Dual Coating of Chitosan and Albumin Negates the Protein Corona-Induced Reduced Vascular Adhesion of Targeted PLGA Microparticles in Human Blood


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Fabrication of Uncoated and CS-Coated PLGA Microparticles
2.2. HSA Conjugation and Staining
2.3. Avidin Conjugation and Targeting Ligand Attachment
2.4. Particle Surface Characterization
2.5. HUVEC Culture
2.6. Blood Preparation
2.7. Parallel Plate Flow Chamber Setup
2.8. SDS-PAGE Gel Preparation
2.9. Statistical Methods
3. Results
3.1. Characterization of HSA and CS-Coated PLGA
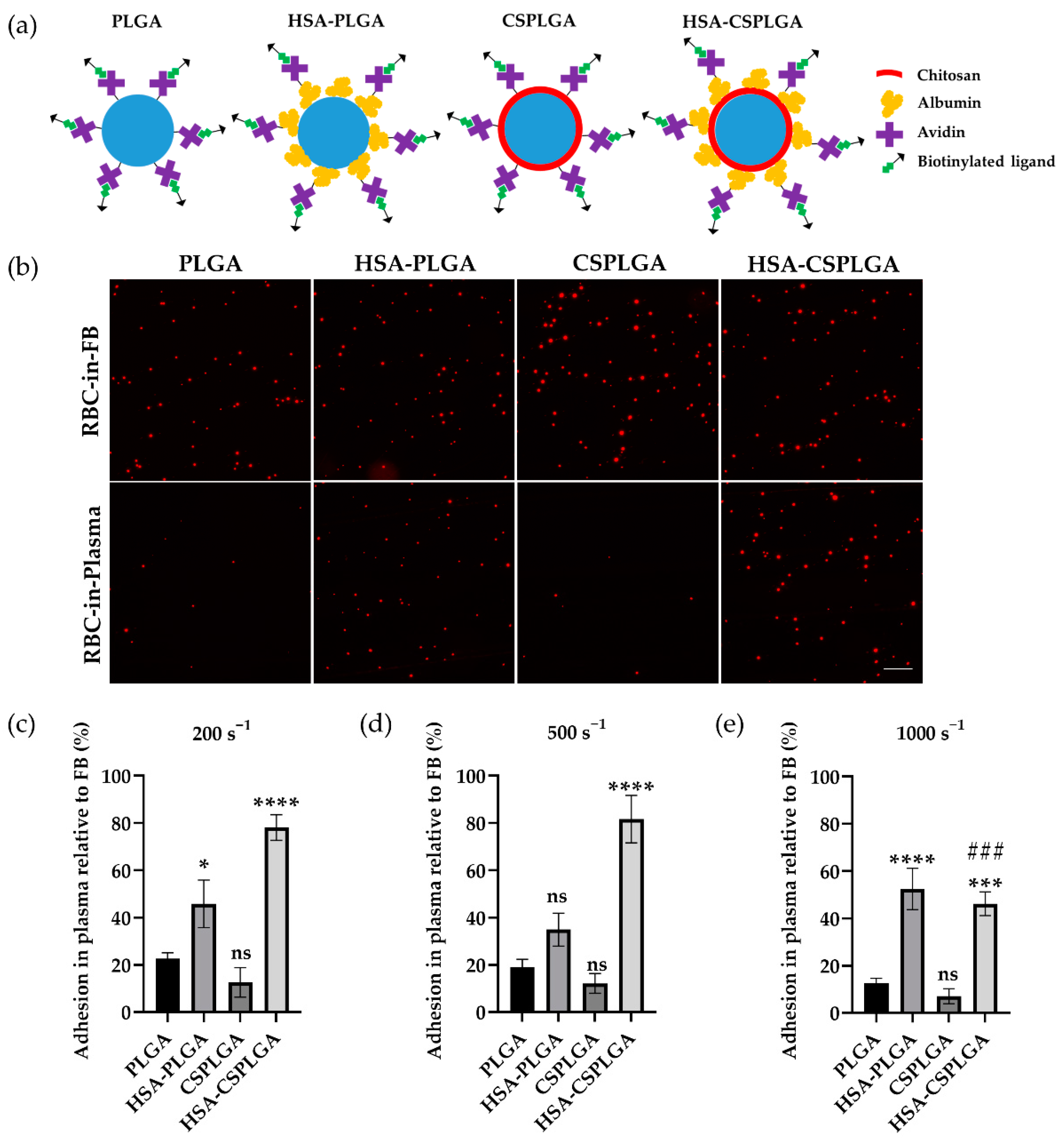
3.2. Adhesion of HSA- and CS-Coated PLGA Particles Targeted with sLeA
3.3. Alternative Ligand Schemes on HSA-CSPLGA to Improve Binding at High Shear
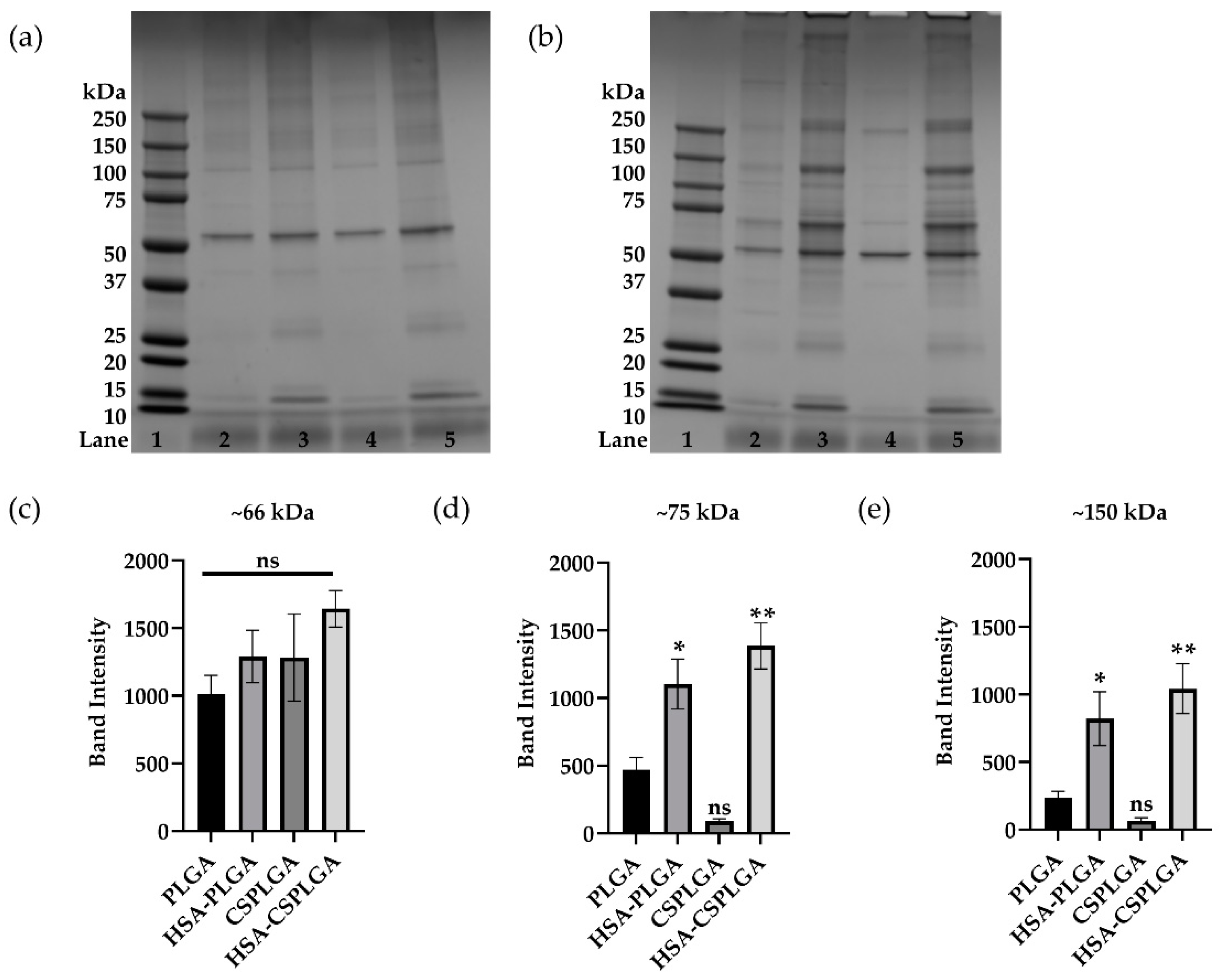
3.4. Characterization of Plasma Protein Corona on Coated PLGA with sLeA
3.5. Impact of Anticoagulant on Binding and Protein Adsorption of Coated PLGA
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hajitou, A.; Pasqualini, R.; Arap, W. Vascular targeting: Recent advances and therapeutic perspectives. Trends Cardiovasc. Med. 2006, 16, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Matoba, T.; Koga, J.I.; Nakano, K.; Egashira, K.; Tsutsui, H. Nanoparticle-mediated drug delivery system for atherosclerotic cardiovascular disease. J. Cardiol. 2017, 70, 206–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal Formulations in Clinical Use: An Updated Review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Peer, D.; Karp, J.M.; Hong, S.; Farokhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2007, 2, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.M.; Cullis, P.R. Liposomal drug delivery systems: From concept to clinical applications. Adv. Drug Deliv. Rev. 2013, 65, 36–48. [Google Scholar] [CrossRef]
- Koren, E.; Torchilin, V.P. Drug carriers for vascular drug delivery. IUBMB Life 2011, 63, 586–595. [Google Scholar] [CrossRef]
- Gupta, A. Sen Nanomedicine approaches in vascular disease: A review. Nanomed. Nanotechnol. Biol. Med. 2011, 7, 763–779. [Google Scholar] [CrossRef]
- Flores, A.M.; Ye, J.; Jarr, K.U.; Hosseini-Nassab, N.; Smith, B.R.; Leeper, N.J. Nanoparticle Therapy for Vascular Diseases. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 635–646. [Google Scholar] [CrossRef]
- Zhong, H.; Chan, G.; Hu, Y.; Hu, H.; Ouyang, D. A Comprehensive Map of FDA-Approved Pharmaceutical Products. Pharmaceutics 2018, 10, 263. [Google Scholar] [CrossRef] [Green Version]
- Kumari, A.; Yadav, S.K.; Yadav, S.C. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf. B Biointerfaces 2010, 75, 1–18. [Google Scholar] [CrossRef]
- Danhier, F.; Ansorena, E.; Silva, J.M.; Coco, R.; Le Breton, A.; Préat, V. PLGA-based nanoparticles: An overview of biomedical applications. J. Control Release 2012, 161, 505–522. [Google Scholar] [CrossRef] [PubMed]
- Vlachopoulos, A.; Karlioti, G.; Balla, E.; Daniilidis, V.; Kalamas, T.; Stefanidou, M.; Bikiaris, N.D.; Christodoulou, E.; Koumentakou, I.; Karavas, E.; et al. Poly(Lactic Acid)-Based Microparticles for Drug Delivery Applications: An Overview of Recent Advances. Pharmaceutics 2022, 14, 359. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Skidmore, S.; Hadar, J.; Garner, J.; Park, H.; Otte, A.; Soh, B.K.; Yoon, G.; Yu, D.; Yun, Y.; et al. Injectable, long-acting PLGA formulations: Analyzing PLGA and understanding microparticle formation. J. Control Release 2019, 304, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Sheffey, V.V.; Siew, E.B.; Tanner, E.E.L.; Eniola-Adefeso, O. PLGA’s Plight and the Role of Stealth Surface Modification Strategies in Its Use for Intravenous Particulate Drug Delivery. Adv. Healthc. Mater. 2022, 11, 2101536. [Google Scholar] [CrossRef]
- Charoenphol, P.; Huang, R.B.; Eniola-Adefeso, O. Potential role of size and hemodynamics in the efficacy of vascular-targeted spherical drug carriers. Biomaterials 2010, 31, 1392–1402. [Google Scholar] [CrossRef]
- Carboni, E.; Tschudi, K.; Nam, J.; Lu, X.; Ma, A.W.K. Particle margination and its implications on intravenous anticancer drug delivery. AAPS PharmSciTech 2014, 15, 762–771. [Google Scholar] [CrossRef] [Green Version]
- Sobczynski, D.J.; Charoenphol, P.; Heslinga, M.J.; Onyskiw, P.J.; Namdee, K.; Thompson, A.J.; Eniola-Adefeso, O. Plasma protein corona modulates the vascular wall interaction of drug carriers in a material and donor specific manner. PLoS ONE 2014, 9, e107408. [Google Scholar] [CrossRef]
- Sobczynski, D.J.; Eniola-Adefeso, O. IgA and IgM protein primarily drive plasma corona-induced adhesion reduction of PLGA nanoparticles in human blood flow. Bioeng. Transl. Med. 2017, 2, 180–190. [Google Scholar] [CrossRef] [Green Version]
- Namdee, K.; Sobczynski, D.J.; Onyskiw, P.J.; Eniola-Adefeso, O. Differential Impact of Plasma Proteins on the Adhesion Efficiency of Vascular-Targeted Carriers (VTCs) in Blood of Common Laboratory Animals. Bioconjug. Chem. 2015, 26, 2419–2428. [Google Scholar] [CrossRef] [Green Version]
- Sobczynski, D.J.; Eniola-Adefeso, O. Effect of anticoagulants on the protein corona-induced reduced drug carrier adhesion efficiency in human blood flow. Acta Biomater. 2017, 48, 186–194. [Google Scholar] [CrossRef] [Green Version]
- Suk, J.S.; Xu, Q.; Kim, N.; Hanes, J.; Ensign, L.M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 2016, 99, 28–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, W.J.; Fromen, C.A.; Lopez-Cazares, G.; Eniola-Adefeso, O. PEGylation of model drug carriers enhances phagocytosis by primary human neutrophils. Acta Biomater. 2018, 79, 283–293. [Google Scholar] [CrossRef] [PubMed]
- George, A.; Shah, P.A.; Shrivastav, P.S. Natural biodegradable polymers based nano-formulations for drug delivery: A review. Int. J. Pharm. 2019, 561, 244–264. [Google Scholar] [CrossRef]
- Frank, L.A.; Onzi, G.R.; Morawski, A.S.; Pohlmann, A.R.; Guterres, S.S.; Contri, R.V. Chitosan as a coating material for nanoparticles intended for biomedical applications. React. Funct. Polym. 2020, 147, 104459. [Google Scholar] [CrossRef]
- Tao, C.; Chuah, Y.J.; Xu, C.; Wang, D.A. Albumin conjugates and assemblies as versatile bio-functional additives and carriers for biomedical applications. J. Mater. Chem. B 2019, 7, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.J.; Zeng, Z.W.; Xiao, R.Z.; Xie, T.; Zhou, G.L.; Zhan, X.R.; Wang, S.L. Recent advances of chitosan nanoparticles as drug carriers. Int. J. Nanomed. 2011, 6, 765–774. [Google Scholar] [CrossRef] [Green Version]
- Kean, T.; Thanou, M. Biodegradation, biodistribution and toxicity of chitosan. Adv. Drug Deliv. Rev. 2010, 62, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Larsen, M.T.; Kuhlmann, M.; Hvam, M.L.; Howard, K.A. Albumin-based drug delivery: Harnessing nature to cure disease. Mol. Cell. Ther. 2016, 4, 3. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Fuentes, M.; Alonso, M.J. Chitosan-based drug nanocarriers: Where do we stand? J. Control Release 2012, 161, 496–504. [Google Scholar] [CrossRef]
- O’Donnell, P.B.; McGinity, J.W. Preparation of microspheres by the solvent evaporation technique. Adv. Drug Deliv. Rev. 1997, 28, 25–42. [Google Scholar] [CrossRef]
- Lu, L.; Duong, V.T.; Shalash, A.O.; Skwarczynski, M.; Toth, I. Chemical Conjugation Strategies for the Development of Protein-Based Subunit Nanovaccines. Vaccines 2021, 9, 563. [Google Scholar] [CrossRef] [PubMed]
- Fish, M.B.; Fromen, C.A.; Lopez-Cazares, G.; Golinski, A.W.; Scott, T.F.; Adili, R.; Holinstat, M.; Eniola-Adefeso, O. Exploring deformable particles in vascular-targeted drug delivery: Softer is only sometimes better. Biomaterials 2017, 124, 169–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, R.B.; Eniola-Adefeso, O. Shear Stress Modulation of IL-1β-Induced E-Selectin Expression in Human Endothelial Cells. PLoS ONE 2012, 7, e31874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, R.C.F.; Ng, T.B.; Wong, J.H.; Chan, W.Y. Chitosan: An Update on Potential Biomedical and Pharmaceutical Applications. Mar. Drugs 2015, 13, 5156–5186. [Google Scholar] [CrossRef]
- Vaezifar, S.; Razavi, S.; Golozar, M.A.; Esfahani, H.Z.; Morshed, M.; Karbasi, S. Characterization of PLGA/Chitosan Electrospun Nano-Biocomposite Fabricated by Two Different Methods. Int. J. Polym. Mater. Polym. Biomater. 2014, 64, 64–75. [Google Scholar] [CrossRef]
- Bobo, D.; Robinson, K.J.; Islam, J.; Thurecht, K.J.; Corrie, S.R. Nanoparticle-Based Medicines: A Review of FDA-Approved Materials and Clinical Trials to Date. Pharm. Res. 2016, 33, 2373–2387. [Google Scholar] [CrossRef]
- Gupta, M.N.; Roy, I. How Corona Formation Impacts Nanomaterials as Drug Carriers. Mol. Pharm. 2020, 17, 725–737. [Google Scholar] [CrossRef]
- Bertrand, N.; Grenier, P.; Mahmoudi, M.; Lima, E.M.; Appel, E.A.; Dormont, F.; Lim, J.-M.; Karnik, R.; Langer, R.; Farokhzad, O.C. Mechanistic understanding of in vivo protein corona formation on polymeric nanoparticles and impact on pharmacokinetics. Nat. Commun. 2017, 8, 777. [Google Scholar] [CrossRef]
- Lee, Y.K.; Choi, E.-J.; Webster, T.J.; Kim, S.-H.; Khang, D. Effect of the protein corona on nanoparticles for modulating cytotoxicity and immunotoxicity. Int. J. Nanomed. 2015, 10, 97–113. [Google Scholar] [CrossRef] [Green Version]
- Lundqvist, M.; Stigler, J.; Elia, G.; Lynch, I.; Cedervall, T.; Dawson, K.A. Nanoparticle size and surface properties determine the protein corona with possible implications for biological impacts. Proc. Natl. Acad. Sci. USA 2008, 105, 14265–14270. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Ukidve, A.; Krishnan, V.; Mitragotri, S. Effect of physicochemical and surface properties on in vivo fate of drug nanocarriers. Adv. Drug Deliv. Rev. 2019, 143, 3–21. [Google Scholar] [CrossRef] [PubMed]
- Schöttler, S.; Becker, G.; Winzen, S.; Steinbach, T.; Mohr, K.; Landfester, K.; Mailänder, V.; Wurm, F.R. Protein adsorption is required for stealth effect of poly(ethylene glycol)- and poly(phosphoester)-coated nanocarriers. Nat. Nanotechnol. 2016, 11, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Shubhra, Q.T.H.; Tóth, J.; Gyenis, J.; Feczkó, T. Surface modification of HSA containing magnetic PLGA nanoparticles by poloxamer to decrease plasma protein adsorption. Colloids Surf. B Biointerfaces 2014, 122, 529–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walkey, C.D.; Olsen, J.B.; Guo, H.; Emili, A.; Chan, W.C.W. Nanoparticle size and surface chemistry determine serum protein adsorption and macrophage uptake. J. Am. Chem. Soc. 2012, 134, 2139–2147. [Google Scholar] [CrossRef] [PubMed]
- Amoozgar, Z.; Park, J.; Lin, Q.; Yeo, Y. Low Molecular-Weight Chitosan as a pH-Sensitive Stealth Coating for Tumor-Specific Drug Delivery. Mol. Pharm. 2012, 9, 1262–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parveen, S.; Sahoo, S.K. Long circulating chitosan/PEG blended PLGA nanoparticle for tumor drug delivery. Eur. J. Pharmacol. 2011, 670, 372–383. [Google Scholar] [CrossRef]
- Manoochehri, S.; Darvishi, B.; Kamalinia, G.; Amini, M.; Fallah, M.; Ostad, S.N.; Atyabi, F.; Dinarvand, R. Surface modification of PLGA nanoparticles via human serum albumin conjugation for controlled delivery of docetaxel. Daru 2013, 21, 58. [Google Scholar] [CrossRef] [Green Version]
- Kohane, D. Microparticles and nanoparticles for drug delivery. Biotechnol. Bioeng. 2007, 96, 203–209. [Google Scholar] [CrossRef]
- Kutscher, H.L.; Chao, P.; Deshmukh, M.; Singh, Y.; Hu, P.; Joseph, L.B.; Reimer, D.C.; Stein, S.; Laskin, D.L.; Sinko, P.J. Threshold size for optimal passive pulmonary targeting and retention of rigid microparticles in rats. J. Control Release 2010, 143, 31. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Li, P.; Kong, L. Chitosan-modified PLGA nanoparticles with versatile surface for improved drug delivery. AAPS PharmSciTech 2013, 14, 585–592. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Gemeinhart, R.A. Understanding the adsorption mechanism of chitosan onto poly(lactide-co-glycolide) particles. Eur. J. Pharm. Biopharm. 2008, 70, 597–604. [Google Scholar] [CrossRef] [Green Version]
- Merlot, A.M.; Kalinowski, D.S.; Richardson, D.R.; Vogt, B.; Pharmacology, M. Unraveling the mysteries of serum albumin-more than just a serum protein. Front. Physiol. 2014, 5, 299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunawan, R.C.; Auguste, D.T. The role of antibody synergy and membrane fluidity in the vascular targeting of immunoliposomes. Biomaterials 2010, 31, 900–907. [Google Scholar] [CrossRef] [PubMed]
- Nelson, R.M.; Dolich, S.; Aruffo, A.; Cecconi, O.; Bevilacqua, M.P. Higher-affinity oligosaccharide ligands for E-selectin. J. Clin. Investig. 1993, 91, 1157. [Google Scholar] [CrossRef] [PubMed]
- Eniola, A.O.; Willcox, P.J.; Hammer, D.A. Interplay between rolling and firm adhesion elucidated with a cell-free system engineered with two distinct receptor-ligand pairs. Biophys. J. 2003, 85, 2720–2731. [Google Scholar] [CrossRef] [Green Version]
- Peng, Q.; Zhang, S.; Yang, Q.; Zhang, T.; Wei, X.Q.; Jiang, L.; Zhang, C.L.; Chen, Q.M.; Zhang, Z.R.; Lin, Y.F. Preformed albumin corona, a protective coating for nanoparticles based drug delivery system. Biomaterials 2013, 34, 8521–8530. [Google Scholar] [CrossRef] [PubMed]
- Cui, T.; Ma, Y.; Yang, J.Y.; Liu, S.; Wang, Z.; Zhang, F.; Wang, J.; Cai, T.; Dong, L.; Hong, J.; et al. Protein corona-guided tumor targeting therapy: Via the surface modulation of low molecular weight PEG. Nanoscale 2021, 13, 5883–5891. [Google Scholar] [CrossRef]
- Poon, I.K.H.; Patel, K.K.; Davis, D.S.; Parish, C.R.; Hulett, M.D. Histidine-rich glycoprotein: The Swiss Army knife of mammalian plasma. Blood. J. Am. Soc. Hematol. 2011, 117, 2093–2101. [Google Scholar] [CrossRef] [Green Version]
- Fedeli, C.; Segat, D.; Tavano, R.; Bubacco, L.; De Franceschi, G.; de Laureto, P.P.; Lubian, E.; Selvestrel, F.; Mancin, F.; Papini, E. The functional dissection of the plasma corona of SiO2-NPs spots histidine rich glycoprotein as a major player able to hamper nanoparticle capture by macrophages. Nanoscale 2015, 7, 17710–17728. [Google Scholar] [CrossRef]
- Gao, S.; Wake, H.; Sakaguchi, M.; Wang, D.; Takahashi, Y.; Teshigawara, K.; Zhong, H.; Mori, S.; Liu, K.; Takahashi, H.; et al. Histidine-Rich Glycoprotein Inhibits High-Mobility Group Box-1-Mediated Pathways in Vascular Endothelial Cells through CLEC-1A. iScience 2020, 23, 101180. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Particle Surface Density (Sites/µm2) | ||||
|---|---|---|---|---|
| Particle Type | HSA [after Avidin] | Avidin | sLeA | aICAM |
| PLGA (sLeA or aICAM) | N/A | 8000 ± 3000 | 1200 ± 300 | 5300 ± 1300 |
| HSA-PLGA (sLeA) | 21,000 ± 8000 [14,000 ± 10,000] | 14,000 ± 12,000 | 1000 ± 300 | N/A |
| CSPLGA (sLeA or aICAM) | N/A | 18,000 ± 8000 | 1200 ± 300 | 5800 ± 1100 |
| HSA-CSPLGA (sLeA or aICAM) | 122,000 ± 32,000 [44,000 ± 11,000] | 32,000 ± 11,000 | 1300 ± 300 | 6000 ± 1300 |
| HSA-CSPLGA (High aICAM) | N/A | 10,000 ± 1500 | ||
| HSA-CSPLGA (sLeA + aICAM) | 4000 ± 1200 | 6300 ± 1400 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lopez-Cazares, G.; Eniola-Adefeso, O. Dual Coating of Chitosan and Albumin Negates the Protein Corona-Induced Reduced Vascular Adhesion of Targeted PLGA Microparticles in Human Blood. Pharmaceutics 2022, 14, 1018. https://doi.org/10.3390/pharmaceutics14051018
Lopez-Cazares G, Eniola-Adefeso O. Dual Coating of Chitosan and Albumin Negates the Protein Corona-Induced Reduced Vascular Adhesion of Targeted PLGA Microparticles in Human Blood. Pharmaceutics. 2022; 14(5):1018. https://doi.org/10.3390/pharmaceutics14051018
Chicago/Turabian StyleLopez-Cazares, Genesis, and Omolola Eniola-Adefeso. 2022. "Dual Coating of Chitosan and Albumin Negates the Protein Corona-Induced Reduced Vascular Adhesion of Targeted PLGA Microparticles in Human Blood" Pharmaceutics 14, no. 5: 1018. https://doi.org/10.3390/pharmaceutics14051018