
Pharmacokinetics of DA-6886, A New 5-HT4 Receptor Agonist, in Rats


Abstract
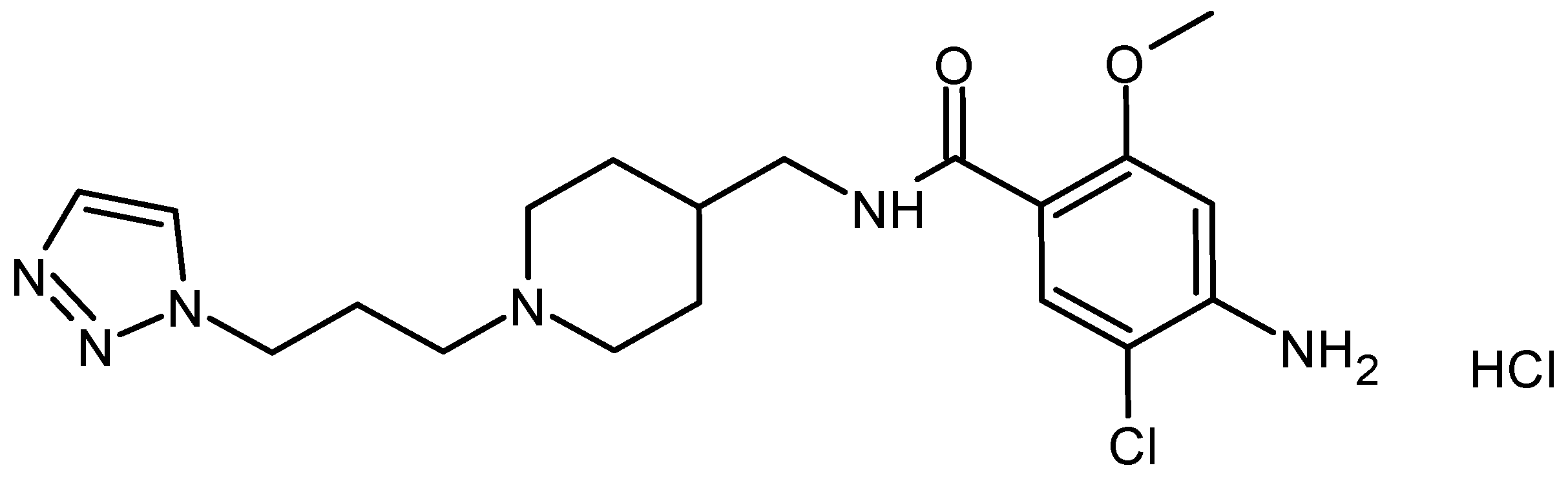
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Animals
2.3. Intravenous and Oral Adminitration
2.4. LC–MS/MS Analysis of DA-6886
2.5. Pharmacokinetic Analysis
2.6. Statistical Analysis
3. Results
3.1. Analytical Method Validation
3.1.1. Linearity, LLOQ, and Specificity
3.1.2. Precision and Accuracy
3.1.3. Matrix Effect and Recovery
3.1.4. Stability of the DA-6886 and Dilution Effect
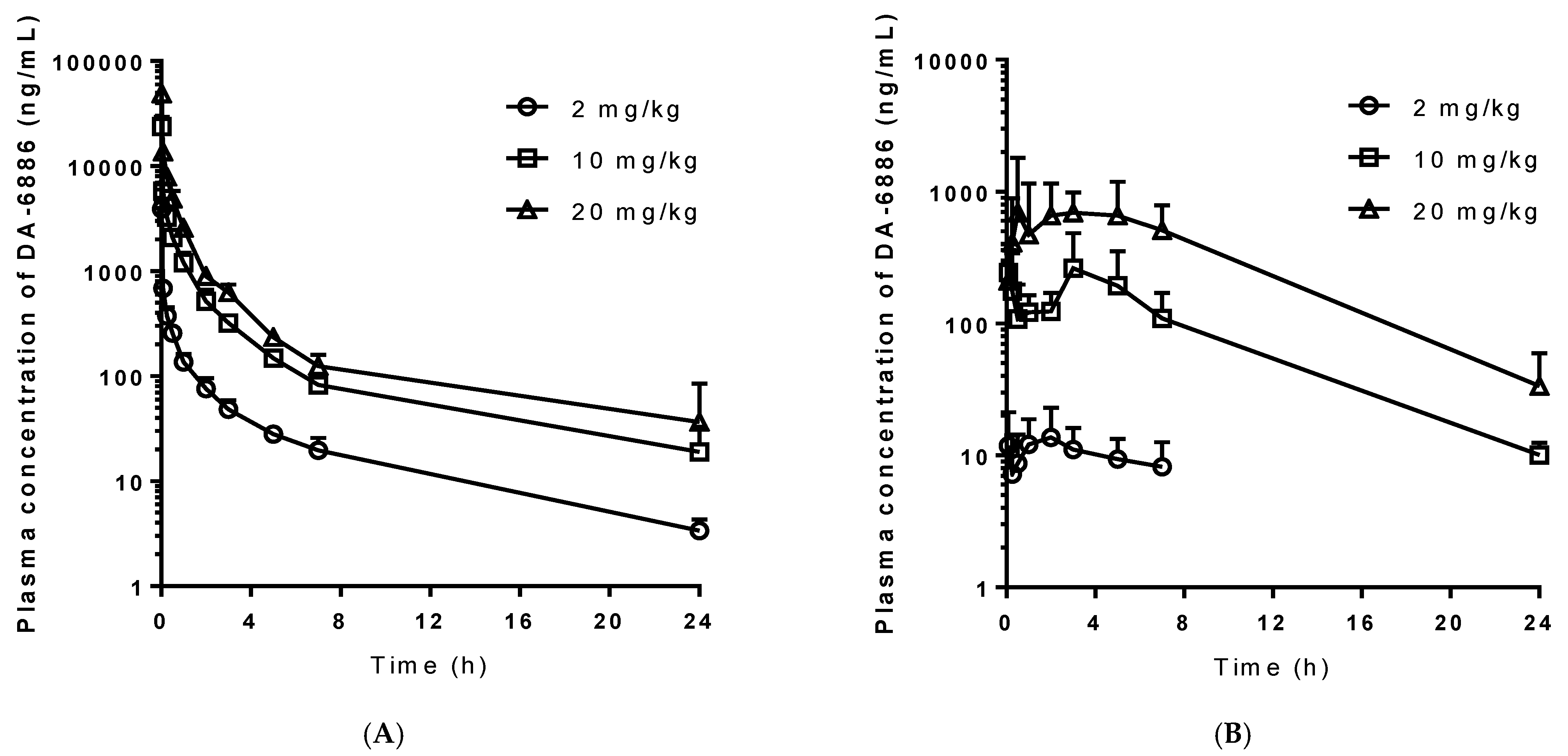
3.2. Pharmacokinetics of DA-6886 following Intravenous Administration
3.3. Pharmacokinetics of DA-6886 following Oral Administration
3.4. Assessment of Dose Proportionality of DA-6886
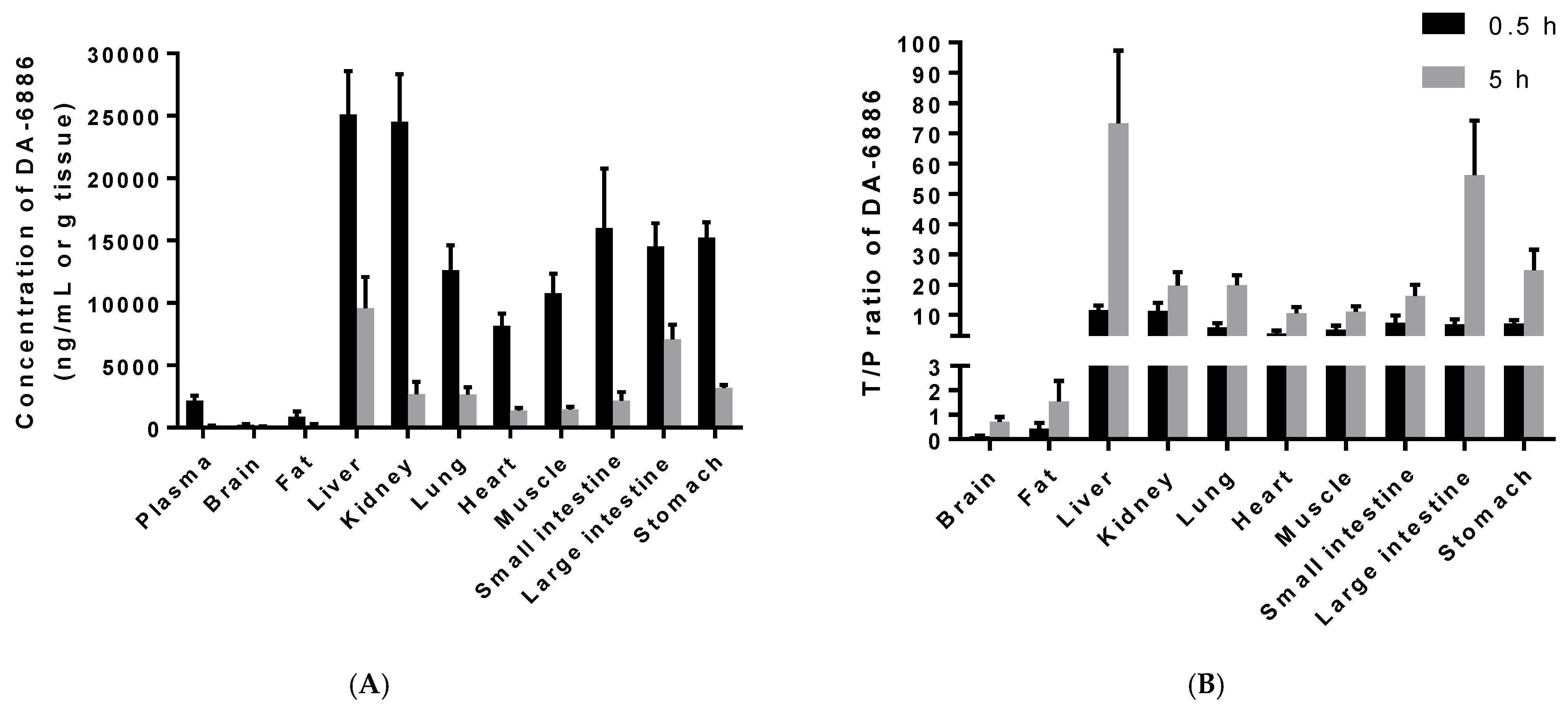
3.5. Tissue Distribution of DA-6886
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Galligan, J.J. Colonic 5-HT4 receptors are targets for novel prokinetic drugs. Neurogastroenterol. Motil. 2021, 33, e14125. [Google Scholar] [CrossRef] [PubMed]
- Manabe, N.; Wong, B.S.; Camilleri, M. New-generation 5-HT4 receptor agonists: Potential for treatment of gastrointestinal motility disorders. Expert Opin. Investig. Drugs 2010, 19, 765–775. [Google Scholar] [CrossRef] [PubMed]
- Tack, J.; Camilleri, M.; Chang, L.; Chey, W.D.; Galligan, J.J.; Lacy, B.E.; Muller-Lissner, S.; Quigley, E.M.; Schuurkes, J.; De Maeyer, J.H.; et al. Systematic review: Cardiovascular safety profile of 5-HT(4) agonists developed for gastrointestinal disorders. Aliment. Pharmacol. Ther. 2012, 35, 745–767. [Google Scholar] [CrossRef] [PubMed]
- Shin, A.; Camilleri, M.; Kolar, G.; Erwin, P.; West, C.P.; Murad, M.H. Systematic review with meta-analysis: Highly selective 5-HT4 agonists (prucalopride, velusetrag or naronapride) in chronic constipation. Aliment. Pharmacol. Ther. 2014, 39, 239–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, C.; Xu, Q.; Wen, X.; Sun, H. Current developments in pharmacological therapeutics for chronic constipation. Acta Pharm. Sin. B 2015, 5, 300–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, J.G.; Foxx-Orenstein, A.E.; Grider, J.R. Propulsion in guinea pig colon induced by 5-hydroxytryptamine (HT) via 5-HT4 and 5-HT3 receptors. J. Pharmacol. Exp. Ther. 1999, 288, 93–97. [Google Scholar] [PubMed]
- Grider, J.R.; Foxx-Orenstein, A.E.; Jin, J.G. 5-Hydroxytryptamine4 receptor agonists initiate the peristaltic reflex in human, rat, and guinea pig intestine. Gastroenterology 1998, 115, 370–380. [Google Scholar] [CrossRef]
- Sinagra, E.; Morreale, G.C.; Mohammadian, G.; Fusco, G.; Guarnotta, V.; Tomasello, G.; Cappello, F.; Rossi, F.; Amvrosiadis, G.; Raimondo, D. New therapeutic perspectives in irritable bowel syndrome: Targeting low-grade inflammation, immuno-neuroendocrine axis, motility, secretion and beyond. World J. Gastroenterol. 2017, 23, 6593–6627. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.J.; Cho, K.H.; Park, H.M.; Sung, H.J.; Choi, S.; Im, W. Pharmacological profile of DA-6886, a novel 5-HT4 receptor agonist to accelerate colonic motor activity in mice. Eur. J. Pharmacol. 2014, 735, 115–122. [Google Scholar] [CrossRef]
- Moon, J.H.; Lee, M.J.; Cho, K.H.; Im, W.; Choi, S.H. Effects of DA-6886 on colonic motility in conscious guinea pigs. Gastroenterology 2014, 146, S357–S358. [Google Scholar] [CrossRef]
- National Library of Medicine (U.S.). Phase I Clinical Trial of DA-6886 in Healthy Male Subjects. Identifier NCT01633723. (2012, August–2014, April). Available online: https://www.clinicaltrials.gov/ct2/show/record/NCT01633723 (accessed on 22 February 2022).
- Kang, H.E.; Jung, H.Y.; Cho, Y.K.; Kim, S.H.; Sohn, S.I.; Baek, S.R.; Lee, M.G. Pharmacokinetics of liquiritigenin in mice, rats, rabbits, and dogs, and animal scale-up. J. Pharm. Sci. 2009, 98, 4327–4342. [Google Scholar] [CrossRef]
- Kwon, M.H.; Lee, D.Y.; Kang, H.E. Development and Validation of an LC-MS/MS Method for Quantification of the Novel Antibacterial Candidate DA-7010 in Plasma and Application to a Preclinical Pharmacokinetic Study. Pharmaceuticals 2021, 14, 163. [Google Scholar] [CrossRef]
- Bioanalytical Method Validation Guidance for Industry. US FDA/Center for Drug Evaluation and Research. 2018. Available online: https://www.fda.gov/media/70858/download (accessed on 8 December 2021).
- Gibaldi, M.; Perrier, D. Pharmacokinetics, 2nd ed.; Marcel-Dekker: New York, NY, USA, 1982. [Google Scholar]
- Chiou, W.L. Critical evaluation of the potential error in pharmacokinetic studies of using the linear trapezoidal rule method for the calculation of the area under the plasma level-time curve. J. Pharmacokinet. BioPharm. 1978, 6, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.P.; Vandenhende, F.R.; DeSante, K.A.; Farid, N.A.; Welch, P.A.; Callaghan, J.T.; Forgue, S.T. Confidence interval criteria for assessment of dose proportionality. Pharm. Res. 2000, 17, 1278–1283. [Google Scholar] [CrossRef] [PubMed]
- Hummel, J.; McKendrick, S.; Brindley, C.; French, R. Exploratory assessment of dose proportionality: Review of current approaches and proposal for a practical criterion. Pharm. Stat. 2009, 8, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Fukuchi, Y.; Toshimoto, K.; Mori, T.; Kakimoto, K.; Tobe, Y.; Sawada, T.; Asaumi, R.; Iwata, T.; Hashimoto, Y.; Nunoya, K.I.; et al. Analysis of Nonlinear Pharmacokinetics of a Highly Albumin-Bound Compound: Contribution of Albumin-Mediated Hepatic Uptake Mechanism. J. Pharm. Sci. 2017, 106, 2704–2714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Gao, Y.; Yang, C.; Xiang, Y.; Zhang, W.; Zhang, T.; Su, R.; Lu, C.; Zhuang, X. Assessment and Confirmation of Species Difference in Nonlinear Pharmacokinetics of Atipamezole with Physiologically Based Pharmacokinetic Modeling. Drug Metab. Dispos. 2020, 48, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Davies, B.; Morris, T. Physiological parameters in laboratory animals and humans. Pharm. Res. 1993, 10, 1093–1095. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Sun, Z.; Wang, Z.; Du, S.; Kong, X.; Li, L.; Yang, J.; Kang, J.; Zhang, X. Pharmacokinetics and tissue distribution study of prucalopride in rats by ultra high performance liquid chromatography with tandem mass spectrometry. J. Pharm. Biomed. Anal. 2016, 131, 246–255. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Nominal Concentration (ng/mL) | Intra-Day (n = 5) | Inter-Day (n = 5) | ||||
|---|---|---|---|---|---|---|
| Measured Concentration (Mean, ng/mL) | Precision (CV, %) | Accuracy (RE, %) | Measured Concentration (Mean, ng/mL) | Precision (CV, %) | Accuracy (RE, %) | |
| 2 (LLOQ) | 1.86 | 5.74 | −6.80 | - | - | - |
| 6 (LQC) | 6.14 | 5.00 | 2.27 | 6.04 | 2.45 | 0.733 |
| 100 | 103 | 2.16 | 2.58 | 102 | 4.05 | 2.40 |
| 1600 (HQC) | 1550 | 3.14 | −3.25 | 1650 | 4.12 | 3.33 |
| Parameters | 2 mg/kg (n = 4) | 10 mg/kg (n = 4) | 20 mg/kg (n = 4) |
|---|---|---|---|
| AUC0–∞ (ng∙h/mL) 1, 2 | 908 ± 153 | 6360 ± 1130 | 13,000 ± 1910 |
| Terminal half-life (h) | 5.20 ± 2.04 | 7.00 ± 2.70 | 6.54 ± 3.83 |
| CL (mL/min/kg) 2 | 37.7 ± 7.60 | 26.8 ± 4.51 | 26.0 ± 3.37 |
| Vss (L/kg) | 7.84 ± 2.21 | 5.64 ± 1.49 | 4.91 ± 3.14 |
| Parameters | 2 mg/kg (n = 4) | 10 mg/kg (n = 4) | 20 mg/kg (n = 4) |
|---|---|---|---|
| AUC0–∞ (ng∙h/mL) 1, 2 | 171 ± 153 | 1960 ± 819 | 7160 ± 2150 |
| Terminal half-life (h) | 7.51 ± 3.95 | 5.93 ± 2.94 | 4.64 ± 1.42 |
| Cmax (ng/mL) 1, 2 | 17.0 ± 6.80 | 393 ± 131 | 1440 ± 653 |
| Tmax (h) 3 | 1.5 (0.5–3) | 1.63 (0.0833–5) | 4 (0.5–7) |
| F (%) | 18.9 | 30.9 | 55.0 |
| Parameters | Slope of the Log-Transformed Parameter Versus Log Dose, 90% CI | Ratio of Dose-Normalized Geometric Mean (Rdnm), 90% CI | Dose Proportionality | Maximal Proportional Dose Ratio 3 | Threshold Dose Ratio to Reject Proportionality 3 |
|---|---|---|---|---|---|
| AUC0–∞ after intravenous administration | 1.066 (0.8513–1.332) | 1.164 (0.7101–2.148) | Inconclusive 1 | 1.96 | - |
| AUC0–∞ after oral administration | 1.858 (1.171–3.124) | 7.211 (1.483–133.0) | Not proportional 2 | 1.11 | 3.68 |
| Cmax after oral administration | 1.875 (0.9649–4.528) | 7.499 (0.9224–3373) | Inconclusive 1 | 1.07 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, D.Y.; Kang, H.E. Pharmacokinetics of DA-6886, A New 5-HT4 Receptor Agonist, in Rats. Pharmaceutics 2022, 14, 702. https://doi.org/10.3390/pharmaceutics14040702
Lee DY, Kang HE. Pharmacokinetics of DA-6886, A New 5-HT4 Receptor Agonist, in Rats. Pharmaceutics. 2022; 14(4):702. https://doi.org/10.3390/pharmaceutics14040702
Chicago/Turabian StyleLee, Dae Young, and Hee Eun Kang. 2022. "Pharmacokinetics of DA-6886, A New 5-HT4 Receptor Agonist, in Rats" Pharmaceutics 14, no. 4: 702. https://doi.org/10.3390/pharmaceutics14040702
APA StyleLee, D. Y., & Kang, H. E. (2022). Pharmacokinetics of DA-6886, A New 5-HT4 Receptor Agonist, in Rats. Pharmaceutics, 14(4), 702. https://doi.org/10.3390/pharmaceutics14040702