshima, Y.; Takase, S. Immunohistochemical study of hyaluronate receptor (CD44) in alcoholic liver disease. Alcohol. Clin. Exp. Res. 2000, 24, 34S–38S. [Google Scholar] [CrossRef]
- Li, W.; Zhou, C.; Fu, Y.; Chen, T.; Liu, X.; Zhang, Z.; Gong, T. Targeted delivery of hyaluronic acid nanomicelles to hepatic stellate cells in hepatic fibrosis rats. Acta Pharm. Sin. B 2020, 10, 693–710. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-Y.; Chiang, T.; Liu, C.-H.; Chern, G.-G.; Lin, T.-T.; Gao, D.-Y.; Chen, Y. Delivery of siRNA using CXCR4-targeted nanoparticles modulates tumor microenvironment and achieves a potent antitumor response in liver cancer. Mol. Ther. 2015, 23, 1772–1782. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-H.; Chan, K.-M.; Chiang, T.; Liu, J.-Y.; Chern, G.-G.; Hsu, F.-F.; Wu, Y.-H.; Liu, Y.-C.; Chen, Y. Dual-functional nanoparticles targeting CXCR4 and delivering antiangiogenic siRNA ameliorate liver fibrosis. Mol. Pharm. 2016, 13, 2253–2262. [Google Scholar] [CrossRef] [PubMed]
- Saiman, Y.; Jiao, J.; Fiel, M.I.; Friedman, S.L.; Aloman, C.; Bansal, M.B. Inhibition of the CXCL12/CXCR4 chemokine axis with AMD3100, a CXCR4 small molecule inhibitor, worsens murine hepatic injury. Hepatol Res. 2015, 45, 794–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mavier, P.; Martin, N.; Couchie, D.; Préaux, A.-M.; Laperche, Y.; Zafrani, E.S. Expression of stromal cell-derived factor-1 and of its receptor CXCR4 in liver regeneration from oval cells in rat. Am. J. Pathol. 2004, 165, 1969–1977. [Google Scholar] [CrossRef] [Green Version]
- Baeck, C.; Wei, X.; Bartneck, M.; Fech, V.; Heymann, F.; Gassler, N.; Hittatiya, K.; Eulberg, D.; Luedde, T.; Trautwein, C.; et al. Pharmacological inhibition of the chemokine C-C motif chemokine ligand 2 (monocyte chemoattractant protein 1) accelerates liver fibrosis regression by suppressing Ly-6C(+) macrophage infiltration in mice. Hepatology 2014, 59, 1060–1072. [Google Scholar] [CrossRef]
- Cubero, F.; Nieto, N. Kupffer cells and alcoholic liver disease. Rev. Esp. De Enferm. Dig. 2006, 98, 460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poilil Surendran, S.; George Thomas, R.; Moon, M.J.; Jeong, Y.Y. Nanoparticles for the treatment of liver fibrosis. Int. J. Nanomed. 2017, 12, 6997–7006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, W.; Zhao, J.; Tang, N.; Zeng, X.; Wu, K.; Ye, C.; Shi, J.; Lu, C.; Ning, B.; Zhang, J.; et al. MicroRNA-155 attenuates activation of hepatic stellate cell by simultaneously preventing EMT process and ERK1 signalling pathway. Liver Int. 2015, 35, 1234–1243. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, C.; Hang, Y.; Tang, W.; Sil, D.; Jensen-Smith, H.C.; Bennett, R.G.; McVicker, B.L.; Oupický, D. Dually Active Polycation/miRNA Nanoparticles for the Treatment of Fibrosis in Alcohol-Associated Liver Disease. Pharmaceutics 2022, 14, 669. https://doi.org/10.3390/pharmaceutics14030669
Zhang C, Hang Y, Tang W, Sil D, Jensen-Smith HC, Bennett RG, McVicker BL, Oupický D. Dually Active Polycation/miRNA Nanoparticles for the Treatment of Fibrosis in Alcohol-Associated Liver Disease. Pharmaceutics. 2022; 14(3):669. https://doi.org/10.3390/pharmaceutics14030669
Chicago/Turabian StyleZhang, Chuhan, Yu Hang, Weimin Tang, Diptesh Sil, Heather C. Jensen-Smith, Robert G. Bennett, Benita L. McVicker, and David Oupický. 2022. "Dually Active Polycation/miRNA Nanoparticles for the Treatment of Fibrosis in Alcohol-Associated Liver Disease" Pharmaceutics 14, no. 3: 669. https://doi.org/10.3390/pharmaceutics14030669
APA StyleZhang, C., Hang, Y., Tang, W., Sil, D., Jensen-Smith, H. C., Bennett, R. G., McVicker, B. L., & Oupický, D. (2022). Dually Active Polycation/miRNA Nanoparticles for the Treatment of Fibrosis in Alcohol-Associated Liver Disease. Pharmaceutics, 14(3), 669. https://doi.org/10.3390/pharmaceutics14030669