
Hot Melt Extrusion-Triggered Amorphization as a Continuous Process for Inducing Extended Supersaturable Drug Immediate-Release from saSMSDs Systems

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of saSMSD/NIT Systems for Short-Term Immediate Release
2.2.1. Preparation of saSMSD/NITCHME Using HME as a Continuous (C) Process
2.2.2. Preparation of saSMSD/NITNCCP Using Co-Precipitation (CP) as a Non-Continuous (NC) Process
2.2.3. Preparation of saSMSD/NITNCFQC Using Fusion/Quench Cooling (FQC) as a Non-Continuous (NC) Process
2.2.4. Preparation of NIT as Physical Mixture (PM) and Pure Amorphous (AM) Forms
2.3. Scanning Electron Microscopy (SEM)
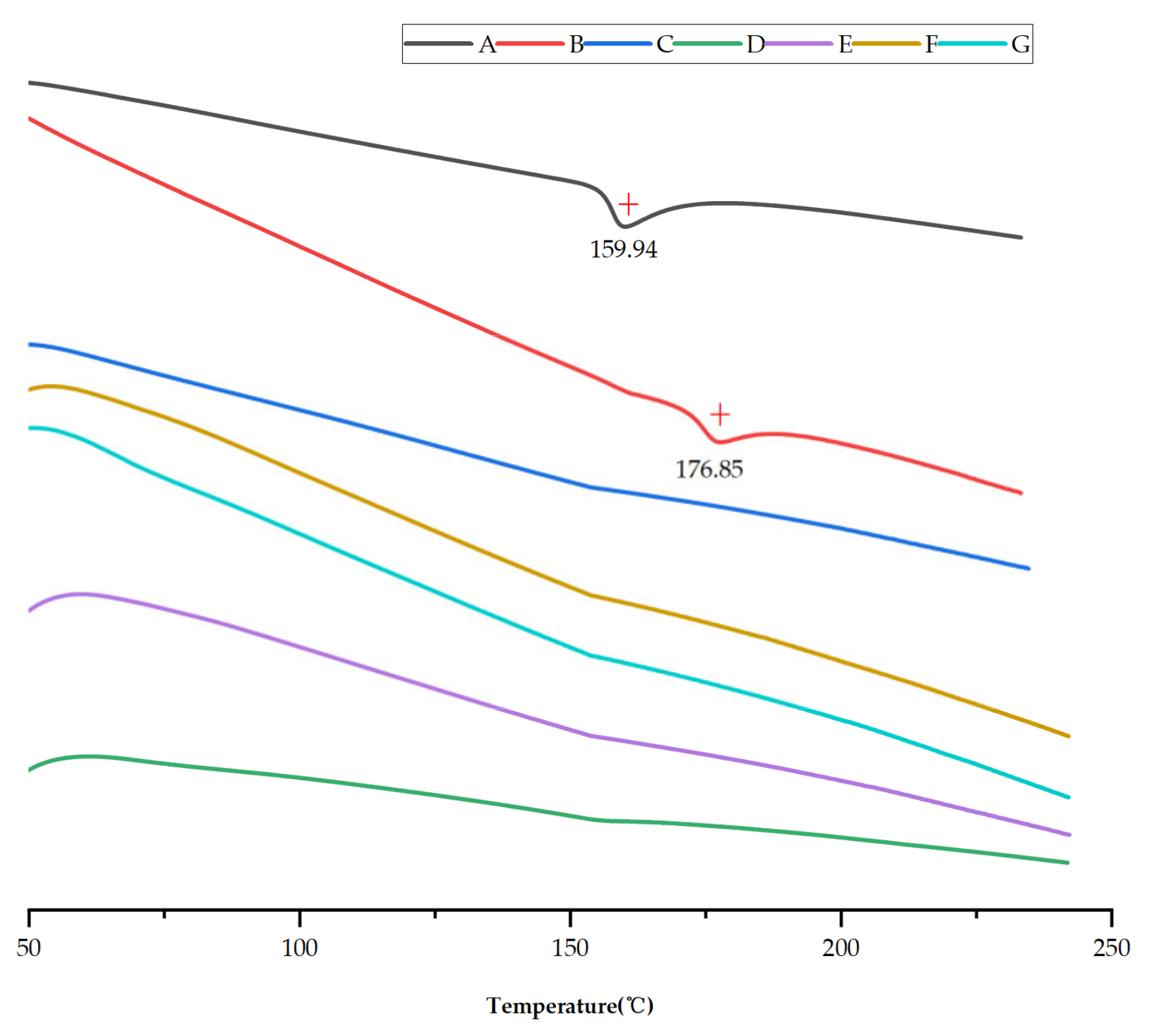
2.4. Differential Scanning Calorimetry (DSC)
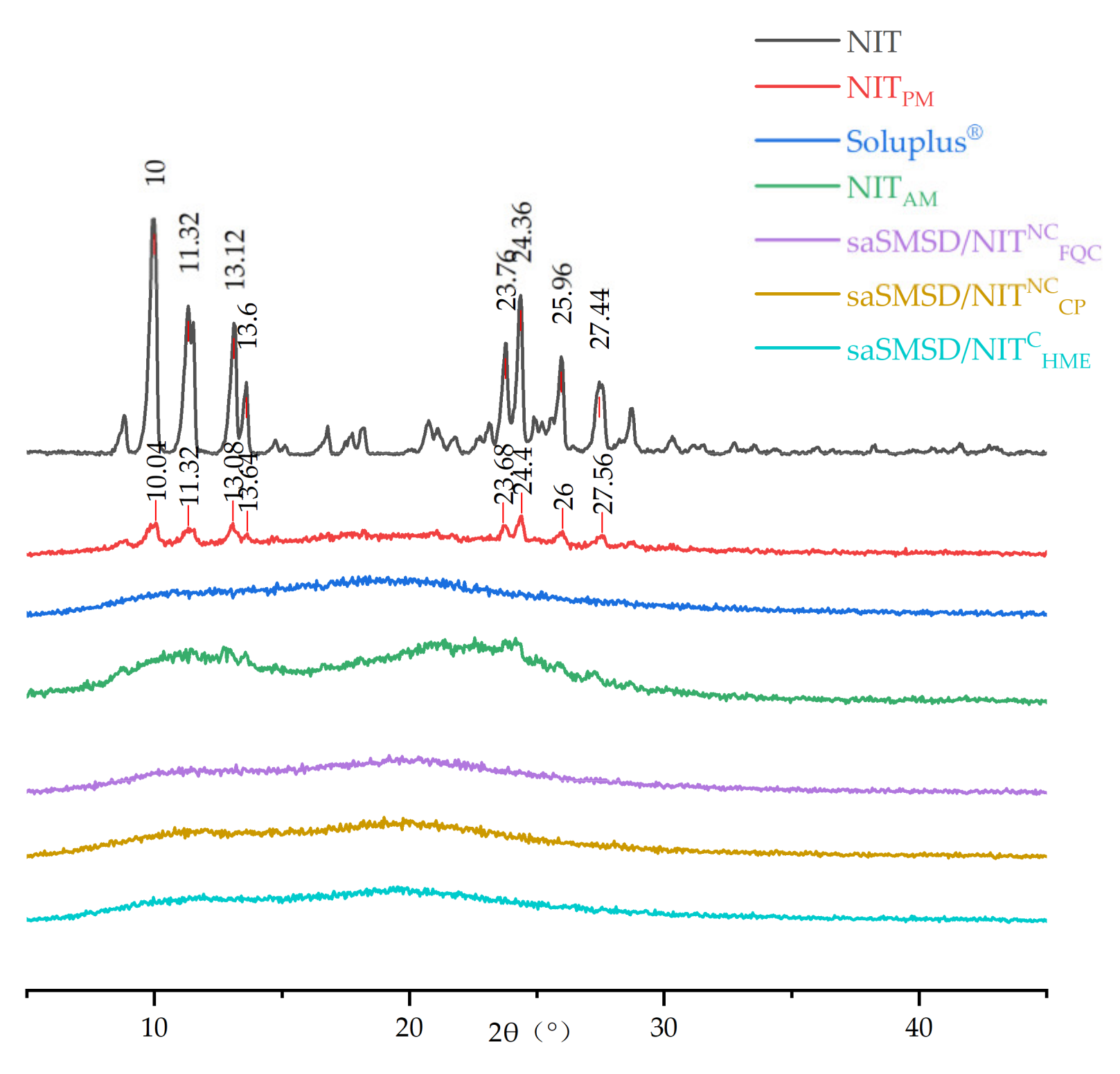
2.5. Powder X-ray Diffraction (PXRD)
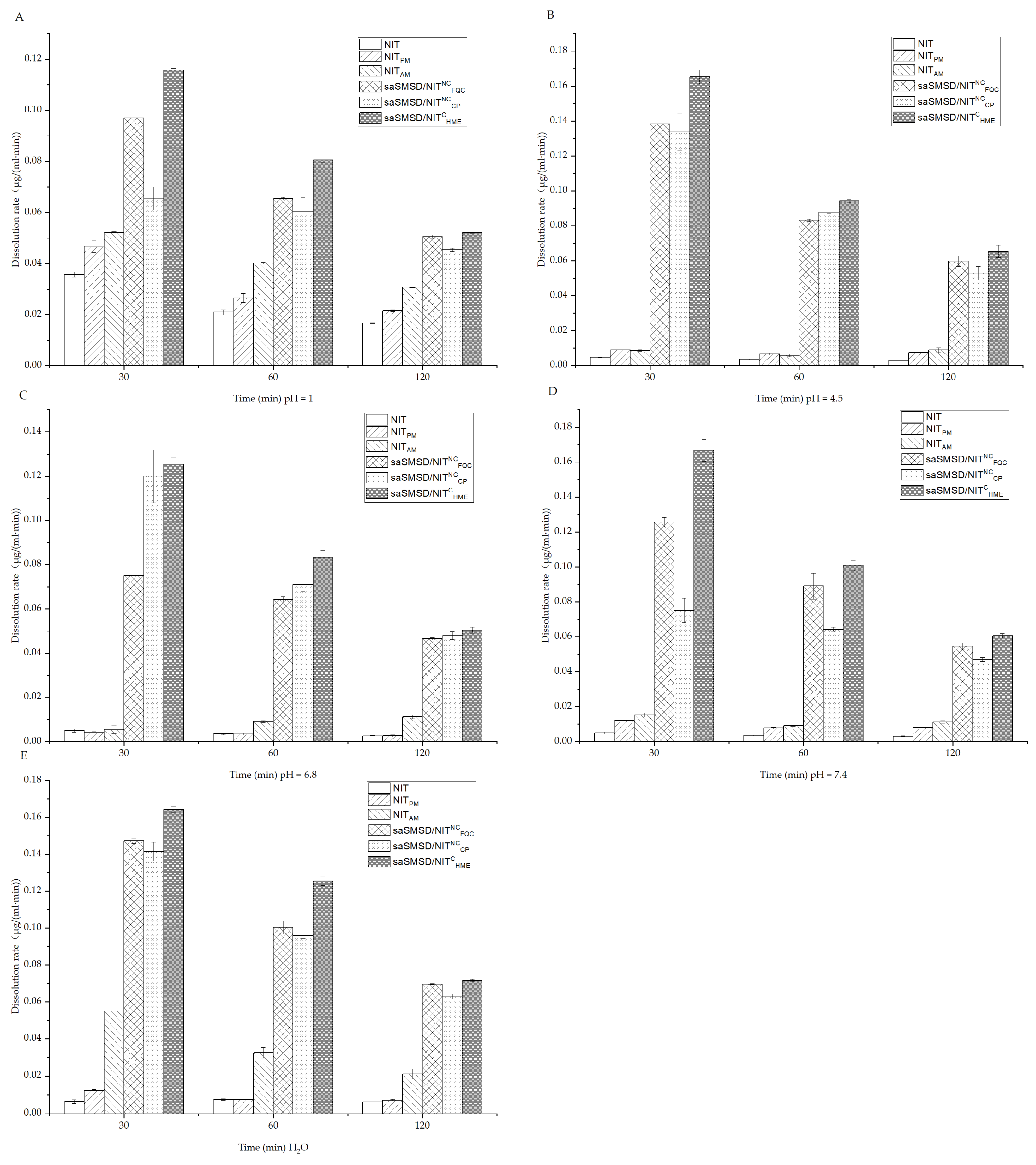
2.6. Measurement of Initial NIT Release Using Dissolution Assays
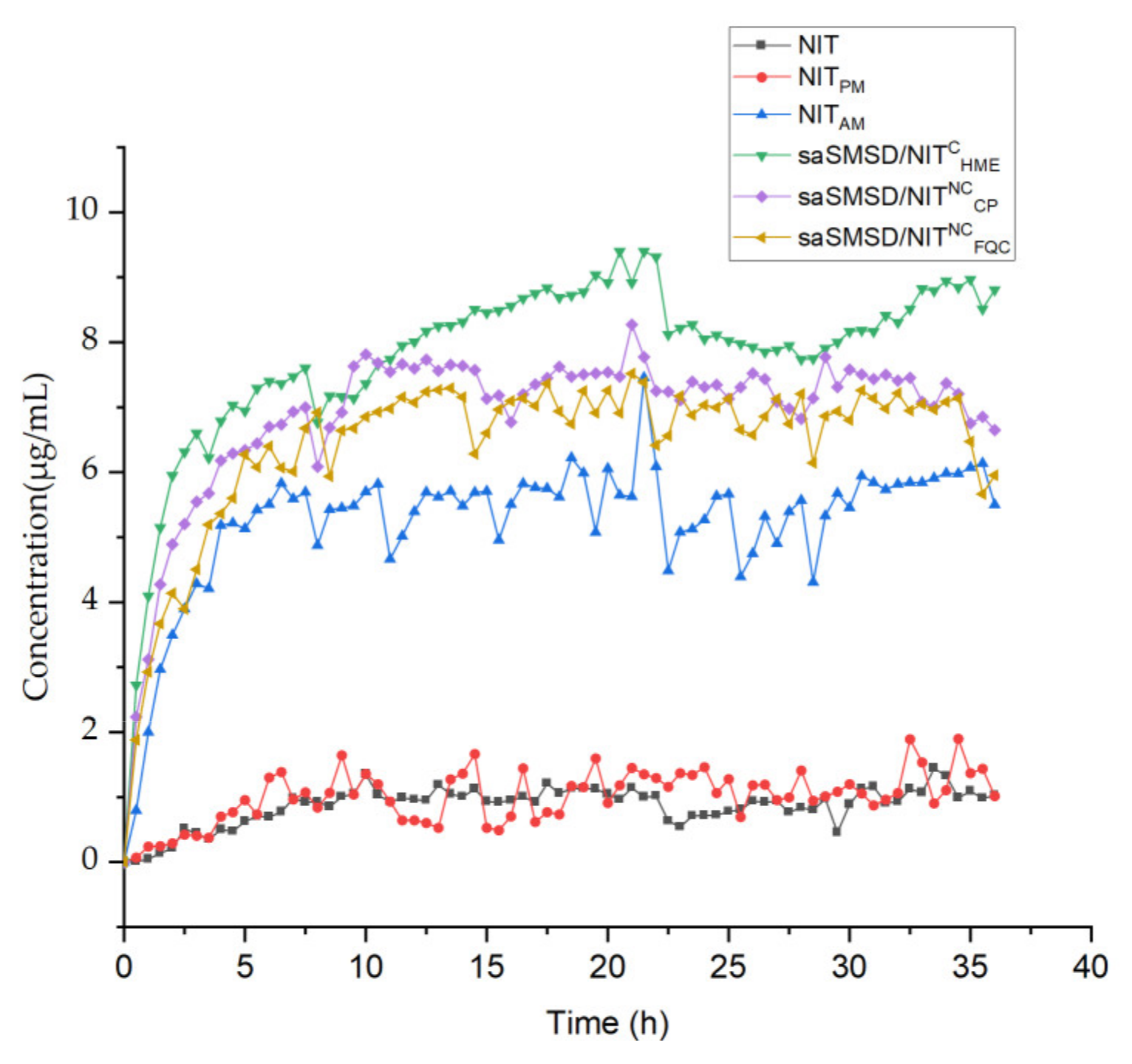
2.7. Evolved Release Stage “Spring-Parachute” Analysis
2.7.1. Semi-Continuous Determinations
2.7.2. Quantification
2.8. Solubility Measurements
2.9. Long-Term Amorphous State Stability Assessments Conducted Using PXRD
2.10. Phase Solubility Measurements
2.11. Fourier Transform-Infrared Spectroscopy Analysis
2.12. Crystallization Inhibition from a Supersaturated State
2.13. Data Analysis
3. Results and Discussion
3.1. Preparation and Characterization of saSMSD/NIT Systems Prepared Using Continuous and Non-Continuous Processes
3.2. SaSMSD/NIT Characteristics: Extended Dissolution, Spring-Parachute and Solubility
3.3. Stability Profiles and Underlying Molecular Mechanisms
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tran, P.; Tran, T.T.-D.; Park, J.-B.; Lee, B.-J. Controlled Release Systems Containing Solid Dispersions: Strategies and Mechanisms. Pharm. Res. 2011, 28, 2353–2378. [Google Scholar] [CrossRef] [PubMed]
- Tran, P.H.; Tran, T.T. Dosage form designs for the controlled drug release of solid dispersions. Int. J. Pharm. 2020, 581, 119274. [Google Scholar] [CrossRef] [PubMed]
- Shi, N.-Q.; Zhou, J.; Walker, J.; Li, L.; Hong, J.K.; Olsen, K.F.; Tang, J.; Ackermann, R.; Wang, Y.; Qin, B.; et al. Microencapsulation of luteinizing hormone-releasing hormone agonist in poly (lactic-co-glycolic acid) microspheres by spray-drying. J. Control Release 2020, 321, 756–772. [Google Scholar] [CrossRef]
- Alqahtani, F.; Belton, P.; Ward, A.; Asare-Addo, K.; Qi, S. An investigation into the use of low quantities of functional additives to control drug release from hot melt extruded solid dispersions for poorly soluble drug delivery. Int. J. Pharm. 2020, 579, 119172. [Google Scholar] [CrossRef] [PubMed]
- Park, K. Drug release mechanisms from amorphous solid dispersions. J. Control Release 2015, 211, 171. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.R.; Ma, Y.; Lee, P.I. Impact of phase separation morphology on release mechanism of amorphous solid dispersions. Eur. J. Pharm. Sci. 2019, 136, 104955. [Google Scholar] [CrossRef]
- Fong, S.Y.K.; Bauer-Brandl, A.; Brandl, M. Oral bioavailability enhancement through supersaturation: An update and meta-analysis. Expert Opin. Drug Deliv. 2017, 14, 403–426. [Google Scholar] [CrossRef]
- Lu, Z.; Yang, Y.; Covington, R.-A.; Bi, Y.V.; Dürig, T.; Ilies, M.A.; Fassihi, R. Supersaturated controlled release matrix using amorphous dispersions of glipizide. Int. J. Pharm. 2016, 511, 957–968. [Google Scholar] [CrossRef]
- Keen, J.M.; Hughey, J.R.; Bennett, R.C.; Jannin, V.; Rosiaux, Y.; Marchaud, D.; McGinity, J.W. Effect of Tablet Structure on Controlled Release from Supersaturating Solid Dispersions Containing Glyceryl Behenate. Mol. Pharm. 2015, 12, 120–126. [Google Scholar] [CrossRef]
- Verma, S.; Rudraraju, V.S. Disintegration Mediated Controlled Release Supersaturating Solid Dispersion Formulation of an Insoluble Drug: Design, Development, Optimization, and In Vitro Evaluation. AAPS PharmSciTech 2015, 16, 85–97. [Google Scholar] [CrossRef] [Green Version]
- Paisana, M.C.; Lino, P.R.; Nunes, P.D.; Pinto, J.F.; Henriques, J.; Paiva, A.M. Laser diffraction as a powerful tool for amorphous solid dispersion screening and dissolution understanding. Eur. J. Pharm. Sci. 2021, 163, 105853. [Google Scholar] [CrossRef] [PubMed]
- Shi, N.-Q.; Yao, J.; Wang, X.-L. Effect of polymers and media type on extending the dissolution of amorphous pioglitazone and inhibiting the recrystallization from a supersaturated state. Drug Dev. Ind. Pharm. 2014, 40, 1112–1122. [Google Scholar] [CrossRef] [PubMed]
- Shi, N.-Q.; Jin, Y.; Zhang, Y.; Che, X.-X.; Xiao, X.; Cui, G.-H.; Chen, Y.-Z.; Feng, B.; Li, Z.-Q.; Qi, X.-R. The Influence of Cellulosic Polymer’s Variables on Dissolution/Solubility of Amorphous Felodipine and Crystallization Inhibition from a Supersaturated State. AAPS PharmSciTech 2018, 20, 12. [Google Scholar] [CrossRef]
- Shi, N.-Q.; Lei, Y.-S.; Song, L.-M.; Yao, J.; Zhang, X.-B.; Wang, X.-L. Impact of amorphous and semicrystalline polymers on the dissolution and crystallization inhibition of pioglitazone solid dispersions. Powder Technol. 2013, 247, 211–221. [Google Scholar] [CrossRef]
- Jog, R.; Burgess, D.J. Pharmaceutical Amorphous Nanoparticles. J. Pharm. Sci. 2017, 106, 39–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Kuminek, G.; Roy, L.; Cavanagh, K.L.; Yin, Q.; Rodríguez-Hornedo, N. Cocrystal Solubility Advantage Diagrams as a Means to Control Dissolution, Supersaturation, and Precipitation. Mol. Pharm. 2019, 16, 3887–3895. [Google Scholar] [CrossRef]
- Goyal, S.; Thorson, M.R.; Schneider, C.L.; Zhang, G.G.Z.; Gong, Y.; Kenis, P.J.A. A Microfluidic Platform for Evaporation-based Salt Screening of Pharmaceutical Parent compounds. Lab. Chip. 2013, 13, 1708–1723. [Google Scholar] [CrossRef]
- Shi, N.-Q.; Zhang, Y.; Li, Y.; Lai, H.-W.; Xiao, X.; Feng, B.; Qi, X.-R. Self-micellizing solid dispersions enhance the properties and therapeutic potential of fenofibrate: Advantages, profiles and mechanisms. Int. J. Pharm. 2017, 528, 563–577. [Google Scholar] [CrossRef]
- Wang, S.; Liu, C.; Chen, H.; Zhu, A.D.; Qian, F. Impact of Surfactants on Polymer Maintained Nifedipine Supersaturation in Aqueous Solution. Pharm. Res. 2020, 37, 113. [Google Scholar] [CrossRef]
- Stappaerts, J.; Augustijns, P. Displacement of itraconazole from cyclodextrin complexes in biorelevant media: In vitro evaluation of supersaturation and precipitation behavior. Int. J. Pharm. 2016, 511, 680–687. [Google Scholar] [CrossRef]
- Hu, C.; Liu, Z.; Liu, C.; Li, J.; Wang, Z.; Xu, L.; Chen, C.; Fan, H.; Qian, F. Enhanced Oral Bioavailability and Anti-Echinococcosis Efficacy of Albendazole Achieved by Optimizing the “Spring” and “Parachute”. Mol. Pharm. 2019, 16, 4978–4986. [Google Scholar] [CrossRef] [PubMed]
- Dressman, J.B.; Herbert, E.; Wieber, A.; Birk, G.; Saal, C.; Lubda, D. Mesoporous silica-based dosage forms improve release characteristics of poorly soluble drugs: Case example fenofibrate. J. Pharm. Pharmacol. 2016, 68, 634–645. [Google Scholar] [CrossRef] [PubMed]
- Taha, N.F.; Emam, M.F.; Emara, L.H. A novel combination of Soluplus((R))/Poloxamer for Meloxicam solid dispersions via hot melt extrusion for rapid onset of action. Part 2: Comparative bioavailability and IVIVC. Drug Dev. Ind. Pharm. 2020, 46, 1362–1372. [Google Scholar] [CrossRef] [PubMed]
- Al-Obaidi, H.; Majumder, M.; Bari, F. Amorphous and Crystalline Particulates: Challenges and Perspectives in Drug Delivery. Curr. Pharm. Des. 2017, 23, 350–361. [Google Scholar] [CrossRef]
- Rautaniemi, K.; Vuorimaa-Laukkanen, E.; Strachan, C.J.; Laaksonen, T. Crystallization Kinetics of an Amorphous Pharmaceutical Compound Using Fluorescence-Lifetime-Imaging Microscopy. Mol. Pharm. 2018, 15, 1964–1971. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Grohganz, H.; Löbmann, K.; Rades, T.; Hempel, N.-J. Co-Amorphous Drug Formulations in Numbers: Recent Advances in Co-Amorphous Drug Formulations with Focus on Co-Formability, Molar Ratio, Preparation Methods, Physical Stability, In Vitro and In Vivo Performance, and New Formulation Strategies. Pharmaceutics 2021, 13, 389. [Google Scholar] [CrossRef]
- Onoue, S.; Suzuki, H.; Kojo, Y.; Matsunaga, S.; Sato, H.; Mizumoto, T.; Yuminoki, K.; Hashimoto, N.; Yamada, S. Self-micellizing solid dispersion of cyclosporine A with improved dissolution and oral bioavailability. Eur. J. Pharm. Sci. 2014, 62, 16–22. [Google Scholar] [CrossRef]
- Shi, N.-Q.; Lai, H.-W.; Zhang, Y.; Feng, B.; Xiao, X.; Zhang, H.-M.; Li, Z.-Q.; Qi, X. On the inherent properties of Soluplus and its application in ibuprofen solid dispersions generated by microwave-quench cooling technology. Pharm. Dev. Technol. 2018, 23, 573–586. [Google Scholar] [CrossRef]
- Feng, X.; Chen, Y.; Li, L.; Zhang, Y.; Zhang, L.; Zhang, Z. Preparation, evaluation and metabolites study in rats of novel amentoflavone-loaded TPGS/soluplus mixed nanomicelles. Drug Deliv. 2020, 27, 137–150. [Google Scholar] [CrossRef]
- Mehra, N.; Aqil, M.; Sultana, Y. A grafted copolymer-based nanomicelles for topical ocular delivery of everolimus: Formulation, characterization, ex-vivo permeation, in-vitro ocular toxicity, and stability study. Eur. J. Pharm. Sci. 2021, 159, 105735. [Google Scholar] [CrossRef]
- Wu, Y.; Woodbine, L.; Carr, A.M.; Pillai, A.R.; Nokhodchi, A.; Maniruzzaman, M. 3D Printed Calcium Phosphate Cement (CPC) Scaffolds for Anti-Cancer Drug Delivery. Pharmaceutics 2020, 12, 1077. [Google Scholar] [CrossRef] [PubMed]
- Torrado-Salmerón, C.; Guarnizo-Herrero, V.; Cerezo-Garreta, J.; Durán, G.T.; Torrado-Santiago, S. Self-Micellizing Technology Improves the Properties of Ezetimibe and Increases Its Effect on Hyperlipidemic Rats. Pharmaceutics 2019, 11, 647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quan, G.; Niu, B.; Singh, V.; Zhou, Y.; Wu, C.-Y.; Pan, X.; Wu, C. Supersaturable solid self-microemulsifying drug delivery system: Precipitation inhibition and bioavailability enhancement. Int. J. Nanomed. 2017, 12, 8801–8811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paudwal, G.; Rawat, N.; Gupta, R.; Baldi, A.; Singh, G.; Gupta, P.N. Recent Advances in Solid Dispersion Technology for Efficient Delivery of Poorly Water-Soluble Drugs. Curr. Pharm. Des. 2019, 25, 1524–1535. [Google Scholar] [CrossRef] [PubMed]
- Simões, M.; Pinto, R.M.; Simões, S. Hot-melt extrusion in the pharmaceutical industry: Toward filing a new drug application. Drug Discov. Today 2019, 24, 1749–1768. [Google Scholar] [CrossRef]
- Gajda, M.; Nartowski, K.P.; Pluta, J.; Karolewicz, B. Continuous, one-step synthesis of pharmaceutical cocrystals via hot melt extrusion from neat to matrix-assisted processing–State of the art. Int. J. Pharm. 2019, 558, 426–440. [Google Scholar] [CrossRef]
- Chivate, A.; Garkal, A.D.; Dhas, N.L.; Mehta, T.A. Hot-Melt Extrusion: An Emerging Technique for Solubility Enhancement of Poorly Water-Soluble Drugs. PDA J. Pharm. Sci. Technol. 2021, 75, 357–373. [Google Scholar] [CrossRef]
- Patil, H.; Tiwari, R.V.; Repka, M.A. Hot-Melt Extrusion: From Theory to Application in Pharmaceutical Formulation. AAPS PharmSciTech 2016, 17, 20–42. [Google Scholar] [CrossRef] [Green Version]
- Kallakunta, V.R.; Sarabu, S.; Bandari, S.; Batra, A.; Bi, V.; Durig, T.; Repka, M.A. Stable amorphous solid dispersions of fenofibrate using hot melt extrusion technology: Effect of formulation and process parameters for a low glass transition temperature drug. J. Drug Deliv. Sci. Technol. 2020, 58, 101395. [Google Scholar] [CrossRef]
- Ma, X.; Huang, S.; Lowinger, M.B.; Liu, X.; Lu, X.; Su, Y.; Williams, R.O. Influence of mechanical and thermal energy on nifedipine amorphous solid dispersions prepared by hot melt extrusion: Preparation and physical stability. Int. J. Pharm. 2019, 561, 324–334. [Google Scholar] [CrossRef]
- Sardelli, L.; Tunesi, M.; Briatico-Vangosa, F.; Petrini, P. 3D-Reactive printing of engineered alginate inks. Soft Matter 2021, 17, 8105–8117. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, R.; Patil, H.; Repka, M.A. Contribution of hot-melt extrusion technology to advance drug delivery in the 21st century. Expert Opin. Drug Deliv. 2016, 13, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Müller, F.B.; Bolli, P.; Erne, P.; Block, L.H.; Kiowski, W.; Bühler, F.R. Antihypertensive therapy with the long-acting calcium antagonist nitrendipine. J. Cardiovasc. Pharmacol. 1984, 6, 1073–1076. [Google Scholar]
- Noyes, A.A.; Whitney, W.R. The Rate of Solution of Solid Substances in their Own SolutioNS. J. Am. Chem. Soc. 1897, 19, 930–934. [Google Scholar] [CrossRef] [Green Version]
- Ainurofiq, A.; Mauludin, R.; Mudhakir, D.; Soewandhi, S.N. A Novel Desloratadine-Benzoic Acid Co-Amorphous Solid: Preparation, Characterization, and Stability Evaluation. Pharmaceutics 2018, 10, 85. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.G.; Kim, D.D.; Jun, H.W.; Yoo, B.K.; Yong, C.S. Improvement of dissolution and bioavailability of nitrendipine by inclusion in hydroxypropyl-beta-cyclodextrin. Drug Dev. Ind. Pharm. 2003, 29, 1085–1094. [Google Scholar] [CrossRef]
- Penumetcha, S.S.; Gutta, L.N.; Dhanala, H.; Yamili, S.; Challa, S.; Rudraraju, S.; Rudraraju, S.; Rudraraju, V. Hot melt extruded Aprepitant-Soluplus solid dispersion: Preformulation considerations, stability and in vitro study. Drug Dev. Ind. Pharm. 2016, 42, 1609–1620. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, L.; Ma, D.; Tang, X.; Zhang, Y.; Yin, T.; Gou, J.; Wang, Y.; He, H. Characterizing and Exploring the Differences in Dissolution and Stability Between Crystalline Solid Dispersion and Amorphous Solid Dispersion. AAPS Pharm. Sci. Tech. 2020, 21, 262. [Google Scholar] [CrossRef]
- Zhu, Z.Y.; Mao, F.F.; Zhu, J.B. Studies on the bioavailability of nitrendipine tablet. Yao Xue Xue Bao 1990, 25, 709–716. [Google Scholar]
- Shuai, S.; Yue, S.; Huang, Q.; Wang, W.; Yang, J.; Lan, K.; Ye, L. Preparation, characterization and in vitro/vivo evaluation of tectorigenin solid dispersion with improved dissolution and bioavailability. Eur. J. Drug Metab. Pharmacokinet. 2016, 41, 413–422. [Google Scholar] [CrossRef]
- Tanida, S.; Kurokawa, T.; Sato, H.; Kadota, K.; Tozuka, Y. Evaluation of the Micellization Mechanism of an Amphipathic Graft Copolymer with Enhanced Solubility of Ipriflavone. Chem. Pharm. Bull. 2016, 64, 68–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Speybroeck, M.; Mellaerts, R.; Mols, R.; Thi, T.D.; Martens, J.; Van Humbeeck, J.; Annaert, P.; Mooter, G.V.D.; Augustijns, P. Enhanced absorption of the poorly soluble drug fenofibrate by tuning its release rate from ordered mesoporous silica. Eur. J. Pharm. Sci. 2010, 41, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Altamimi, M.A.; Neau, S.H. Investigation of the in vitro performance difference of drug-Soluplus(R) and drug-PEG 6000 dispersions when prepared using spray drying or lyophilization. Saudi Pharm. J. 2017, 25, 419–439. [Google Scholar] [CrossRef] [Green Version]
- Sarpal, K.; Munson, E.J. Amorphous Solid Dispersions of Felodipine and Nifedipine with Soluplus(R): Drug-Polymer Miscibility and Intermolecular Interactions. J. Pharm. Sci. 2021, 110, 1457–1469. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, T.; Connors, K.A. Phase solubility techniques. Adv. Anal. Chem. Instrum. 1965, 4, 117–212. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Abbreviation | Molecular Weight (Da) |
|---|---|---|
| Nitrendipine | NIT | 360.36 |
| Soluplus® | So | 90,000–140,000 |
| Physical mixtures | NITPM | N/A |
| Pure amorphous nitrendipine | NITAM | N/A |
| saSMSD made by HME technique | saSMSD/NITCHME | N/A |
| saSMSD made by CP techniques | saSMSD/NITNCCP | N/A |
| saSMSD made by FQC techniques | saSMSD/NITNCFQC | N/A |
| Concentration (mg/mL) of Pre-Dissolved Soluplus® | Initial NIT Concentration (μg/mL) in Supersaturated Solution | Half-Time (min) of NIT Crystallization from a Supersaturated Sate |
|---|---|---|
| 0 | 33.33 | 30.55 ± 9.38 |
| 0.25 | 33.33 | 43.13 ± 4.45 |
| 0.1 | 33.33 | 80.51 ± 3.92 |
| 0.15 | 33.33 | 112.95 ± 1.98 |
| 0.35 | 33.33 | 170.22 ± 3.42 |
| Concentration (mg/mL) of Soluplus® | Drug | ΔGtr° (KJ/mol) |
|---|---|---|
| 0.1 | NIT | −3.27 ± 0.13 |
| 0.3 | NIT | −4.62 ± 0.06 |
| 0.5 | NIT | −6.10 ± 0.41 |
| 0.7 | NIT | −6.53 ± 0.62 |
| 0.9 | NIT | −6.84 ± 0.14 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, H.; Zhang, Y.; Ma, Y.; Zhang, H.; Hao, C.; Zhang, Y.; Li, Z.; Qi, X.; Shi, N. Hot Melt Extrusion-Triggered Amorphization as a Continuous Process for Inducing Extended Supersaturable Drug Immediate-Release from saSMSDs Systems. Pharmaceutics 2022, 14, 765. https://doi.org/10.3390/pharmaceutics14040765
Yu H, Zhang Y, Ma Y, Zhang H, Hao C, Zhang Y, Li Z, Qi X, Shi N. Hot Melt Extrusion-Triggered Amorphization as a Continuous Process for Inducing Extended Supersaturable Drug Immediate-Release from saSMSDs Systems. Pharmaceutics. 2022; 14(4):765. https://doi.org/10.3390/pharmaceutics14040765
Chicago/Turabian StyleYu, Huan, Yanfei Zhang, Yinghui Ma, Huifeng Zhang, Chengyi Hao, Yong Zhang, Zhengqiang Li, Xianrong Qi, and Nianqiu Shi. 2022. "Hot Melt Extrusion-Triggered Amorphization as a Continuous Process for Inducing Extended Supersaturable Drug Immediate-Release from saSMSDs Systems" Pharmaceutics 14, no. 4: 765. https://doi.org/10.3390/pharmaceutics14040765