
Application of CRISPR-Cas9 System to Study Biological Barriers to Drug Delivery





Abstract
:1. Introduction
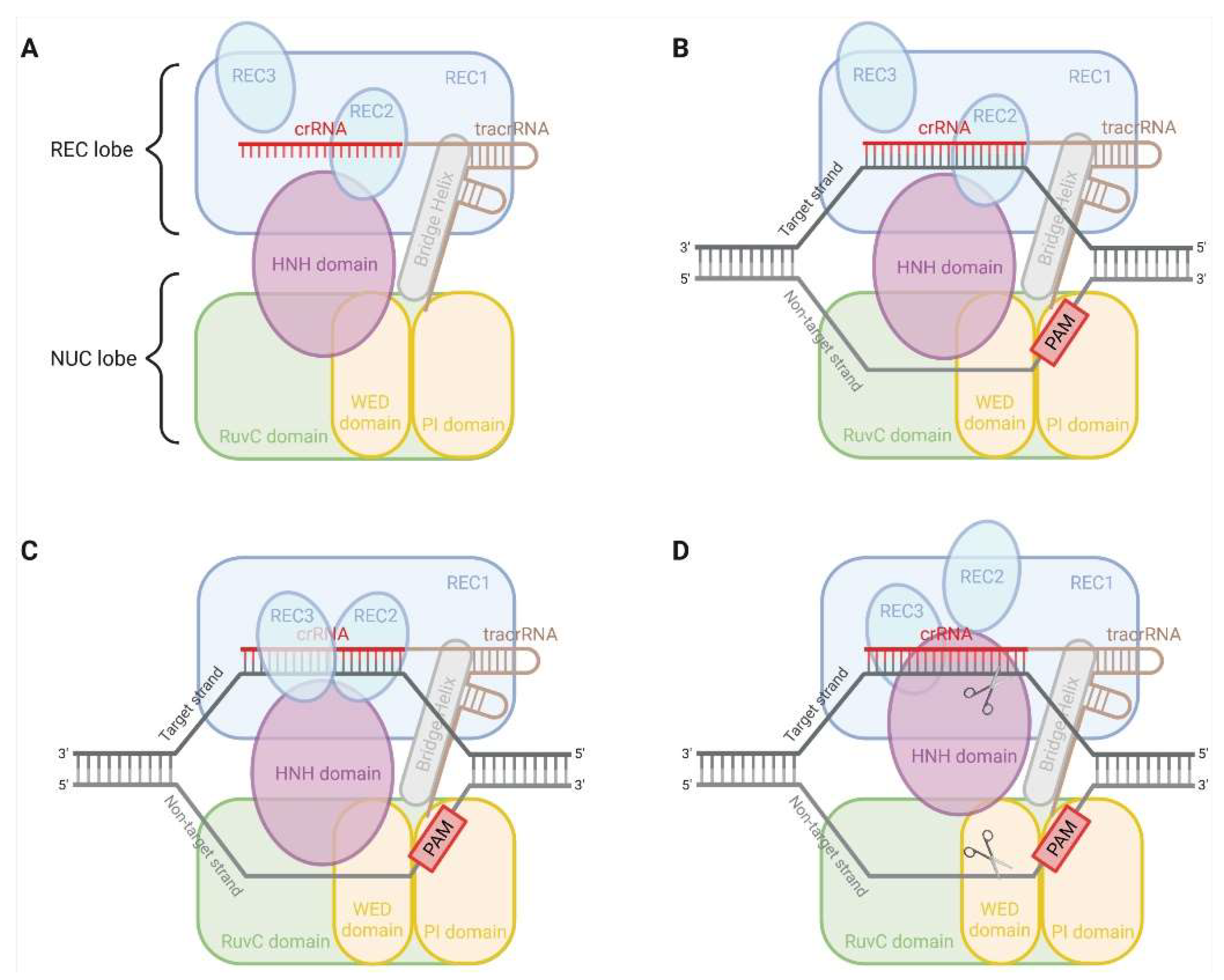
2. CRISPR-Cas9 System
3. Application of CRISPR-Cas9 in Drug Delivery Barrier Studies
3.1. Intestinal Barriers to Drug Delivery
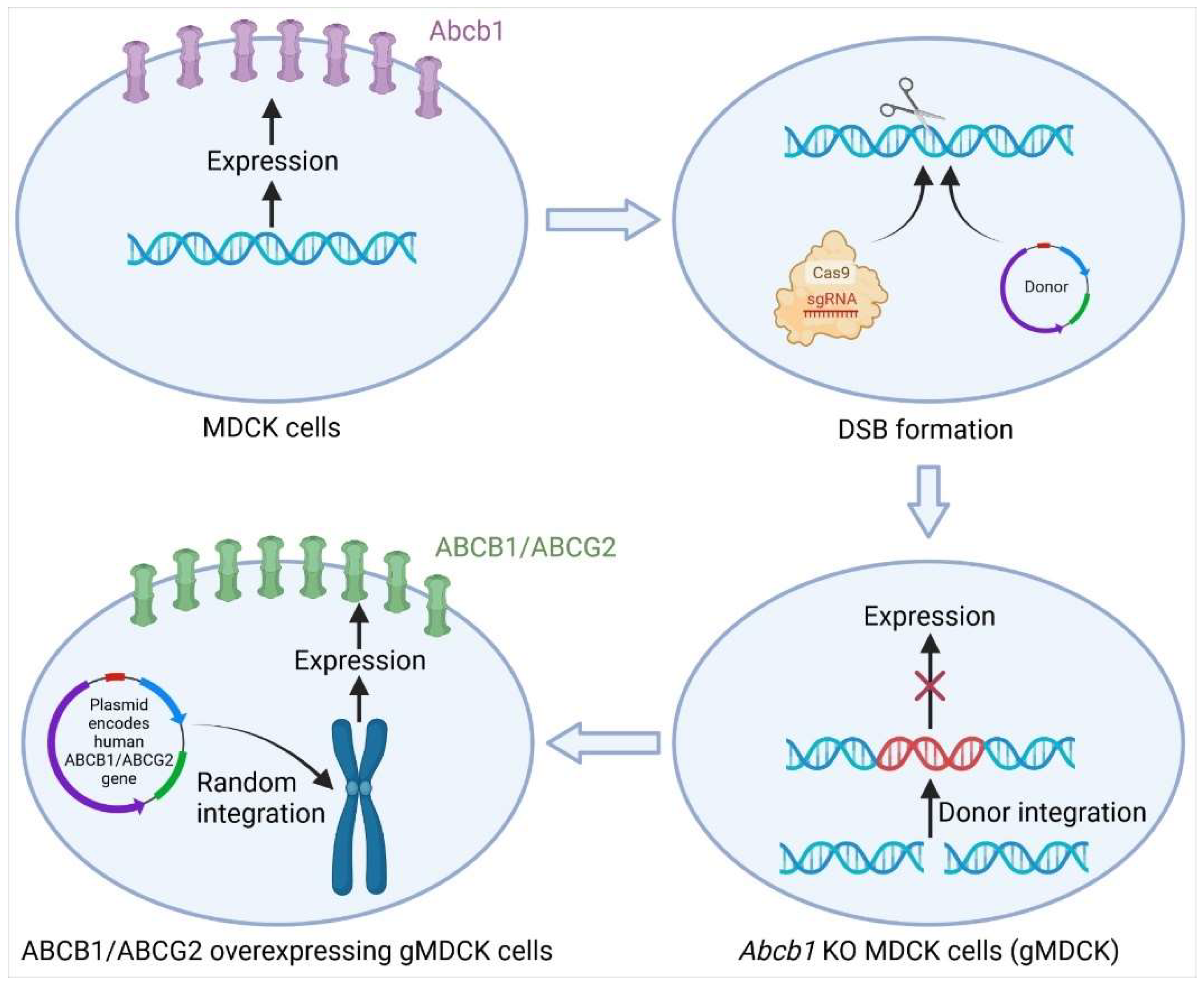
3.1.1. Knockout of Abcb1 in MDCK Cells by CRISPR-Cas9
3.1.2. Mdr1a/b Double-Knockout Rat Models
3.2. Biological Barriers to Anticancer Drugs
3.2.1. Knockout and Regulation of ABC Transporter Genes in Cancer Cells by CRISPR-Cas9
3.2.2. Genome-Wide CRISPR-Cas9 Knockout Screen
3.2.3. Novel Mechanisms of Platinum Accumulation in Cancer Cells
3.3. Blood-Brain Barriers
3.4. Regulation of Transporter Genes
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- International Transporter Consortium; Giacomini, K.M.; Huang, S.M.; Tweedie, D.J.; Benet, L.Z.; Brouwer, K.L.; Chu, X.; Dahlin, A.; Evers, R.; Fischer, V.; et al. Membrane transporters in drug development. Nat. Rev. Drug Discov. 2010, 9, 215–236. [Google Scholar] [CrossRef] [PubMed]
- Zamek-Gliszczynski, M.J.; Taub, M.E.; Chothe, P.P.; Chu, X.; Giacomini, K.M.; Kim, R.B.; Ray, A.S.; Stocker, S.L.; Unadkat, J.D.; Wittwer, M.B.; et al. Transporters in Drug Development: 2018 ITC Recommendations for Transporters of Emerging Clinical Importance. Clin. Pharmacol. Ther. 2018, 104, 890–899. [Google Scholar] [CrossRef] [PubMed]
- Evers, R.; Piquette-Miller, M.; Polli, J.W.; Russel, F.G.M.; Sprowl, J.A.; Tohyama, K.; Ware, J.A.; de Wildt, S.N.; Xie, W.; Brouwer, K.L.R.; et al. Disease-Associated Changes in Drug Transporters May Impact the Pharmacokinetics and/or Toxicity of Drugs: A White Paper From the International Transporter Consortium. Clin. Pharmacol. Ther. 2018, 104, 900–915. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Revalde, J.; Paxton, J.W. The effects of dietary and herbal phytochemicals on drug transporters. Adv. Drug Deliv. Rev. 2017, 116, 45–62. [Google Scholar] [CrossRef]
- Mao, Q.; Lai, Y.; Wang, J. Drug Transporters in Xenobiotic Disposition and Pharmacokinetic Prediction. Drug Metab. Dispos. 2018, 46, 561–566. [Google Scholar] [CrossRef] [Green Version]
- Nakanishi, T.; Tamai, I. Interaction of Drug or Food with Drug Transporters in Intestine and Liver. Curr. Drug Metab. 2015, 16, 753–764. [Google Scholar] [CrossRef]
- Jansen, R.; Embden, J.D.; Gaastra, W.; Schouls, L.M. Identification of genes that are associated with DNA repeats in prokaryotes. Mol. Microbiol. 2002, 43, 1565–1575. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Koonin, E.V.; Makarova, K.S.; Zhang, F. Diversity, classification and evolution of CRISPR-Cas systems. Curr. Opin. Microbiol. 2017, 37, 67–78. [Google Scholar] [CrossRef]
- Liu, Z.; Dong, H.; Cui, Y.; Cong, L.; Zhang, D. Application of different types of CRISPR/Cas-based systems in bacteria. Microb. Cell Fact. 2020, 19, 172. [Google Scholar] [CrossRef] [PubMed]
- Shmakov, S.; Smargon, A.; Scott, D.; Cox, D.; Pyzocha, N.; Yan, W.; Abudayyeh, O.O.; Gootenberg, J.S.; Makarova, K.S.; Wolf, Y.I.; et al. Diversity and evolution of class 2 CRISPR-Cas systems. Nat. Rev. Microbiol. 2017, 15, 169–182. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Huang, X.; Fang, X.; Zhang, Y.; Wang, W. CRISPR-Cas9 System as a Versatile Tool for Genome Engineering in Human Cells. Mol. Ther. Nucleic. Acids 2016, 5, e388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimasu, H.; Ran, F.A.; Hsu, P.D.; Konermann, S.; Shehata, S.I.; Dohmae, N.; Ishitani, R.; Zhang, F.; Nureki, O. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 2014, 156, 935–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bratovic, M.; Fonfara, I.; Chylinski, K.; Galvez, E.J.C.; Sullivan, T.J.; Boerno, S.; Timmermann, B.; Boettcher, M.; Charpentier, E. Bridge helix arginines play a critical role in Cas9 sensitivity to mismatches. Nat. Chem. Biol. 2020, 16, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Hirano, H.; Gootenberg, J.S.; Horii, T.; Abudayyeh, O.O.; Kimura, M.; Hsu, P.D.; Nakane, T.; Ishitani, R.; Hatada, I.; Zhang, F.; et al. Structure and Engineering of Francisella novicida Cas9. Cell 2016, 164, 950–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deltcheva, E.; Chylinski, K.; Sharma, C.M.; Gonzales, K.; Chao, Y.; Pirzada, Z.A.; Eckert, M.R.; Vogel, J.; Charpentier, E. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 2011, 471, 602–607. [Google Scholar] [CrossRef] [Green Version]
- Szczelkun, M.D.; Tikhomirova, M.S.; Sinkunas, T.; Gasiunas, G.; Karvelis, T.; Pschera, P.; Siksnys, V.; Seidel, R. Direct observation of R-loop formation by single RNA-guided Cas9 and Cascade effector complexes. Proc. Natl. Acad. Sci. USA 2014, 111, 9798–9803. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.S.; Dagdas, Y.S.; Kleinstiver, B.P.; Welch, M.M.; Sousa, A.A.; Harrington, L.B.; Sternberg, S.H.; Joung, J.K.; Yildiz, A.; Doudna, J.A. Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature 2017, 550, 407–410. [Google Scholar] [CrossRef] [Green Version]
- Palermo, G.; Miao, Y.; Walker, R.C.; Jinek, M.; Mc Cammon, J.A. CRISPR-Cas9 conformational activation as elucidated from enhanced molecular simulations. Proc. Natl. Acad. Sci. USA 2017, 114, 7260–7265. [Google Scholar] [CrossRef] [Green Version]
- Dagdas, Y.S.; Chen, J.S.; Sternberg, S.H.; Doudna, J.A.; Yildiz, A. A conformational checkpoint between DNA binding and cleavage by CRISPR-Cas9. Sci. Adv. 2017, 3, eaao0027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell. Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosicki, M.; Tomberg, K.; Bradley, A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018, 36, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Binda, C.S.; Klaver, B.; Berkhout, B.; Das, A.T. CRISPR-Cas9 Dual-gRNA Attack Causes Mutation, Excision and Inversion of the HIV-1 Proviral DNA. Viruses 2020, 12, 330. [Google Scholar] [CrossRef] [Green Version]
- Janssen, J.M.; Chen, X.; Liu, J.; Goncalves, M. The Chromatin Structure of CRISPR-Cas9 Target DNA Controls the Balance between Mutagenic and Homology-Directed Gene-Editing Events. Mol. Ther. Nucleic. Acids 2019, 16, 141–154. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Li, Z. CRISPR-Cas systems: Overview, innovations and applications in human disease research and gene therapy. Comput. Struct. Biotechnol. J. 2020, 18, 2401–2415. [Google Scholar] [CrossRef]
- Yamano, T.; Zetsche, B.; Ishitani, R.; Zhang, F.; Nishimasu, H.; Nureki, O. Structural Basis for the Canonical and Non-canonical PAM Recognition by CRISPR-Cpf1. Mol. Cell 2017, 67, 633–645.e3. [Google Scholar] [CrossRef] [Green Version]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; van der Oost, J.; Regev, A.; et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef] [Green Version]
- Strecker, J.; Jones, S.; Koopal, B.; Schmid-Burgk, J.; Zetsche, B.; Gao, L.; Makarova, K.S.; Koonin, E.V.; Zhang, F. Engineering of CRISPR-Cas12b for human genome editing. Nat. Commun. 2019, 10, 212. [Google Scholar] [CrossRef]
- Teng, F.; Cui, T.; Feng, G.; Guo, L.; Xu, K.; Gao, Q.; Li, T.; Li, J.; Zhou, Q.; Li, W. Repurposing CRISPR-Cas12b for mammalian genome engineering. Cell Discov. 2018, 4, 63. [Google Scholar] [CrossRef]
- Bigelyte, G.; Young, J.K.; Karvelis, T.; Budre, K.; Zedaveinyte, R.; Djukanovic, V.; Van Ginkel, E.; Paulraj, S.; Gasior, S.; Jones, S.; et al. Miniature type V-F CRISPR-Cas nucleases enable targeted DNA modification in cells. Nat. Commun. 2021, 12, 6191. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.J.; Orlova, N.; Oakes, B.L.; Ma, E.; Spinner, H.B.; Baney, K.L.M.; Chuck, J.; Tan, D.; Knott, G.J.; Harrington, L.B.; et al. CasX enzymes comprise a distinct family of RNA-guided genome editors. Nature 2019, 566, 218–223. [Google Scholar] [CrossRef]
- Ran, F.A.; Cong, L.; Yan, W.X.; Scott, D.A.; Gootenberg, J.S.; Kriz, A.J.; Zetsche, B.; Shalem, O.; Wu, X.; Makarova, K.S.; et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 2015, 520, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Koo, T.; Park, S.W.; Kim, D.; Kim, K.; Cho, H.Y.; Song, D.W.; Lee, K.J.; Jung, M.H.; Kim, S.; et al. In vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni. Nat. Commun. 2017, 8, 14500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esvelt, K.M.; Mali, P.; Braff, J.L.; Moosburner, M.; Yaung, S.J.; Church, G.M. Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nat. Methods 2013, 10, 1116–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dugar, G.; Leenay, R.T.; Eisenbart, S.K.; Bischler, T.; Aul, B.U.; Beisel, C.L.; Sharma, C.M. CRISPR RNA-Dependent Binding and Cleavage of Endogenous RNAs by the Campylobacter jejuni Cas9. Mol. Cell 2018, 69, 893–905.e7. [Google Scholar] [CrossRef] [Green Version]
- Ma, E.; Harrington, L.B.; O'Connell, M.R.; Zhou, K.; Doudna, J.A. Single-Stranded DNA Cleavage by Divergent CRISPR-Cas9 Enzymes. Mol. Cell 2015, 60, 398–407. [Google Scholar] [CrossRef] [Green Version]
- Muller, M.; Lee, C.M.; Gasiunas, G.; Davis, T.H.; Cradick, T.J.; Siksnys, V.; Bao, G.; Cathomen, T.; Mussolino, C. Streptococcus thermophilus CRISPR-Cas9 Systems Enable Specific Editing of the Human Genome. Mol. Ther. 2016, 24, 636–644. [Google Scholar] [CrossRef] [Green Version]
- Swiech, L.; Heidenreich, M.; Banerjee, A.; Habib, N.; Li, Y.; Trombetta, J.; Sur, M.; Zhang, F. In vivo interrogation of gene function in the mammalian brain using CRISPR-Cas9. Nat. Biotechnol. 2015, 33, 102–106. [Google Scholar] [CrossRef]
- Agudelo, D.; Carter, S.; Velimirovic, M.; Duringer, A.; Rivest, J.F.; Levesque, S.; Loehr, J.; Mouchiroud, M.; Cyr, D.; Waters, P.J.; et al. Versatile and robust genome editing with Streptococcus thermophilus CRISPR1-Cas9. Genome Res. 2020, 30, 107–117. [Google Scholar] [CrossRef]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Park, J.E.; Paa, P.; Rajakumar, P.D.; Prekop, H.T.; Chew, Y.T.; Manivannan, S.N.; Chew, W.L. Programmable C:G to G:C genome editing with CRISPR-Cas9-directed base excision repair proteins. Nat. Commun. 2021, 12, 1384. [Google Scholar] [CrossRef] [PubMed]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Homma, A.; Sayadi, J.; Yang, S.; Ohashi, J.; Takumi, T. Sequence features associated with the cleavage efficiency of CRISPR/Cas9 system. Sci. Rep. 2016, 6, 19675. [Google Scholar] [CrossRef] [PubMed]
- Rahdar, M.; McMahon, M.A.; Prakash, T.P.; Swayze, E.E.; Bennett, C.F.; Cleveland, D.W. Synthetic CRISPR RNA-Cas9-guided genome editing in human cells. Proc. Natl. Acad. Sci. USA 2015, 112, E7110–E7117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef]
- Verkuijl, S.A.; Rots, M.G. The influence of eukaryotic chromatin state on CRISPR-Cas9 editing efficiencies. Curr. Opin. Biotechnol. 2019, 55, 68–73. [Google Scholar] [CrossRef] [Green Version]
- Daer, R.M.; Cutts, J.P.; Brafman, D.A.; Haynes, K.A. The Impact of Chromatin Dynamics on Cas9-Mediated Genome Editing in Human Cells. ACS Synth. Biol. 2017, 6, 428–438. [Google Scholar] [CrossRef] [Green Version]
- Smits, A.H.; Ziebell, F.; Joberty, G.; Zinn, N.; Mueller, W.F.; Clauder-Munster, S.; Eberhard, D.; Falth Savitski, M.; Grandi, P.; Jakob, P.; et al. Biological plasticity rescues target activity in CRISPR knock outs. Nat. Methods 2019, 16, 1087–1093. [Google Scholar] [CrossRef]
- Conti, A.; Di Micco, R. p53 activation: A checkpoint for precision genome editing? Genome Med. 2018, 10, 66. [Google Scholar] [CrossRef]
- Sinha, S.; Barbosa, K.; Cheng, K.; Leiserson, M.D.M.; Jain, P.; Deshpande, A.; Wilson, D.M., 3rd; Ryan, B.M.; Luo, J.; Ronai, Z.A.; et al. A systematic genome-wide mapping of oncogenic mutation selection during CRISPR-Cas9 genome editing. Nat. Commun. 2021, 12, 6512. [Google Scholar] [CrossRef] [PubMed]
- Mirgayazova, R.; Khadiullina, R.; Chasov, V.; Mingaleeva, R.; Miftakhova, R.; Rizvanov, A.; Bulatov, E. Therapeutic Editing of the TP53 Gene: Is CRISPR/Cas9 an Option? Genes 2020, 11, 704. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Fortunati, E.; Liu, D.X.; Li, Y. Pleiotropic Roles of ABC Transporters in Breast Cancer. Int. J. Mol. Sci. 2021, 22, 3199. [Google Scholar] [CrossRef] [PubMed]
- Papathanasiou, S.; Markoulaki, S.; Blaine, L.J.; Leibowitz, M.L.; Zhang, C.Z.; Jaenisch, R.; Pellman, D. Whole chromosome loss and genomic instability in mouse embryos after CRISPR-Cas9 genome editing. Nat. Commun. 2021, 12, 5855. [Google Scholar] [CrossRef]
- Doudna, J.A.; Charpentier, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef]
- Moreira, D.; Pereira, A.M.; Lopes, A.L.; Coimbra, S. The best CRISPR/Cas9 versus RNA interference approaches for Arabinogalactan proteins' study. Mol. Biol. Rep. 2020, 47, 2315–2325. [Google Scholar] [CrossRef] [Green Version]
- Boettcher, M.; McManus, M.T. Choosing the Right Tool for the Job: RNAi, TALEN, or CRISPR. Mol. Cell 2015, 58, 575–585. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, U.I.; Gramatte, T.; Krappweis, J.; Oertel, R.; Kirch, W. P-glycoprotein inhibitor erythromycin increases oral bioavailability of talinolol in humans. Int. J. Clin. Pharmacol. Ther. 2000, 38, 161–167. [Google Scholar] [CrossRef]
- Meerum Terwogt, J.M.; Beijnen, J.H.; ten Bokkel Huinink, W.W.; Rosing, H.; Schellens, J.H. Co-administration of cyclosporin enables oral therapy with paclitaxel. Lancet 1998, 352, 285. [Google Scholar] [CrossRef]
- Malingre, M.M.; Richel, D.J.; Beijnen, J.H.; Rosing, H.; Koopman, F.J.; Ten Bokkel Huinink, W.W.; Schot, M.E.; Schellens, J.H. Coadministration of cyclosporine strongly enhances the oral bioavailability of docetaxel. J. Clin. Oncol. 2001, 19, 1160–1166. [Google Scholar] [CrossRef]
- Simoff, I.; Karlgren, M.; Backlund, M.; Lindstrom, A.C.; Gaugaz, F.Z.; Matsson, P.; Artursson, P. Complete Knockout of Endogenous Mdr1 (Abcb1) in MDCK Cells by CRISPR-Cas9. J. Pharm. Sci. 2016, 105, 1017–1021. [Google Scholar] [CrossRef] [Green Version]
- Karlgren, M.; Simoff, I.; Backlund, M.; Wegler, C.; Keiser, M.; Handin, N.; Muller, J.; Lundquist, P.; Jareborg, A.C.; Oswald, S.; et al. A CRISPR-Cas9 Generated MDCK Cell Line Expressing Human MDR1 Without Endogenous Canine MDR1 (cABCB1): An Improved Tool for Drug Efflux Studies. J. Pharm. Sci. 2017, 106, 2909–2913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, E.C.; Broccatelli, F.; Plise, E.; Chen, B.; Liu, L.; Cheong, J.; Zhang, S.; Jorski, J.; Gaffney, K.; Umemoto, K.K.; et al. Evaluating the Utility of Canine Mdr1 Knockout Madin-Darby Canine Kidney I Cells in Permeability Screening and Efflux Substrate Determination. Mol. Pharm. 2018, 15, 5103–5113. [Google Scholar] [CrossRef]
- Wegler, C.; Gazit, M.; Issa, K.; Subramaniam, S.; Artursson, P.; Karlgren, M. Expanding the Efflux In Vitro Assay Toolbox: A CRISPR-Cas9 Edited MDCK Cell Line with Human BCRP and Completely Lacking Canine MDR1. J. Pharm. Sci. 2021, 110, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Sparreboom, A.; van Asperen, J.; Mayer, U.; Schinkel, A.H.; Smit, J.W.; Meijer, D.K.; Borst, P.; Nooijen, W.J.; Beijnen, J.H.; van Tellingen, O. Limited oral bioavailability and active epithelial excretion of paclitaxel (Taxol) caused by P-glycoprotein in the intestine. Proc. Natl. Acad. Sci. USA 1997, 94, 2031–2035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, U.; Wagenaar, E.; Beijnen, J.H.; Smit, J.W.; Meijer, D.K.; van Asperen, J.; Borst, P.; Schinkel, A.H. Substantial excretion of digoxin via the intestinal mucosa and prevention of long-term digoxin accumulation in the brain by the mdr 1a P-glycoprotein. Br. J. Pharmacol. 1996, 119, 1038–1044. [Google Scholar] [CrossRef] [PubMed]
- Miyake, T.; Tsutsui, H.; Haraya, K.; Tachibana, T.; Morimoto, K.; Takehara, S.; Ayabe, M.; Kobayashi, K.; Kazuki, Y. Quantitative prediction of P-glycoprotein-mediated drug-drug interactions and intestinal absorption using humanized mice. Br. J. Pharmacol. 2021, 178, 4335–4351. [Google Scholar] [CrossRef]
- Liang, C.; Zhao, J.; Lu, J.; Zhang, Y.; Ma, X.; Shang, X.; Li, Y.; Ma, X.; Liu, M.; Wang, X. Development and Characterization of MDR1 (Mdr1a/b) CRISPR/Cas9 Knockout Rat Model. Drug Metab. Dispos. 2019, 47, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Norouzi-Barough, L.; Sarookhani, M.; Salehi, R.; Sharifi, M.; Moghbelinejad, S. CRISPR/Cas9, a new approach to successful knockdown of ABCB1/P-glycoprotein and reversal of chemosensitivity in human epithelial ovarian cancer cell line. Iran. J. Basic. Med. Sci. 2018, 21, 181–187. [Google Scholar] [CrossRef]
- Ha, J.S.; Byun, J.; Ahn, D.R. Overcoming doxorubicin resistance of cancer cells by Cas9-mediated gene disruption. Sci. Rep. 2016, 6, 22847. [Google Scholar] [CrossRef]
- Liu, T.; Li, Z.; Zhang, Q.; De Amorim Bernstein, K.; Lozano-Calderon, S.; Choy, E.; Hornicek, F.J.; Duan, Z. Targeting ABCB1 (MDR1) in multi-drug resistant osteosarcoma cells using the CRISPR-Cas9 system to reverse drug resistance. Oncotarget 2016, 7, 83502–83513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Qiu, J.G.; Li, Y.; Di, J.M.; Zhang, W.J.; Jiang, Q.W.; Zheng, D.W.; Chen, Y.; Wei, M.N.; Huang, J.R.; et al. Targeting ABCB1-mediated tumor multidrug resistance by CRISPR/Cas9-based genome editing. Am. J. Transl. Res. 2016, 8, 3986–3994. [Google Scholar]
- Takahashi, K.; Inukai, T.; Imamura, T.; Yano, M.; Tomoyasu, C.; Lucas, D.M.; Nemoto, A.; Sato, H.; Huang, M.; Abe, M.; et al. Anti-leukemic activity of bortezomib and carfilzomib on B-cell precursor ALL cell lines. PLoS ONE 2017, 12, e0188680. [Google Scholar] [CrossRef] [PubMed]
- Barghout, S.H.; Aman, A.; Nouri, K.; Blatman, Z.; Arevalo, K.; Thomas, G.E.; MacLean, N.; Hurren, R.; Ketela, T.; Saini, M.; et al. A genome-wide CRISPR/Cas9 screen in acute myeloid leukemia cells identifies regulators of TAK-243 sensitivity. JCI Insight 2021, 6, e141518. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Lin, Y.; Song, Z.; Xiao, W.; Chen, L.; Yin, J.; Zhou, Y.; Barta, S.K.; Petrus, M.; Waldmann, T.A.; et al. A20 and RBX1 Regulate Brentuximab Vedotin Sensitivity in Hodgkin Lymphoma Models. Clin. Cancer. Res. 2020, 26, 4093–4106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.S.L.; Yusa, K. Genome-wide CRISPR-Cas9 screening in mammalian cells. Methods 2019, 164, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelson, T.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef] [Green Version]
- Doench, J.G.; Fusi, N.; Sullender, M.; Hegde, M.; Vaimberg, E.W.; Donovan, K.F.; Smith, I.; Tothova, Z.; Wilen, C.; Orchard, R.; et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016, 34, 184–191. [Google Scholar] [CrossRef] [Green Version]
- Sheng, Q.; D'Alessio, J.A.; Menezes, D.L.; Karim, C.; Tang, Y.; Tam, A.; Clark, S.; Ying, C.; Connor, A.; Mansfield, K.G.; et al. PCA062, a P-cadherin Targeting Antibody-Drug Conjugate, Displays Potent Antitumor Activity Against P-cadherin-expressing Malignancies. Mol. Cancer Ther. 2021, 20, 1270–1282. [Google Scholar] [CrossRef]
- Loganzo, F.; Tan, X.; Sung, M.; Jin, G.; Myers, J.S.; Melamud, E.; Wang, F.; Diesl, V.; Follettie, M.T.; Musto, S.; et al. Tumor cells chronically treated with a trastuzumab-maytansinoid antibody-drug conjugate develop varied resistance mechanisms but respond to alternate treatments. Mol. Cancer Ther. 2015, 14, 952–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamblett, K.J.; Jacob, A.P.; Gurgel, J.L.; Tometsko, M.E.; Rock, B.M.; Patel, S.K.; Milburn, R.R.; Siu, S.; Ragan, S.P.; Rock, D.A.; et al. SLC46A3 Is Required to Transport Catabolites of Noncleavable Antibody Maytansine Conjugates from the Lysosome to the Cytoplasm. Cancer Res. 2015, 75, 5329–5340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinneer, K.; Meekin, J.; Tiberghien, A.C.; Tai, Y.T.; Phipps, S.; Kiefer, C.M.; Rebelatto, M.C.; Dimasi, N.; Moriarty, A.; Papadopoulos, K.P.; et al. SLC46A3 as a Potential Predictive Biomarker for Antibody-Drug Conjugates Bearing Noncleavable Linked Maytansinoid and Pyrrolobenzodiazepine Warheads. Clin. Cancer Res. 2018, 24, 6570–6582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Nagel, R.; Zaal, E.A.; Ugalde, A.P.; Han, R.; Proost, N.; Song, J.Y.; Pataskar, A.; Burylo, A.; Fu, H.; et al. SLC1A3 contributes to L-asparaginase resistance in solid tumors. EMBO J. 2019, 38, e102147. [Google Scholar] [CrossRef] [PubMed]
- Abada, P.; Howell, S.B. Regulation of Cisplatin cytotoxicity by cu influx transporters. Met. Based Drugs 2010, 2010, 317581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bompiani, K.M.; Tsai, C.Y.; Achatz, F.P.; Liebig, J.K.; Howell, S.B. Copper transporters and chaperones CTR1, CTR2, ATOX1, and CCS as determinants of cisplatin sensitivity. Metallomics 2016, 8, 951–962. [Google Scholar] [CrossRef] [Green Version]
- Planells-Cases, R.; Lutter, D.; Guyader, C.; Gerhards, N.M.; Ullrich, F.; Elger, D.A.; Kucukosmanoglu, A.; Xu, G.; Voss, F.K.; Reincke, S.M.; et al. Subunit composition of VRAC channels determines substrate specificity and cellular resistance to Pt-based anti-cancer drugs. EMBO J. 2015, 34, 2993–3008. [Google Scholar] [CrossRef]
- He, Y.J.; Meghani, K.; Caron, M.C.; Yang, C.; Ronato, D.A.; Bian, J.; Sharma, A.; Moore, J.; Niraj, J.; Detappe, A.; et al. DYNLL1 binds to MRE11 to limit DNA end resection in BRCA1-deficient cells. Nature 2018, 563, 522–526. [Google Scholar] [CrossRef]
- Al-Majdoub, Z.M.; Achour, B.; Couto, N.; Howard, M.; Elmorsi, Y.; Scotcher, D.; Alrubia, S.; El-Khateeb, E.; Vasilogianni, A.M.; Alohali, N.; et al. Mass spectrometry-based abundance atlas of ABC transporters in human liver, gut, kidney, brain and skin. FEBS Lett. 2020, 594, 4134–4150. [Google Scholar] [CrossRef]
- Henderson, J.T.; Piquette-Miller, M. Blood-brain barrier: An impediment to neuropharmaceuticals. Clin. Pharmacol. Ther. 2015, 97, 308–313. [Google Scholar] [CrossRef]
- Dong, X. Current Strategies for Brain Drug Delivery. Theranostics 2018, 8, 1481–1493. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, T.; Mizuno, T.; Kurosawa, T.; Yamaguchi, T.; Higuchi, K.; Tega, Y.; Nozaki, Y.; Kawabata, K.; Deguchi, Y.; Kusuhara, H. Functional Investigation of Solute Carrier Family 35, Member F2, in Three Cellular Models of the Primate Blood-Brain Barrier. Drug Metab. Dispos. 2021, 49, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Winter, G.E.; Radic, B.; Mayor-Ruiz, C.; Blomen, V.A.; Trefzer, C.; Kandasamy, R.K.; Huber, K.V.M.; Gridling, M.; Chen, D.; Klampfl, T.; et al. The solute carrier SLC35F2 enables YM155-mediated DNA damage toxicity. Nat. Chem. Biol. 2014, 10, 768–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, B.; Hagenbuch, B.; Kullak-Ublick, G.A.; Benke, D.; Aguzzi, A.; Meier, P.J. Organic anion-transporting polypeptides mediate transport of opioid peptides across blood-brain barrier. J. Pharmacol. Exp. Ther. 2000, 294, 73–79. [Google Scholar]
- Lee, W.; Glaeser, H.; Smith, L.H.; Roberts, R.L.; Moeckel, G.W.; Gervasini, G.; Leake, B.F.; Kim, R.B. Polymorphisms in human organic anion-transporting polypeptide 1A2 (OATP1A2): Implications for altered drug disposition and central nervous system drug entry. J. Biol. Chem. 2005, 280, 9610–9617. [Google Scholar] [CrossRef] [Green Version]
- Bronger, H.; Konig, J.; Kopplow, K.; Steiner, H.H.; Ahmadi, R.; Herold-Mende, C.; Keppler, D.; Nies, A.T. ABCC drug efflux pumps and organic anion uptake transporters in human gliomas and the blood-tumor barrier. Cancer Res. 2005, 65, 11419–11428. [Google Scholar] [CrossRef] [Green Version]
- Sano, Y.; Mizuno, T.; Mochizuki, T.; Uchida, Y.; Umetsu, M.; Terasaki, T.; Kusuhara, H. Evaluation of Organic Anion Transporter 1A2-knock-in Mice as a Model of Human Blood-brain Barrier. Drug Metab. Dispos. 2018, 46, 1767–1775. [Google Scholar] [CrossRef]
- Saxonov, S.; Berg, P.; Brutlag, D.L. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc. Natl. Acad. Sci. USA 2006, 103, 1412–1417. [Google Scholar] [CrossRef] [Green Version]
- Takechi, T.; Hirota, T.; Sakai, T.; Maeda, N.; Kobayashi, D.; Ieiri, I. Interindividual Differences in the Expression of ATP-Binding Cassette and Solute Carrier Family Transporters in Human Skin: DNA Methylation Regulates Transcriptional Activity of the Human ABCC3 Gene. Drug Metab. Dispos. 2018, 46, 628–635. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Class 2 | Subtype | Effector Nuclease | Size (aa) | Target | TracrRNA Requirement | Seed Sequence Requirement | PAM Sequence | Cleavage Product |
|---|---|---|---|---|---|---|---|---|
| Type II | A | SpyCas9 | 1368 | dsDNA (or ssDNA/ssRNA with PAMmers) | Yes | Yes | NGG | DSB (blunt end)/SSB |
| A | St1Cas9 | 1121 | dsDNA | Yes | Yes | NNRGAA | DSB (blunt end) | |
| A | St3Cas9 | 1388 | dsDNA | Yes | Yes | NGGNG | DSB (blunt end) | |
| A | SauCas9 | 1053 | dsDNA/ssRNA | Yes | Yes | NNAGAAW/- | DSB (blunt end)/SSB | |
| B | FnoCas9 | 1629 | dsDNA/ssRNA | Yes | Yes | NGG/- | DSB (blunt end)/SSB | |
| C | CjeCas9 | 984 | dsDNA/ssRNA | Yes | Yes | NNNVRYM/- | DSB (blunt end)/SSB | |
| C | NmeCas9 | 1082 | dsDNA/ssDNA | Yes/No | Yes | NNNNGATT/- | DSB (blunt end)/SSB | |
| Type V | A | Cas12a | 1200–1500 | dsDNA/ssDNA | No | Yes | Optimal 5′ T-rich and suboptimal C-containing PAMs/- | DSB (sticky end with 5-nt 5′-overhang)/SSB |
| B | Cas12b | 1100–1300 | dsDNA/ssDNA | Yes | Yes | Optimal 5′ T-rich and suboptimal C-containing PAMs/- | DSB (sticky end with 6-nt 5′-overhang)/SSB | |
| E | Cas12e | <1000 | dsDNA | Yes | Unknown | 5′ T-rich PAMs | DSB (sticky end with 10-nt 5′-overhang) | |
| F | Cas12f | 400–600 | dsDNA/ssDNA | Yes | Unknown | 5′ T-rich PAMs/- | DSB (sticky end with 5’-overhang) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, J.; Biswas, R.; Bugde, P.; Li, J.; Liu, D.-X.; Li, Y. Application of CRISPR-Cas9 System to Study Biological Barriers to Drug Delivery. Pharmaceutics 2022, 14, 894. https://doi.org/10.3390/pharmaceutics14050894
He J, Biswas R, Bugde P, Li J, Liu D-X, Li Y. Application of CRISPR-Cas9 System to Study Biological Barriers to Drug Delivery. Pharmaceutics. 2022; 14(5):894. https://doi.org/10.3390/pharmaceutics14050894
Chicago/Turabian StyleHe, Ji, Riya Biswas, Piyush Bugde, Jiawei Li, Dong-Xu Liu, and Yan Li. 2022. "Application of CRISPR-Cas9 System to Study Biological Barriers to Drug Delivery" Pharmaceutics 14, no. 5: 894. https://doi.org/10.3390/pharmaceutics14050894
APA StyleHe, J., Biswas, R., Bugde, P., Li, J., Liu, D.-X., & Li, Y. (2022). Application of CRISPR-Cas9 System to Study Biological Barriers to Drug Delivery. Pharmaceutics, 14(5), 894. https://doi.org/10.3390/pharmaceutics14050894