
Interaction of Antifungal Drugs with CYP3A- and OATP1B-Mediated Venetoclax Elimination

 , ,
, ,
Abstract
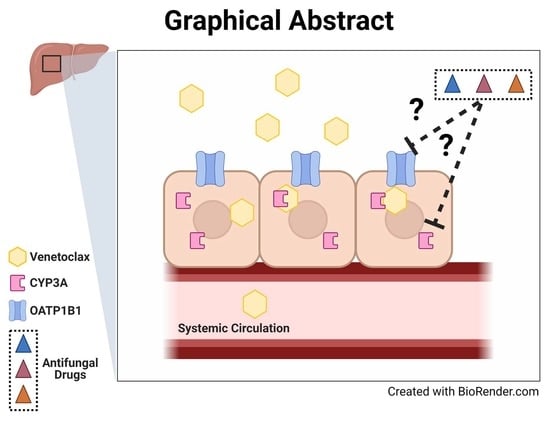
:
1. Introduction
2. Materials and Methods
2.1. Chemical and Reagents
2.2. In Vitro Metabolism Studies
2.3. In Vivo Pharmacokinetic Studies
2.4. Pharmacokinetic Data Analysis
2.5. In Vitro Transport Assays
2.6. In Vitro to In Vivo Extrapolation
2.7. Real-Time Polymerase Chain Reaction (qPCR)
2.8. Statistical Analysis
3. Results
3.1. Contribution of CYP3A to the Interaction between Ketoconazole and Venetoclax
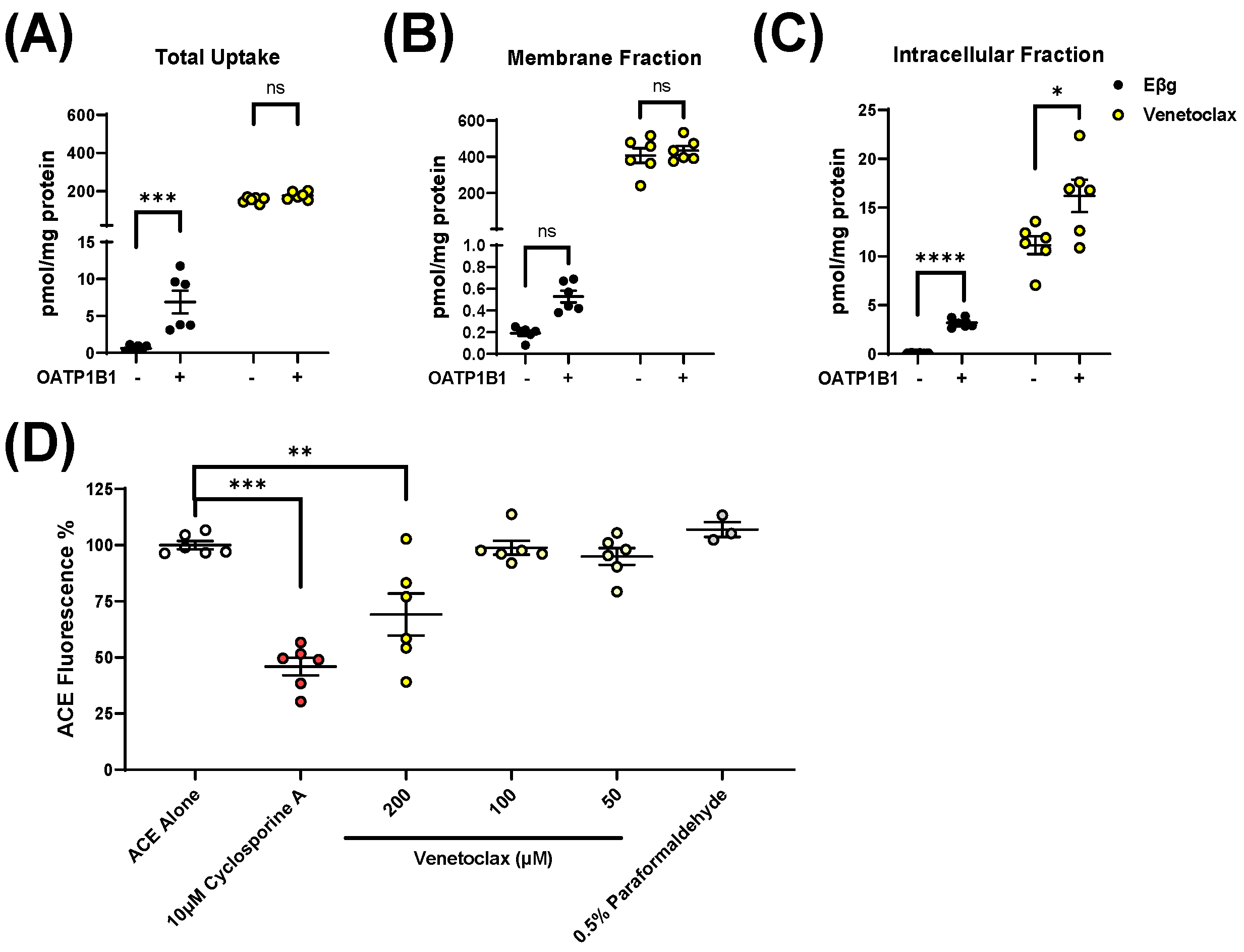
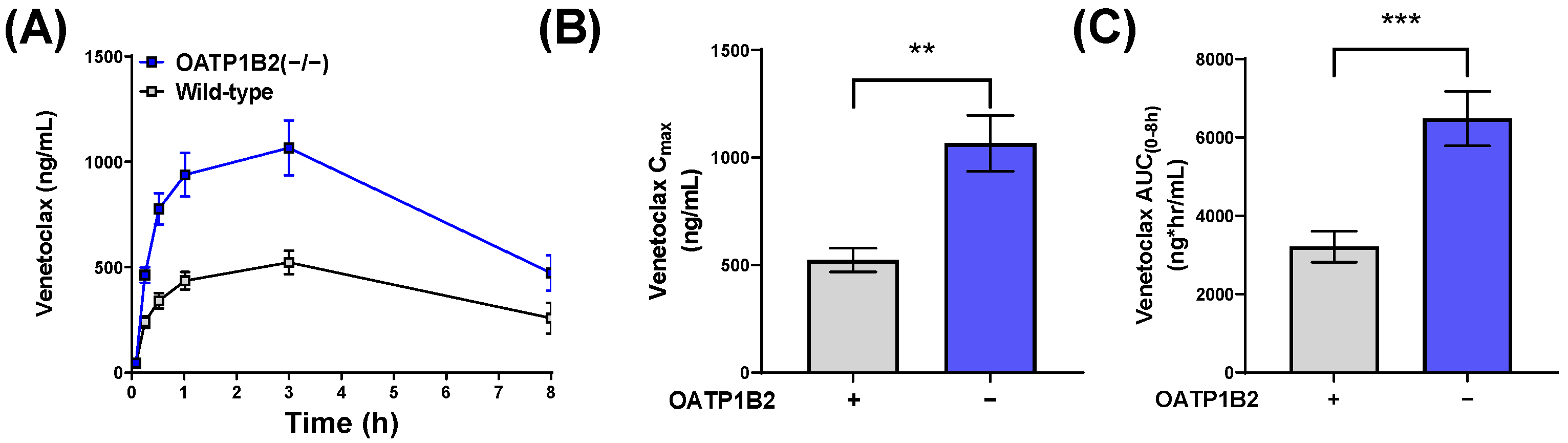
3.2. Influence of OATP1B1 and OATP1B2 on Venetoclax Uptake and Exposure
3.3. Impact of Clinically-Relevant Antifungal Drugs on OATP1B1 and OATP1B2 Function
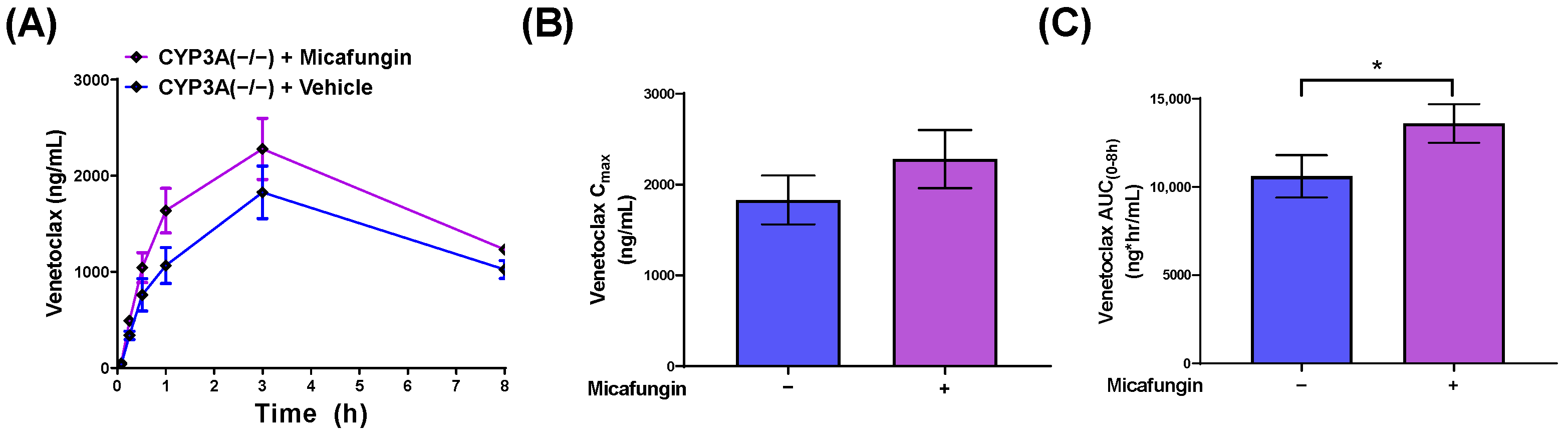
3.4. Impact of Micafungin on Venetoclax Exposure
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pollyea, D.A.; Amaya, M.; Strati, P.; Konopleva, M.Y. Venetoclax for AML: Changing the Treatment Paradigm. Blood Adv. 2019, 3, 4326–4335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Davids, M.S.; Roberts, A.W.; Seymour, J.F.; Pagel, J.M.; Kahl, B.S.; Wierda, W.G.; Puvvada, S.; Kipps, T.J.; Anderson, M.A.; Salem, A.H.; et al. Phase I First-in-Human Study of Venetoclax in Patients with Relapsed or Refractory Non-Hodgkin Lymphoma. J. Clin. Oncol. 2017, 35, 826–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Michmerhuizen, M.J.; Lao, Y.; Wan, K.; Salem, A.H.; Sawicki, J.; Serby, M.; Vaidyanathan, S.; Wong, S.L.; Agarwal, S.; et al. Metabolism and Disposition of a Novel B-Cell Lymphoma-2 Inhibitor Venetoclax in Humans and Characterization of Its Unusual Metabolites. Drug Metab. Dispos. 2017, 45, 294–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, S.K.; Salem, A.H.; Danilov, A.V.; Hu, B.; Puvvada, S.; Gutierrez, M.; Chien, D.; Lewis, L.D.; Wong, S.L. Effect of Ketoconazole, a Strong CYP3A Inhibitor, on the Pharmacokinetics of Venetoclax, a BCL-2 Inhibitor, in Patients with Non-Hodgkin Lymphoma. Br. J. Clin. Pharmacol. 2017, 83, 846–854. [Google Scholar] [CrossRef]
- Agarwal, S.K.; DiNardo, C.D.; Potluri, J.; Dunbar, M.; Kantarjian, H.M.; Humerickhouse, R.A.; Wong, S.L.; Menon, R.M.; Konopleva, M.Y.; Salem, A.H. Management of Venetoclax-Posaconazole Interaction in Acute Myeloid Leukemia Patients: Evaluation of Dose Adjustments. Clin. Ther. 2017, 39, 359–367. [Google Scholar] [CrossRef]
- Vermeer, L.M.M.; Isringhausen, C.D.; Ogilvie, B.W.; Buckley, D.B. Evaluation of Ketoconazole and Its Alternative Clinical CYP3A4/5 Inhibitors as Inhibitors of Drug Transporters: The in Vitro Effects of Ketoconazole, Ritonavir, Clarithromycin, and Itraconazole on 13 Clinically-Relevant Drug Transporters. Drug Metab. Dispos. 2016, 44, 453–459. [Google Scholar] [CrossRef] [Green Version]
- Garrison, D.A.; Talebi, Z.; Eisenmann, E.D.; Sparreboom, A.; Baker, S.D. Role of OATP1B1 and OATP1B3 in Drug-Drug Interactions Mediated by Tyrosine Kinase Inhibitors. Pharmaceutics 2020, 12, 856. [Google Scholar] [CrossRef]
- Berking, S.; Doedens, D.; Horns, H.; Fiegl, M.; Ostermann, H.; Rieger, C.T. Antifungal Prophylaxis in Newly Diagnosed AML Patients-Adherence to Guidelines and Feasibility in a Real Life Setting. Mycoses 2017, 60, 600–606. [Google Scholar] [CrossRef]
- Eisenmann, E.D.; Talebi, Z.; Sparreboom, A.; Baker, S.D. Boosting the Oral Bioavailability of Anticancer Drugs through Intentional Drug–Drug Interactions. Basic Clin. Pharmacol. Toxicol. 2021, bcpt.13623. [Google Scholar] [CrossRef]
- Xie, F.; Ding, X.; Zhang, Q.-Y. An Update on the Role of Intestinal Cytochrome P450 Enzymes in Drug Disposition. Acta Pharm. Sin. B 2016, 6, 374–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmerman, E.I.; Hu, S.; Roberts, J.L.; Gibson, A.A.; Orwick, S.J.; Li, L.; Sparreboom, A.; Baker, S.D. Contribution of OATP1B1 and OATP1B3 to the Disposition of Sorafenib and Sorafenib-Glucuronide. Clin. Cancer Res. 2013, 19, 1458–1466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenmann, E.D.; Jin, Y.; Weber, R.H.; Sparreboom, A.; Baker, S.D. Development and Validation of a Sensitive UHPLC-MS/MS Analytical Method for Venetoclax in Mouse Plasma, and Its Application to Pharmacokinetic Studies. J. Chromatogr. B 2020, 1152, 122176. [Google Scholar] [CrossRef] [PubMed]
- van Herwaarden, A.E.; Wagenaar, E.; van der Kruijssen, C.M.M.; van Waterschoot, R.A.B.; Smit, J.W.; Song, J.-Y.; van der Valk, M.A.; van Tellingen, O.; van der Hoorn, J.W.A.; Rosing, H.; et al. Knockout of Cytochrome P450 3A Yields New Mouse Models for Understanding Xenobiotic Metabolism. J. Clin. Investig. 2007, 117, 3583–3592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaher, H.; zu Schwabedissen, H.E.M.; Tirona, R.G.; Cox, M.L.; Obert, L.A.; Agrawal, N.; Palandra, J.; Stock, J.L.; Kim, R.B.; Ware, J.A. Targeted Disruption of Murine Organic Anion-Transporting Polypeptide 1b2 (Oatp1b2/Slco1b2) Significantly Alters Disposition of Prototypical Drug Substrates Pravastatin and Rifampin. Mol. Pharmacol. 2008, 74, 320–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leblanc, A.F.; Huang, K.M.; Uddin, M.E.; Anderson, J.T.; Chen, M.; Hu, S. Murine Pharmacokinetic Studies. Bio-Protocol 2018, 8, e3056. [Google Scholar] [CrossRef]
- Lancaster, C.S.; Bruun, G.H.; Peer, C.J.; Mikkelsen, T.S.; Corydon, T.J.; Gibson, A.A.; Hu, S.; Orwick, S.J.; Mathijssen, R.H.J.; Figg, W.D.; et al. OATP1B1 Polymorphism as a Determinant of Erythromycin Disposition. Clin. Pharmacol. Ther. 2012, 92, 642–650. [Google Scholar] [CrossRef]
- Ramsey, L.B.; Bruun, G.H.; Yang, W.; Treviño, L.R.; Vattathil, S.; Scheet, P.; Cheng, C.; Rosner, G.L.; Giacomini, K.M.; Fan, Y.; et al. Rare versus Common Variants in Pharmacogenetics: SLCO1B1 Variation and Methotrexate Disposition. Genome Res. 2012, 22, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Neul, C.; Schaeffeler, E.; Frisch, F.; Winter, S.; Schwab, M.; Koepsell, H.; Hu, S.; Laufer, S.; Baker, S.D.; et al. Sorafenib Activity and Disposition in Liver Cancer Does Not Depend on Organic Cation Transporter 1. Clin. Pharmacol. Ther. 2019, 107, 227–237. [Google Scholar] [CrossRef]
- Ungvári, O.; Király, L.; Bakos, É.; Özvegy-Laczka, C. 8-Acetoxy-Trisulfopyrene as the First Activatable Fluorogenic Probe for Add-and-Read Assessment of Organic Anion-Transporting Polypeptides, OATP1B1, OATP1B3, and OATP2B1. FASEB J. 2021, 35, e21863. [Google Scholar] [CrossRef]
- FDA Center for Drug Evaluation and Research. In Vitro Drug Interaction Studies—Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions Guidance for Industry. 2020. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/vitro-drug-interaction-studies-cytochrome-p450-enzyme-and-transporter-mediated-drug-interactions (accessed on 9 March 2022).
- Parkinson, A. Regulatory Recommendations for Calculating the Unbound Maximum Hepatic Inlet Concentration: A Complicated Story with a Surprising and Happy Ending. Drug Metab. Dispos. 2019, 47, 779–784. [Google Scholar] [CrossRef] [PubMed]
- FDA Center for Drug Evaluation and Research Application Number: 208573Orig1s000. Clinical Pharmacology and Biopharmaceutics Review(s). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/208573Orig1s000ClinPharmR.pdf (accessed on 9 March 2022).
- Taplitz, R.A.; Kennedy, E.B.; Bow, E.J.; Crews, J.; Gleason, C.; Hawley, D.K.; Langston, A.A.; Nastoupil, L.J.; Rajotte, M.; Rolston, K.V.; et al. Antimicrobial Prophylaxis for Adult Patients with Cancer-Related Immunosuppression: ASCO and IDSA Clinical Practice Guideline Update. J. Clin. Oncol. 2018, 36, 3043–3054. [Google Scholar] [CrossRef] [PubMed]
- Kalliokoski, A.; Niemi, M. Impact of OATP Transporters on Pharmacokinetics. Br. J. Pharmacol. 2009, 158, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Custodio, J.M.; Wang, H.; Hao, J.; Lepist, E.-I.; Ray, A.S.; Andrews, J.; Ling, K.H.J.; Cheng, A.; Kearney, B.P.; Ramanathan, S. Pharmacokinetics of Cobicistat Boosted-Elvitegravir Administered in Combination with Rosuvastatin. J. Clin. Pharmacol. 2014, 54, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Sherman, E.M.; Worley, M.V.; Unger, N.R.; Gauthier, T.P.; Schafer, J.J. Cobicistat: Review of a Pharmacokinetic Enhancer for HIV Infection. Clin. Ther. 2015, 37, 1876–1893. [Google Scholar] [CrossRef] [PubMed]
- la Porte, C.J.L.; Colbers, E.P.H.; Bertz, R.; Voncken, D.S.; Wikstrom, K.; Boeree, M.J.; Koopmans, P.P.; Hekster, Y.A.; Burger, D.M. Pharmacokinetics of Adjusted-Dose Lopinavir-Ritonavir Combined with Rifampin in Healthy Volunteers. Antimicrob. Agents Chemother. 2004, 48, 1553–1560. [Google Scholar] [CrossRef] [Green Version]
- Yunivita, V.; Dian, S.; Ganiem, A.R.; Hayati, E.; Achmad, T.H.; Dewi, A.P.; Teulen, M.; Meijerhof-Jager, P.; van Crevel, R.; Aarnoutse, R.; et al. Pharmacokinetics and Safety/Tolerability of Higher Oral and Intravenous Doses of Rifampicin in Adult Tuberculous Meningitis Patients. Int. J. Antimicrob. Agents 2016, 48, 415–421. [Google Scholar] [CrossRef]
- Templeton, I.E.; Thummel, K.E.; Kharasch, E.D.; Kunze, K.L.; Hoffer, C.; Nelson, W.L.; Isoherranen, N. Contribution of Itraconazole Metabolites to Inhibition of CYP3A4 in Vivo. Clin. Pharmacol. Ther. 2008, 83, 77–85. [Google Scholar] [CrossRef] [Green Version]
- Houšť, J.; Spížek, J.; Havlíček, V. Antifungal Drugs. Metabolites 2020, 10, 106. [Google Scholar] [CrossRef] [Green Version]
- Geist, M.J.P.; Egerer, G.; Burhenne, J.; Riedel, K.-D.; Weiss, J.; Mikus, G. Steady-State Pharmacokinetics and Metabolism of Voriconazole in Patients. J. Antimicrob. Chemother. 2013, 68, 2592–2599. [Google Scholar] [CrossRef] [Green Version]
- Grant, S.M.; Clissold, S.P. Fluconazole. A Review of Its Pharmacodynamic and Pharmacokinetic Properties, and Therapeutic Potential in Superficial and Systemic Mycoses. Drugs 1990, 39, 877–916. [Google Scholar] [CrossRef] [PubMed]
- Cornely, O.A.; Böhme, A.; Schmitt-Hoffmann, A.; Ullmann, A.J. Safety and Pharmacokinetics of Isavuconazole as Antifungal Prophylaxis in Acute Myeloid Leukemia Patients with Neutropenia: Results of a Phase 2, Dose Escalation Study. Antimicrob. Agents Chemother. 2015, 59, 2078–2085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wasmann, R.E.; Muilwijk, E.W.; Burger, D.M.; Verweij, P.E.; Knibbe, C.A.; Brüggemann, R.J. Clinical Pharmacokinetics and Pharmacodynamics of Micafungin. Clin. Pharmacokinet. 2018, 57, 267–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, J.A.; Holland, S.D.; Wickersham, P.J.; Sterrett, A.; Schwartz, M.; Bonfiglio, C.; Hesney, M.; Winchell, G.A.; Deutsch, P.J.; Greenberg, H.; et al. Single- and Multiple-Dose Pharmacokinetics of Caspofungin in Healthy Men. Antimicrob. Agents Chemother. 2002, 46, 739–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kofla, G.; Ruhnke, M. Pharmacology and Metabolism of Anidulafungin, Caspofungin and Micafungin in the Treatment of Invasive Candidosis: Review of the Literature. Eur. J. Med. Res. 2011, 16, 159–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riedmaier, A.E.; Lindley, D.J.; Hall, J.A.; Castleberry, S.; Slade, R.T.; Stuart, P.; Carr, R.A.; Borchardt, T.B.; Bow, D.A.J.; Nijsen, M. Mechanistic Physiologically Based Pharmacokinetic Modeling of the Dissolution and Food Effect of a Biopharmaceutics Classification System IV Compound-the Venetoclax Story. J. Pharm. Sci. 2018, 107, 495–502. [Google Scholar] [CrossRef] [Green Version]
- FDA Center for Drug Evaluation and Research Application Number: 208573Orig1s000. Pharmacology Review(s). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/208573Orig1s000PharmR.pdf (accessed on 9 March 2022).
- Johnson, B.A.; Cheang, M.S.; Goldenberg, G.J. Comparison of Adriamycin Uptake in Chick Embryo Heart and Liver Cells an Murine L5178Y Lymphoblasts in Vitro: Role of Drug Uptake in Cardiotoxicity. Cancer Res. 1986, 46, 218–223. [Google Scholar]
- Burns, K.; Nair, P.C.; Rowland, A.; Mackenzie, P.I.; Knights, K.M.; Miners, J.O. The Nonspecific Binding of Tyrosine Kinase Inhibitors to Human Liver Microsomes. Drug Metab. Dispos. 2015, 43, 1934–1937. [Google Scholar] [CrossRef] [Green Version]
- Kotsampasakou, E.; Brenner, S.; Jäger, W.; Ecker, G.F. Identification of Novel Inhibitors of Organic Anion Transporting Polypeptides 1B1 and 1B3 (OATP1B1 and OATP1B3) Using a Consensus Vote of Six Classification Models. Mol. Pharm. 2015, 12, 4395–4404. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, T.; Desai, A.; Goldwater, R.; Han, D.; Howieson, C.; Akhtar, S.; Kowalski, D.; Lademacher, C.; Pearlman, H.; Rammelsberg, D.; et al. Pharmacokinetic Effects of Isavuconazole Coadministration with the Cytochrome P450 Enzyme Substrates Bupropion, Repaglinide, Caffeine, Dextromethorphan, and Methadone in Healthy Subjects: Clinical Pharmacology in Drug Development. Clin. Pharmacol. Drug Dev. 2017, 6, 54–65. [Google Scholar] [CrossRef]
- Yamazaki, T.; Desai, A.; Goldwater, R.; Han, D.; Lasseter, K.C.; Howieson, C.; Akhtar, S.; Kowalski, D.; Lademacher, C.; Rammelsberg, D.; et al. Pharmacokinetic Interactions between Isavuconazole and the Drug Transporter Substrates Atorvastatin, Digoxin, Metformin, and Methotrexate in Healthy Subjects: Clinical Pharmacology in Drug Development. Clin. Pharmacol. Drug Dev. 2017, 6, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Krishna, G.; Ma, L.; Prasad, P.; Moton, A.; Martinho, M.; O’Mara, E. Effect of Posaconazole on the Pharmacokinetics of Simvastatin and Midazolam in Healthy Volunteers. Expert Opin. Drug Metab. Toxicol. 2012, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lempers, V.J.C.; van den Heuvel, J.J.M.W.; Russel, F.G.M.; Aarnoutse, R.E.; Burger, D.M.; Brüggemann, R.J.; Koenderink, J.B. Inhibitory Potential of Antifungal Drugs on ATP-Binding Cassette Transporters P-Glycoprotein, MRP1 to MRP5, BCRP, and BSEP. Antimicrob. Agents Chemother. 2016, 60, 3372–3379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inaba, H.; Panetta, J.C.; Pounds, S.; Wang, L.; Li, L.; Navid, F.; Federico, S.M.; Eisenmann, E.D.; Vasilyeva, A.; Wang, Y.-D.; et al. Sorafenib Population Pharmacokinetics and Skin Toxicities in Children and Adolescents with Refractory/Relapsed Leukemia or Solid Tumor Malignancies. Clin. Cancer Res. 2019, 25, 7320–7330. [Google Scholar] [CrossRef] [Green Version]
- Lipp, H.-P. Antifungal Agents—Clinical Pharmacokinetics and Drug Interactions. Mycoses 2008, 51, 7–18. [Google Scholar] [CrossRef]
- Agarwal, S.K.; Hu, B.; Chien, D.; Wong, S.L.; Salem, A.H. Evaluation of Rifampin’s Transporter Inhibitory and CYP3A Inductive Effects on the Pharmacokinetics of Venetoclax, a BCL-2 Inhibitor: Results of a Single- and Multiple-Dose Study. J. Clin. Pharmacol. 2016, 56, 1335–1343. [Google Scholar] [CrossRef]
- Agarwal, S.K.; Tong, B.; Bueno, O.F.; Menon, R.M.; Salem, A.H. Effect of Azithromycin on Venetoclax Pharmacokinetics in Healthy Volunteers: Implications for Dosing Venetoclax with P-Gp Inhibitors. Adv. Ther. 2018, 35, 2015–2023. [Google Scholar] [CrossRef]
- Lachowiez, C.; DiNardo, C.D.; Konopleva, M. Venetoclax in Acute Myeloid Leukemia—Current and Future Directions. Leuk. Lymphoma 2020, 61, 1313–1322. [Google Scholar] [CrossRef]
- Garrison, D.; Talebi, Z.; Jin, Y.; Uddin, M.E.; Gibson, A.; Sparreboom, A.; Baker, S. Interaction of the FLT3 Inhibitor Gilteritinib with Xenobiotic Uptake Transporters. FASEB J. 2021, 35. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mouse Genotype | Sex | N | Co-Treatment (Dose mg/kg) | Venetoclax Cmax (ng/mL) | Venetoclax AUC(0–8h) (ng*h/mL) | Venetoclax AUC Fold Increase |
|---|---|---|---|---|---|---|
| Wild-type FVB | F | 5 | PEG400 | 1030 (140) | 6530 (720) | |
| Wild-type FVB | F | 5 | Ketoconazole (50) | 3080 (290) ** | 19,700 (1600) *** | 3.0 (versus vehicle-treated wild-type FVB) |
| CYP3A(−/−) | F | 5 | PEG 400 | 1570 (310) | 10,600 (1900) | 1.6 (versus vehicle-treated wild-type FVB) |
| CYP3A(−/−) | F | 5 | Ketoconazole (50) | 2700 (570) * | 17,300 (3300) ** | 1.6 (versus vehicle-treated wild-type CYP3A(−/−)) |
| Wild-type DBA | F | 10 | None | 523 (55) | 3211 (395) | |
| OATP1B2(−/−) | F | 10 | None | 1066 (130) ** | 6481 (695) *** | 2.0 (versus wild-type DBA) |
| CYP3A(−/−) | F | 5 | None | 1830 (270) | 10,600 (1200) | |
| CYP3A(−/−) | F | 5 | Micafungin (100) | 2280 (320) | 13,600 (1100) * | 1.3 (versus vehicle-treated wild-type CYP3A(−/−)) |
| Inhibitor | OATP1B1 IC50 (µM) | 95% CI | OATP1B2 IC50 (µM) | 95% CI | Cmax,ss (µM) a | Citation | Cmax,ss/IC50 (>0.1 Considered Clinically Relevant) |
|---|---|---|---|---|---|---|---|
| Ketoconazole | 1.5 | (1.2, 1.7) | 2.2 | (1.4, 3.4) | 1.88 | [7] | 1.25 |
| Cobicistat | 0.31 | (0.24, 0.40) | 0.7 | (0.57, 0.83) | 1.29 | [27] | 1.84 |
| Ritonavir | 0.34 | (0.29, 0.40) | ND | ND | 1.94 | [28] | 2.71 |
| Rifampin | 1.2 | (0.9, 2.2) | 2.1 | (1.5, 3) | 29 | [29] | 24.2 |
| Itraconazole | 37 | (31, 45) | >50 | ND | 1.15 | [30] | 0.03 |
| Hydroxy-ITZ | 3.5 | (3, 3.9) | >2 | ND | 0.6 | [30] | 0.17 |
| Keto-ITZ | 8.3 | (6.9, 10) | ND | ND | 0.02 | [30] | 0.002 |
| Posaconazole | 1.9 | (1.6, 2.2) | >20 | ND | 2.75 | [31] | 1.45 |
| Voriconazole | 80 | (62, 104) | >200 | ND | 10.2 | [32] | 0.13 |
| Fluconazole | 4100 | (3200, 5300) | >5000 | ND | 61.7 | [33] | 0.06 |
| Isavuconazole | 2.5 | (2.1, 3.0) | 2.5 | (1.1, 5.8) | 8.23 | [34] | 3.29 |
| Micafungin | 2.1 | (1.8, 2.4) | 4.9 | (4.2, 5.7) | 17.3 | [35] | 8.24 |
| Caspofungin | 9.2 | (8.0, 11) | >100 | ND | 7.99 | [36] | 0.87 |
| Anidulafungin | 42 | (31, 57) | ND | ND | 6.14 | [37] | 0.15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eisenmann, E.D.; Garrison, D.A.; Talebi, Z.; Jin, Y.; Silvaroli, J.A.; Kim, J.-G.; Sparreboom, A.; Savona, M.R.; Mims, A.S.; Baker, S.D. Interaction of Antifungal Drugs with CYP3A- and OATP1B-Mediated Venetoclax Elimination. Pharmaceutics 2022, 14, 694. https://doi.org/10.3390/pharmaceutics14040694
Eisenmann ED, Garrison DA, Talebi Z, Jin Y, Silvaroli JA, Kim J-G, Sparreboom A, Savona MR, Mims AS, Baker SD. Interaction of Antifungal Drugs with CYP3A- and OATP1B-Mediated Venetoclax Elimination. Pharmaceutics. 2022; 14(4):694. https://doi.org/10.3390/pharmaceutics14040694
Chicago/Turabian StyleEisenmann, Eric D., Dominique A. Garrison, Zahra Talebi, Yan Jin, Josie A. Silvaroli, Jin-Gyu Kim, Alex Sparreboom, Michael R. Savona, Alice S. Mims, and Sharyn D. Baker. 2022. "Interaction of Antifungal Drugs with CYP3A- and OATP1B-Mediated Venetoclax Elimination" Pharmaceutics 14, no. 4: 694. https://doi.org/10.3390/pharmaceutics14040694