
Redesigning of Cell-Penetrating Peptides to Improve Their Efficacy as a Drug Delivery System

 ,
, 
Abstract
:1. Introduction
2. Different Methods to Study the Internalization of CPPs
3. Mechanism of Internalization
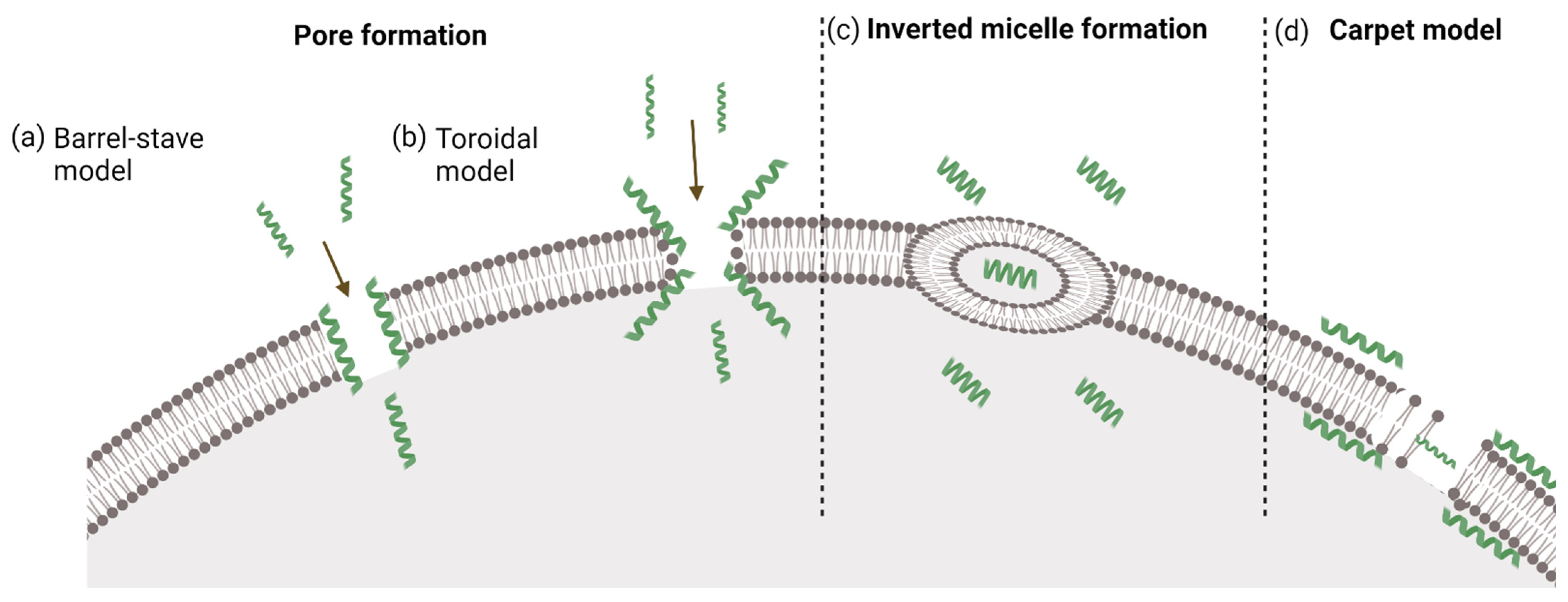
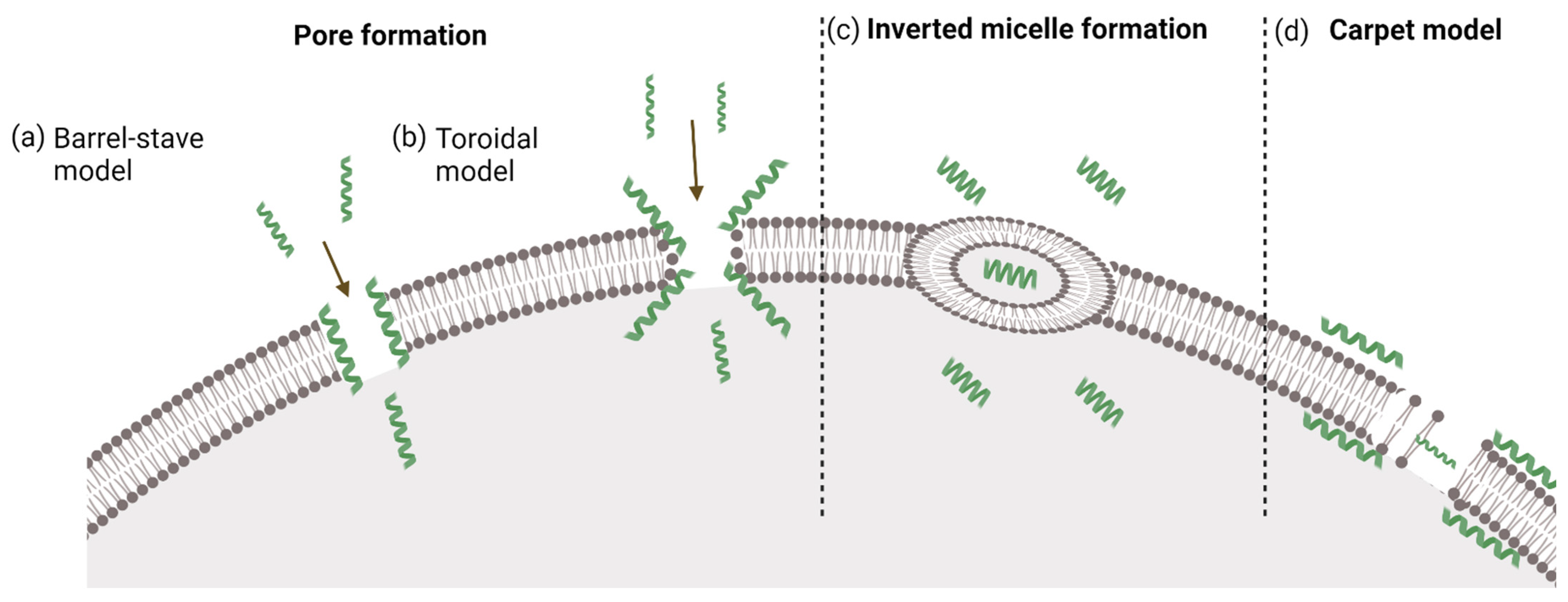
3.1. Direct Penetration
3.1.1. Inverted Micelle Formation
3.1.2. Pore Formation
3.1.3. The Carpet Model
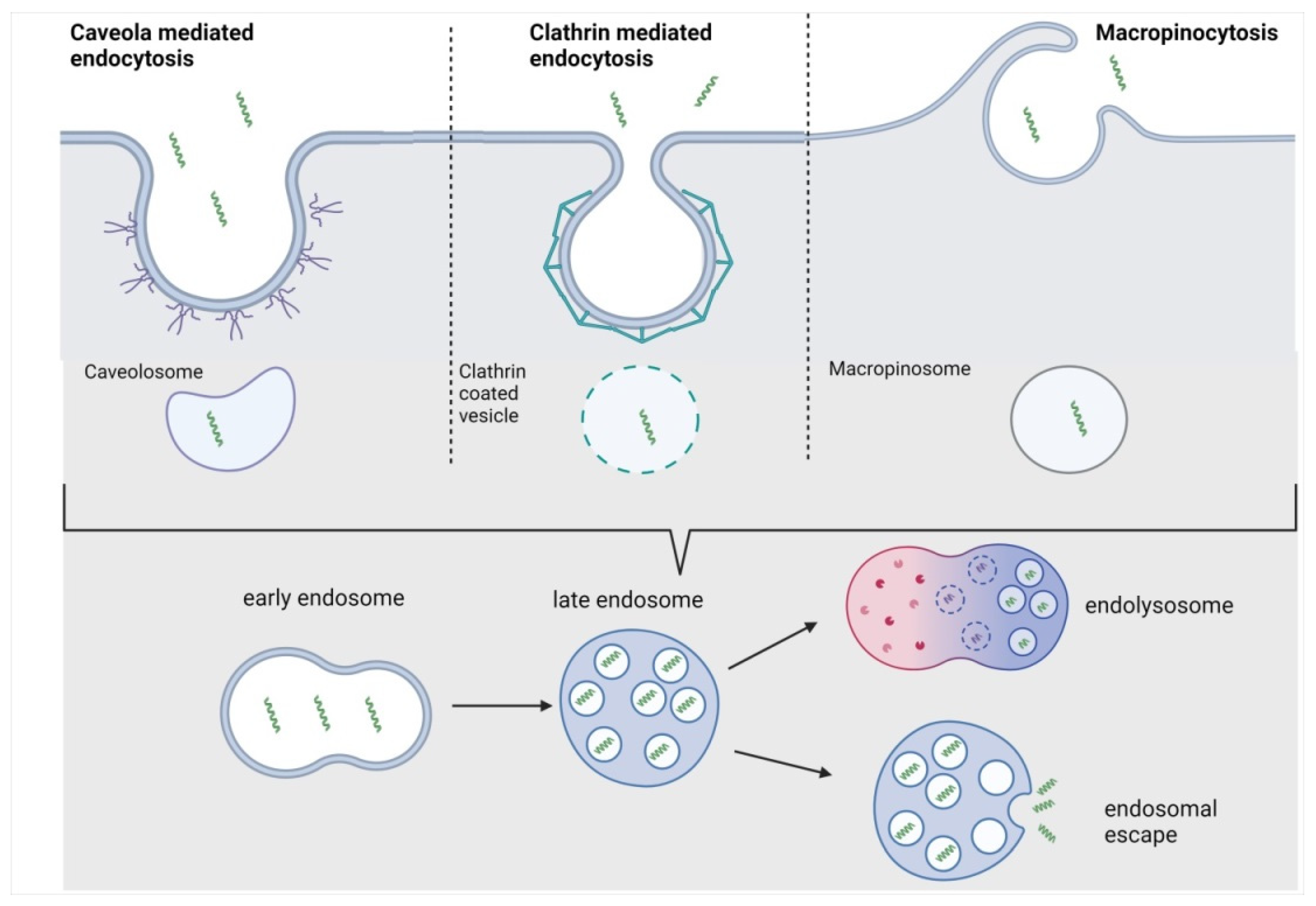
3.2. Endocytosis
3.2.1. Macropinocytosis
3.2.2. Clathrin-Mediated Endocytosis (CME)
3.2.3. Caveolae-Mediated Endocytosis (CvME)
4. Modification of CPPs
4.1. Improving the Stability of CPPs
4.1.1. Replacement of L-Amino Acids by Their D-Variant or Other Unnatural Amino Acids
4.1.2. PEGylation
4.2. Improving the Stability and/or the Internalization of CPP
4.2.1. Conformational Constraints
4.2.2. Stapled Peptides
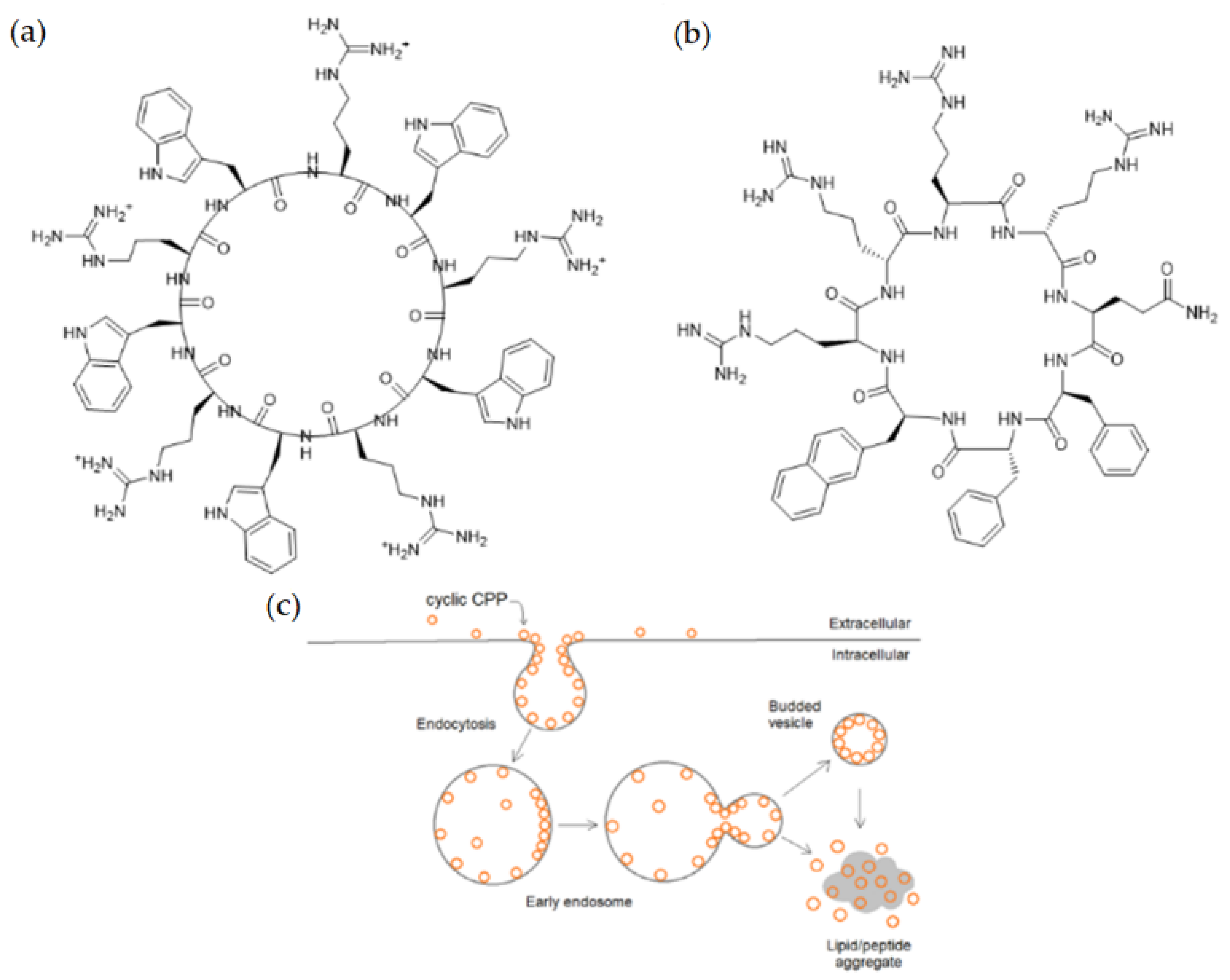
4.2.3. Cyclization
4.2.4. N-Alkylation
4.2.5. Increasing the Hydrophobicity by Aromatic Ring(s)
4.2.6. Modifications to the Peptide Backbone or Side Chain to Enhance Cellular Uptake
4.3. Promoting Endosomal Escape
4.3.1. Exploitation of the Proton Sponge Effect
4.3.2. N-Terminal Stearylation
4.3.3. Application or Conjugation of Endosomolytic Compounds
4.3.4. Conjugation of Endosomolytic Peptides or Endosomal Escape Domains (EEDs)
4.4. Facilitating Direct Translocation
4.5. Enhancing the Selectivity of CPPs
4.5.1. Cell-Penetrating Homing Peptides (CPHP)
4.5.2. Modification of CPPs to Enhance Selectivity
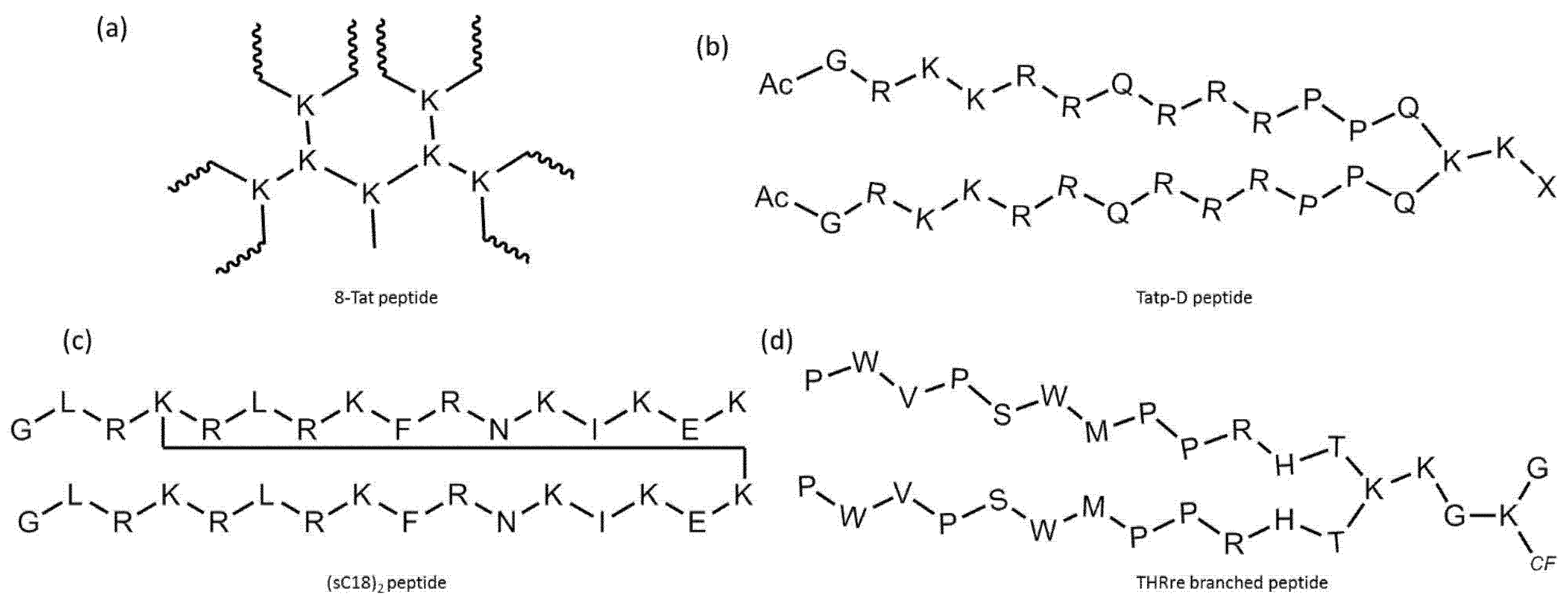
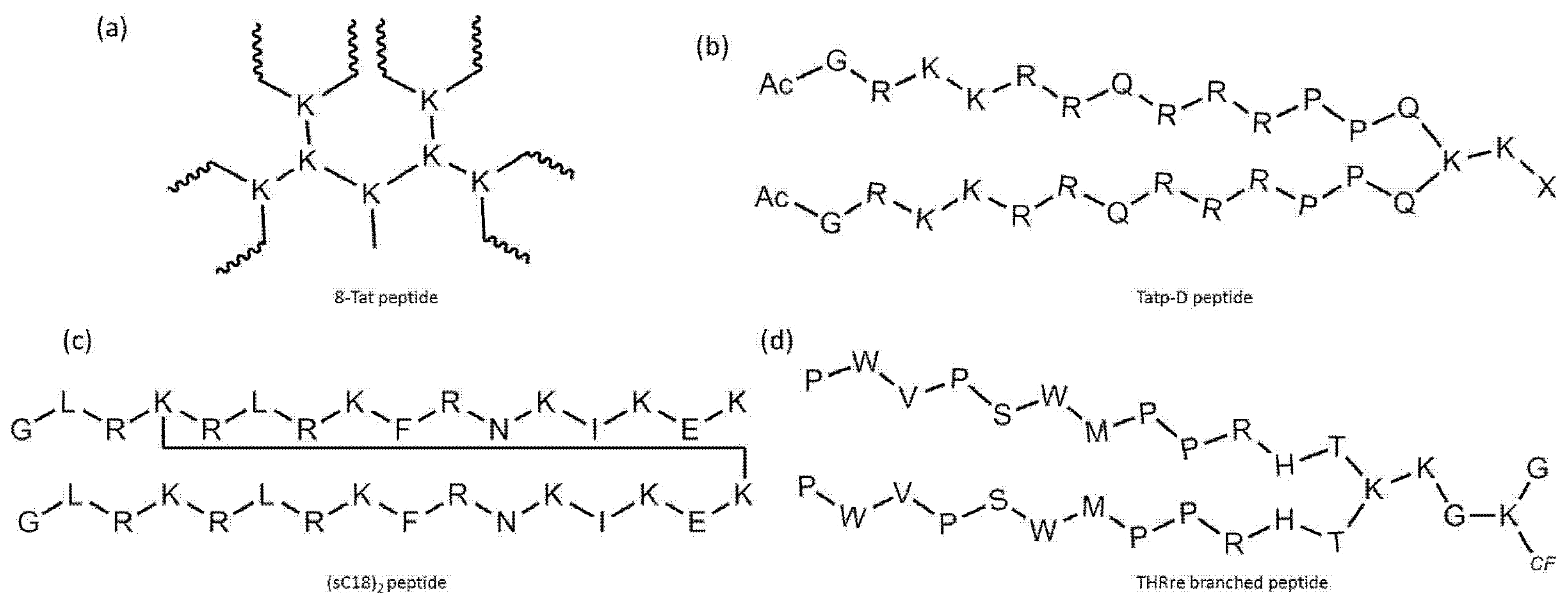
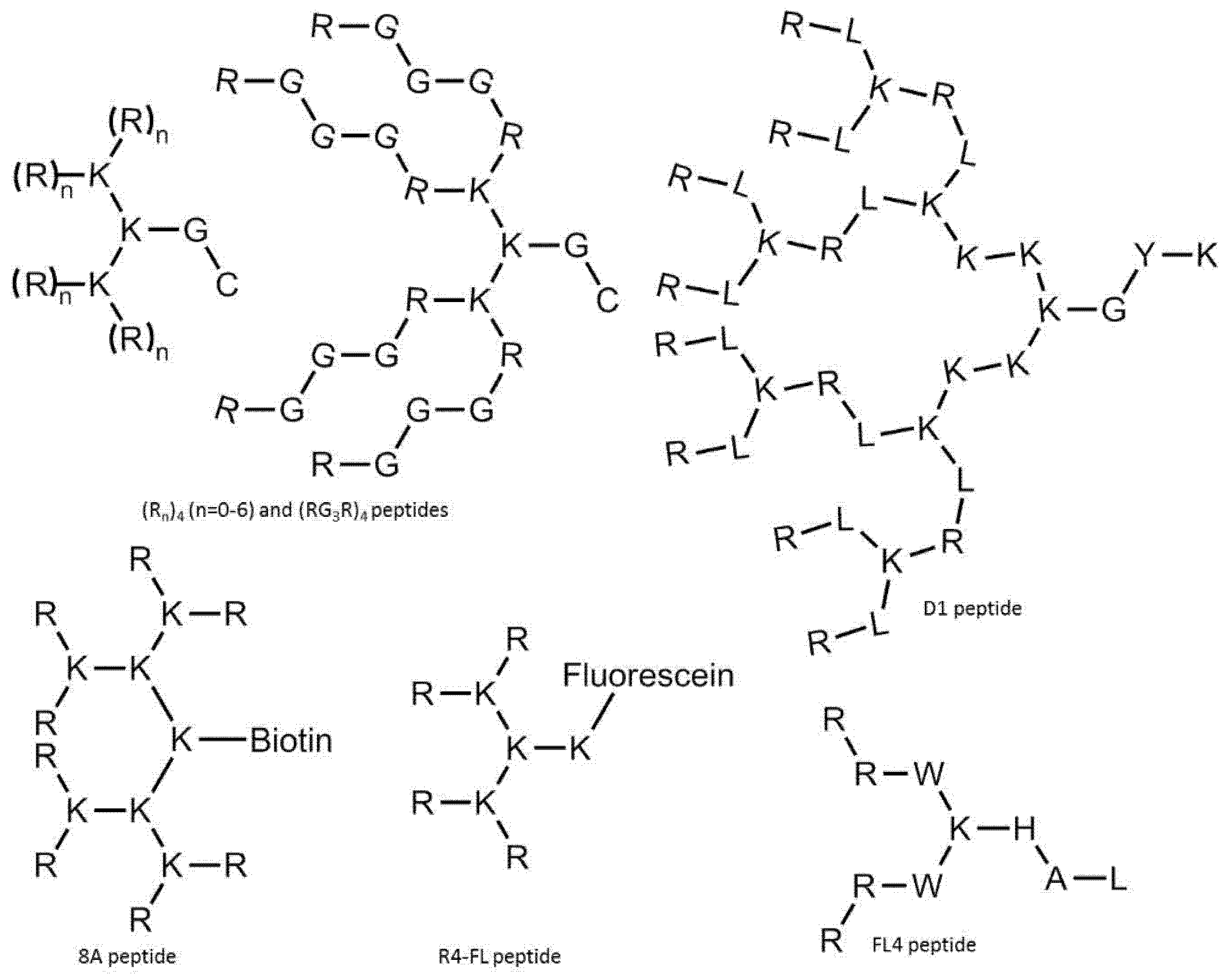
5. Cell-Penetrating Peptide Derivatives with Branching Structure
5.1. Branched Peptides Built from Linear CPPs
5.2. Branched Construct That Behave as a Cell-Penetrating Peptide
6. Clinical Applications
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Green, M.; Loewenstein, P.M. Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans-activator protein. Cell 1988, 55, 1179–1188. [Google Scholar] [CrossRef]
- Vivès, E.; Brodin, P.; Lebleu, B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J. Biol. Chem. 1997, 272, 16010–16017. [Google Scholar] [CrossRef] [Green Version]
- Derossi, D.; Joliot, A.H.; Chassaing, G.; Prochiantz, A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J. Biol. Chem. 1994, 269, 10444–10450. [Google Scholar] [CrossRef]
- Hudecz, F.; Bánóczi, Z.; Csík, G. Medium-sized peptides as built in carriers for biologically active compounds. Med. Res. Rev. 2005, 25, 679–736. [Google Scholar] [CrossRef]
- Futaki, S.; Arafiles, J.V.V.; Hirose, H. Peptide-assisted intracellular delivery of biomacromolecules. Chem. Lett. 2020, 49, 1088–1094. [Google Scholar] [CrossRef]
- Bánoczi, Z.; Tantos, Á.; Farkas, A.; Tompa, P.; Friedrich, P.; Hudecz, F. Synthesis of cell-penetrating conjugates of calpain activator peptides. Bioconjug. Chem. 2007, 18, 130–137. [Google Scholar] [CrossRef]
- Szabó, I.; Orbán, E.; Schlosser, G.; Hudecz, F.; Bánóczi, Z. Cell-penetrating conjugates of pentaglutamylated methotrexate as potential anticancer drugs against resistant tumor cells. Eur. J. Med. Chem. 2016, 115, 361–368. [Google Scholar] [CrossRef] [Green Version]
- Bánóczi, Z.; Alexa, A.; Farkas, A.; Friedrich, P.; Hudecz, F. Novel cell-penetrating calpain substrate. Bioconjug. Chem. 2008, 19, 1375–1381. [Google Scholar] [CrossRef]
- Miklán, Z.; Orbán, E.; Bánóczi, Z.; Hudecz, F. New pemetrexed-peptide conjugates: Synthesis, characterization and in vitro cytostatic effect on non-small cell lung carcinoma (NCI-H358) and human leukemia (HL-60) cells. J. Pept. Sci. 2011, 17, 805–811. [Google Scholar] [CrossRef]
- Di Pisa, M.; Chassaing, G.; Swiecicki, J.M. Translocation mechanism(s) of cell-penetrating peptides: Biophysical studies using artificial membrane bilayers. Biochemistry 2015, 54, 194–207. [Google Scholar] [CrossRef]
- Lee, C.C.; Sun, Y.; Huang, H.W. Membrane-mediated peptide conformation change from α-monomers to β-aggregates. Biophys. J. 2010, 98, 2236–2245. [Google Scholar] [CrossRef] [Green Version]
- Stalmans, S.; Bracke, N.; Wynendaele, E.; Gevaert, B.; Peremans, K.; Burvenich, C.; Polis, I.; De Spiegeleer, B. Cell-penetrating peptides selectively cross the blood-brain barrier in vivo. PLoS ONE 2015, 10, e0139652. [Google Scholar] [CrossRef] [Green Version]
- Ruczyński, J.; Rusiecka, I.; Turecka, K.; Kozłowska, A.; Alenowicz, M.; Gągało, I.; Kawiak, A.; Rekowski, P.; Waleron, K.; Kocić, I. Transportan 10 improves the pharmacokinetics and pharmacodynamics of vancomycin. Sci. Rep. 2019, 9, 3247. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.Y.; Cao, X.W.; Fu, L.Y.; Zhang, T.Z.; Wang, F.J.; Zhao, J. Screening and characterization of a novel high-efficiency tumor-homing cell-penetrating peptide from the buffalo cathelicidin family. J. Pept. Sci. 2019, 25, e3201. [Google Scholar] [CrossRef]
- Maity, S.K.; Stahl, P.; Hensel, A.; Knauer, S.; Hirschhäuser, C.; Schmuck, C. Cancer-Cell-Specific Drug Delivery by a Tumor-Homing CPP-Gossypol Conjugate Employing a Tracelessly Cleavable Linker. Chem.—A Eur. J. 2020, 26, 3010–3015. [Google Scholar] [CrossRef]
- Splith, K.; Neundorf, I. Antimicrobial peptides with cell-penetrating peptide properties and vice versa. Eur. Biophys. J. 2011, 40, 387–397. [Google Scholar] [CrossRef]
- Ponnappan, N.; Budagavi, D.P.; Chugh, A. CyLoP-1: Membrane-active peptide with cell-penetrating and antimicrobial properties. Biochim. Biophys. Acta—Biomembr. 2017, 1859, 167–176. [Google Scholar] [CrossRef]
- Lundberg, M.; Johansson, M. Is VP22 nuclear homing an artifact? Nat. Biotechnol. 2001, 19, 713–714. [Google Scholar] [CrossRef]
- Lundberg, M.; Johansson, M. Positively charged DNA-binding proteins cause apparent cell membrane translocation. Biochem. Biophys. Res. Commun. 2002, 291, 367–371. [Google Scholar] [CrossRef]
- Richard, J.P.; Melikov, K.; Vives, E.; Ramos, C.; Verbeure, B.; Gait, M.J.; Chernomordik, L.V.; Lebleu, B. Cell-penetrating peptides: A reevaluation of the mechanism of cellular uptake. J. Biol. Chem. 2003, 278, 585–590. [Google Scholar] [CrossRef] [Green Version]
- Lundberg, M.; Wikström, S.; Johansson, M. Cell surface adherence and endocytosis of protein transduction domains. Mol. Ther. 2003, 8, 143–150. [Google Scholar] [CrossRef]
- Baranyai, Z.; Biri-Kovács, B.; Krátký, M.; Szeder, B.; Debreczeni, M.L.; Budai, J.; Kovács, B.; Horváth, L.; Pári, E.; Németh, Z.; et al. Cellular Internalization and Inhibition Capacity of New Anti-Glioma Peptide Conjugates: Physicochemical Characterization and Evaluation on Various Monolayer- and 3D-Spheroid-Based in Vitro Platforms. J. Med. Chem. 2021, 64, 2982–3005. [Google Scholar] [CrossRef]
- Moghal, M.M.R.; Islam, M.Z.; Hossain, F.; Saha, S.K.; Yamazaki, M. Role of Membrane Potential on Entry of Cell-Penetrating Peptide Transportan 10 into Single Vesicles. Biophys. J. 2020, 118, 57–69. [Google Scholar] [CrossRef]
- Masuda, T.; Hirose, H.; Baba, K.; Walrant, A.; Sagan, S.; Inagaki, N.; Fujimoto, T.; Futaki, S. An Artificial Amphiphilic Peptide Promotes Endocytic Uptake by Inducing Membrane Curvature. Bioconjug. Chem. 2020, 31, 1611–1615. [Google Scholar] [CrossRef]
- Takeuchi, T.; Futaki, S. Current understanding of direct translocation of arginine-rich cell-penetrating peptides and its internalization mechanisms. Chem. Pharm. Bull. 2016, 64, 1431–1437. [Google Scholar] [CrossRef] [Green Version]
- Nakase, I.; Niwa, M.; Takeuchi, T.; Sonomura, K.; Kawabata, N.; Koike, Y.; Takehashi, M.; Tanaka, S.; Ueda, K.; Simpson, J.C.; et al. Cellular uptake of arginine-rich peptides: Roles for macropinocytosis and actin rearrangement. Mol. Ther. 2004, 10, 1011–1022. [Google Scholar] [CrossRef]
- Kosuge, M.; Takeuchi, T.; Nakase, I.; Jones, A.T.; Futaki, S. Cellular internalization and distribution of arginine-rich peptides as a function of extracellular peptide concentration, serum, and plasma membrane associated proteoglycans. Bioconjug. Chem. 2008, 19, 656–664. [Google Scholar] [CrossRef]
- Al-Taei, S.; Penning, N.A.; Simpson, J.C.; Futaki, S.; Takeuchi, T.; Nakase, I.; Jones, A.T. Intracellular Traffic and Fate of Protein Transduction Domains HIV-1 TAT Peptide and Octaarginine. Implications for Their Utilization as Drug Delivery Vectors. Bioconjug. Chem. 2006, 17, 90–100. [Google Scholar] [CrossRef]
- Duchardt, F.; Fotin-Mleczek, M.; Schwarz, H.; Fischer, R.; Brock, R. A comprehensive model for the cellular uptake of cationic cell-penetrating peptides. Traffic 2007, 8, 848–866. [Google Scholar] [CrossRef]
- Fretz, M.M.; Penning, N.A.; Al-Taei, S.; Futaki, S.; Takeuchi, T.; Nakase, I.; Storm, G.; Jones, A.T. Temperature-, concentration- and cholesterol-dependent translocation of L- and D-octa-arginine across the plasma and nuclear membrane of CD34+ leukaemia cells. Biochem. J. 2007, 403, 335–342. [Google Scholar] [CrossRef] [Green Version]
- Jiao, C.Y.; Delaroche, D.; Burlina, F.; Alves, I.D.; Chassaing, G.; Sagan, S. Translocation and endocytosis for cell-penetrating peptide internalization. J. Biol. Chem. 2009, 284, 33957–33965. [Google Scholar] [CrossRef] [Green Version]
- Illien, F.; Rodriguez, N.; Amoura, M.; Joliot, A.; Pallerla, M.; Cribier, S.; Burlina, F.; Sagan, S. Quantitative fluorescence spectroscopy and flow cytometry analyses of cell-penetrating peptides internalization pathways: Optimization, pitfalls, comparison with mass spectrometry quantification. Sci. Rep. 2016, 6, 36938. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Afshar, S. In Vitro Assays: Friends or Foes of Cell-Penetrating Peptides. Int. J. Mol. Sci. 2020, 21, 4719. [Google Scholar] [CrossRef]
- Ruseska, I.; Zimmer, A. Internalization mechanisms of cell-penetrating peptides. Beilstein J. Nanotechnol. 2020, 11, 101–123. [Google Scholar] [CrossRef]
- Walrant, A.; Bechara, C.; Alves, D.I.; Sagan, S. Molecular partners for interaction and cell internalization of cell-penetrating peptides: How identical are they? Nanomedicine 2012, 7, 133–143. [Google Scholar] [CrossRef]
- Gräslund, A.; Madani, F.; Lindberg, S.; Langel, Ü.; Futaki, S. Mechanisms of cellular uptake of cell-penetrating peptides. J. Biophys. 2011, 2011, 414729. [Google Scholar]
- Allolio, C.; Magarkar, A.; Jurkiewicz, P.; Baxová, K.; Javanainen, M.; Mason, P.E.; Šachl, R.; Cebecauer, M.; Hof, M.; Horinek, D.; et al. Arginine-rich cell-penetrating peptides induce membrane multilamellarity and subsequently enter via formation of a fusion pore. Proc. Natl. Acad. Sci. USA 2018, 115, 11923–11928. [Google Scholar] [CrossRef] [Green Version]
- Szabó, I.; Illien, F.; Dókus, L.E.; Yousef, M.; Baranyai, Z.; Bősze, S.; Ise, S.; Kawano, K.; Sagan, S.; Futaki, S.; et al. Influence of the Dabcyl group on the cellular uptake of cationic peptides: Short oligoarginines as efficient cell-penetrating peptides. Amino Acids 2021, 53, 1033–1049. [Google Scholar] [CrossRef]
- Ziegler, A.; Nervi, P.; Dürrenberger, M.; Seelig, J. The cationic cell-penetrating peptide CPPTAT derived from the HIV-1 protein TAT is rapidly transported into living fibroblasts: Optical, biophysical, and metabolic evidence. Biochemistry 2005, 44, 138–148. [Google Scholar] [CrossRef]
- Watkins, C.L.; Schmaljohann, D.; Futaki, S.; Jones, A.T. Low concentration thresholds of plasma membranes for rapid energy-independent translocation of a cell-penetrating peptide. Biochem. J. 2009, 420, 179–189. [Google Scholar] [CrossRef] [Green Version]
- Ruczynski, J.; Wierzbicki, P.M.; Kogut-Wierzbicka, M.; Mucha, P.; Siedlecka-Kroplewska, K.; Rekowski, P. Cell-penetrating peptides as a promising tool for delivery of various molecules into the cells. Folia Histochem. Cytobiol. 2014, 52, 257–269. [Google Scholar] [CrossRef] [Green Version]
- Alves, I.D.; Jiao, C.Y.; Aubry, S.; Aussedat, B.; Burlina, F.; Chassaing, G.; Sagan, S. Cell biology meets biophysics to unveil the different mechanisms of penetratin internalization in cells. Biochim. Biophys. Acta—Biomembr. 2010, 1798, 2231–2239. [Google Scholar] [CrossRef]
- Deshayes, S.; Morris, M.C.; Divita, G.; Heitz, F. Interactions of amphipathic CPPs with model membranes. Biochim. Biophys. Acta—Biomembr. 2006, 1758, 328–335. [Google Scholar] [CrossRef] [Green Version]
- Deshayes, S.; Heitz, A.; Morris, M.C.; Charnet, P.; Divita, G.; Heitz, F. Insight into the Mechanism of Internalization of the Cell-Penetrating Carrier Peptide Pep-1 through Conformational Analysis. Biochemistry 2004, 43, 1449–1457. [Google Scholar] [CrossRef]
- Herce, H.D.; Garcia, A.E. Molecular dynamics simulations suggest a mechanism for translocation of the HIV-1 TAT peptide across lipid membranes. Proc. Natl. Acad. Sci. USA 2007, 104, 20805–20810. [Google Scholar] [CrossRef] [Green Version]
- Herce, H.D.; Garcia, A.E.; Litt, J.; Kane, R.S.; Martin, P.; Enrique, N.; Rebolledo, A.; Milesi, V. Arginine-rich peptides destabilize the plasma membrane, consistent with a pore formation translocation mechanism of cell-penetrating peptides. Biophys. J. 2009, 97, 1917–1925. [Google Scholar] [CrossRef] [Green Version]
- Shai, Y. Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by α-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim. Biophys. Acta—Biomembr. 1999, 1462, 55–70. [Google Scholar] [CrossRef] [Green Version]
- Pieta, P.; Mirza, J.; Lipkowski, J. Direct visualization of the alamethicin pore formed in a planar phospholipid matrix. Proc. Natl. Acad. Sci. USA 2012, 109, 21223–21227. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Harroun, T.A.; Weiss, T.M.; Ding, L.; Huang, H.W. Barrel-stave model or toroidal model? A case study on melittin pores. Biophys. J. 2001, 81, 1475–1485. [Google Scholar] [CrossRef] [Green Version]
- López-Meza, J.E.; Ochoa-Zarzosa, A.; Aguilar, J.A.; Loeza-Lara, P.D. Antimicrobial Peptides: Diversity and Perspectives for Their Biomedical Application. In Biomedical Engineering, Trends, Research and Technologies; InTech: Rijeka, Croatia, 2011. [Google Scholar]
- Pouny, Y.; Rapaport, D.; Shai, Y.; Mor, A.; Nicolas, P. Interaction of Antimicrobial Dermaseptin and its Fluorescently Labeled Analogs with Phospholipid Membranes. Biochemistry 1992, 31, 12416–12423. [Google Scholar] [CrossRef]
- Mudhakir, D.; Harashima, H. Learning from the viral journey: How to enter cells and how to overcome intracellular barriers to reach the nucleus. AAPS J. 2009, 11, 65–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thennarasu, S.; Tan, A.; Penumatchu, R.; Shelburne, C.E.; Heyl, D.L.; Ramamoorthy, A. Antimicrobial and membrane disrupting activities of a peptide derived from the human cathelicidin antimicrobial peptide ll37. Biophys. J. 2010, 98, 248–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludtke, S.; He, K.; Huang, H. Membrane Thinning Caused by Magainin 2+. Biochemistry 1995, 34, 16764–16769. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.C.; Zhang, J.; Min, K.A.; Lee, K.; Byun, Y.; David, A.E.; He, H.; Yang, V.C. Cell-penetrating peptides: Achievements and challenges in application for cancer treatment. J. Biomed. Mater. Res.—Part A 2014, 102, 575–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, F.; Zhao, Y.; Liu, Y.; Chang, X.; Chen, C.; Zhao, Y. Cellular uptake, intracellular trafficking, and cytotoxicity of nanomaterials. Small 2011, 7, 1322–1337. [Google Scholar] [CrossRef]
- Conner, S.D.; Schmid, S.L. Regulated portals of entry into the cell. Nature 2003, 422, 37–44. [Google Scholar] [CrossRef]
- Wadia, J.S.; Stan, R.V.; Dowdy, S.F. Transducible TAT-HA fusogenic peptide enhances escape of TAT-fusion proteins after lipid raft macropinocytosis. Nat. Med. 2004, 10, 310–315. [Google Scholar] [CrossRef]
- Norbury, C.C.; Hewlett, L.J.; Prescott, A.R.; Shastri, N.; Watts, C. Class I MHC presentation of exogenous soluble antigen via macropinocytosis in bone marrow macrophages. Immunity 1995, 3, 783–791. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.P.; Gleeson, P.A. Macropinocytosis: An endocytic pathway for internalising large gulps. Immunol. Cell Biol. 2011, 89, 836–843. [Google Scholar] [CrossRef]
- Casella, J.F.; Flanagan, M.D.; Lin, S. Cytochalasin D inhibits actin polymerization and induces depolymerization of actin filaments formed during platelet shape change. Nature 1981, 293, 302–305. [Google Scholar] [CrossRef]
- Kim, G.C.; Cheon, D.H.; Lee, Y. Challenge to overcome current limitations of cell-penetrating peptides. Biochim. Biophys. Acta—Proteins Proteom. 2021, 1869, 140604. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, G.; Nakase, I.; Fukuda, Y.; Masuda, R.; Oishi, S.; Shimura, K.; Kawaguchi, Y.; Takatani-Nakase, T.; Langel, Ü.; Gräslund, A.; et al. CXCR4 stimulates macropinocytosis: Implications for cellular uptake of arginine-rich cell-penetrating peptides and HIV. Chem. Biol. 2012, 19, 1437–1446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplan, I.M.; Wadia, J.S.; Dowdy, S.F. Cationic TAT peptide transduction domain enters cells by macropinocytosis. J. Control. Release 2005, 102, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Nakase, I.; Tadokoro, A.; Kawabata, N.; Takeuchi, T.; Katoh, H.; Hiramoto, K.; Negishi, M.; Nomizu, M.; Sugiura, Y.; Futaki, S. Interaction of arginine-rich peptides with membrane-associated proteoglycans is crucial for induction of actin organization and macropinocytosis. Biochemistry 2007, 46, 492–501. [Google Scholar] [CrossRef]
- Pang, H.B.; Braun, G.B.; Ruoslahti, E. Neuropilin-1 and heparan sulfate proteoglycans cooperate in cellular uptake of nanoparticles functionalized by cationic cell-penetrating peptides. Sci. Adv. 2015, 1, e1500821. [Google Scholar] [CrossRef] [Green Version]
- Nakase, I.; Noguchi, K.; Aoki, A.; Takatani-Nakase, T.; Fujii, I.; Futaki, S. Arginine-rich cell-penetrating peptide-modified extracellular vesicles for active macropinocytosis induction and efficient intracellular delivery. Sci. Rep. 2017, 7, 1991. [Google Scholar] [CrossRef]
- Nakase, I.; Osaki, K.; Tanaka, G.; Utani, A.; Futaki, S. Molecular interplays involved in the cellular uptake of octaarginine on cell surfaces and the importance of syndecan-4 cytoplasmic v domain for the activation of protein kinase Cα. Biochem. Biophys. Res. Commun. 2014, 446, 857–862. [Google Scholar] [CrossRef]
- Ezzat, K.; Helmfors, H.; Tudoran, O.; Juks, C.; Lindberg, S.; Padari, K.; El-Andaloussi, S.; Pooga, M.; Langel, Ü. Scavenger receptor-mediated uptake of cell-penetrating peptide nanocomplexes with oligonucleotides. FASEB J. 2012, 26, 1172–1180. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Tanaka, G.; Nakase, I.; Imanishi, M.; Chiba, J.; Hatanaka, Y.; Futaki, S. Identification of cellular proteins interacting with octaarginine (R8) cell-penetrating peptide by photo-crosslinking. Bioorganic Med. Chem. Lett. 2013, 23, 3738–3740. [Google Scholar] [CrossRef]
- Arukuusk, P.; Pärnaste, L.; Margus, H.; Eriksson, N.K.J.; Vasconcelos, L.; Padari, K.; Pooga, M.; Langel, Ü. Differential endosomal pathways for radically modified peptide vectors. Bioconjug. Chem. 2013, 24, 1721–1732. [Google Scholar] [CrossRef]
- Xiang, S.; Tong, H.; Shi, Q.; Fernandes, J.C.; Jin, T.; Dai, K.; Zhang, X. Uptake mechanisms of non-viral gene delivery. J. Control. Release 2012, 158, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Kirchhausen, T. Imaging endocytic clathrin structures in living cells. Trends Cell Biol. 2009, 19, 596–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forrester, J.V.; Dick, A.D.; McMenamin, P.G.; Roberts, F.; Pearlman, E. Biochemistry and cell biology. In The Eye; Elsevier: Amsterdam, The Netherlands, 2016; pp. 157–268.e4. [Google Scholar]
- Schmidt, N.; Mishra, A.; Lai, G.H.; Wong, G.C.L. Arginine-rich cell-penetrating peptides. FEBS Lett. 2010, 584, 1806–1813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richard, J.P.; Melikov, K.; Brooks, H.; Prevot, P.; Lebleu, B.; Chernomordik, L.V. Cellular uptake of unconjugated TAT peptide involves clathrin-dependent endocytosis and heparan sulfate receptors. J. Biol. Chem. 2005, 280, 15300–15306. [Google Scholar] [CrossRef] [Green Version]
- Veldhoen, S.; Laufer, S.D.; Trampe, A.; Restle, T. Cellular delivery of small interfering RNA by a non-covalently attached cell-penetrating peptide: Quantitative analysis of uptake and biological effect. Nucleic Acids Res. 2006, 34, 6561–6573. [Google Scholar] [CrossRef] [Green Version]
- Futaki, S.; Nakase, I. Cell-Surface Interactions on Arginine-Rich Cell-Penetrating Peptides Allow for Multiplex Modes of Internalization. Acc. Chem. Res. 2017, 50, 2449–2456. [Google Scholar] [CrossRef]
- Palade, G.E. Fine structure of blood capillaries. J. Appl. Phys. 1953, 24, 1424. [Google Scholar]
- Smart, E.J.; Graf, G.A.; McNiven, M.A.; Sessa, W.C.; Engelman, J.A.; Scherer, P.E.; Okamoto, T.; Lisanti, M.P. Caveolins, Liquid-Ordered Domains, and Signal Transduction. Mol. Cell. Biol. 1999, 19, 7289–7304. [Google Scholar] [CrossRef] [Green Version]
- Branza-Nichita, N.; Macovei, A.; Lazar, C. Caveolae-Dependent Endocytosis in Viral Infection. In Molecular Regulation of Endocytosis; InTech: Rijeka, Croatia, 2012. [Google Scholar]
- Kiss, A.L.; Botos, E. Endocytosis via caveolae: Alternative pathway with distinct cellular compartments to avoid lysosomal degradation? J. Cell. Mol. Med. 2009, 13, 1228–1237. [Google Scholar] [CrossRef] [Green Version]
- Nabi, I.R.; Le, P.U. Caveolae/raft-dependent endocytosis. J. Cell Biol. 2003, 161, 673–677. [Google Scholar] [CrossRef]
- Cleal, K.; He, L.; Watson, P.D.; Jones, A.T. Endocytosis, Intracellular Traffic and Fate of Cell Penetrating Peptide Based Conjugates and Nanoparticles. Curr. Pharm. Des. 2013, 19, 2878–2894. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, N.; Gomez, G.A.; Howes, M.T.; Lo, H.P.; McMahon, K.A.; Rae, J.A.; Schieber, N.L.; Hill, M.M.; Gaus, K.; Yap, A.S.; et al. Endocytic Crosstalk: Cavins, Caveolins, and Caveolae Regulate Clathrin-Independent Endocytosis. PLoS Biol. 2014, 12, 1001832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhee, M.; Davis, P. Mechanism of uptake of C105Y, a novel cell-penetrating peptide. J. Biol. Chem. 2006, 281, 1233–1240. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, A.; Pellegrini, V.; Arcangeli, C.; Fittipaldi, A.; Giacca, M.; Beltram, F. Caveolae-mediated internalization of extracellular HIV-1 Tat fusion proteins visualized in real time. Mol. Ther. 2003, 8, 284–294. [Google Scholar] [CrossRef]
- Pujals, S.; Giralt, E. Proline-rich, amphipathic cell-penetrating peptides. Adv. Drug Deliv. Rev. 2008, 60, 473–484. [Google Scholar] [CrossRef]
- Veiman, K.L.; Mäger, I.; Ezzat, K.; Margus, H.; Lehto, T.; Langel, K.; Kurrikoff, K.; Arukuusk, P.; Suhorutšenko, J.; Padari, K.; et al. PepFect14 peptide vector for efficient gene delivery in cell cultures. Mol. Pharm. 2013, 10, 199–210. [Google Scholar] [CrossRef]
- Taylor, B.N.; Mehta, R.R.; Yamada, T.; Lekmine, F.; Christov, K.; Chakrabarty, A.M.; Green, A.; Bratescu, L.; Shilkaitis, A.; Beattie, C.W.; et al. Noncationic peptides obtained from azurin preferentially enter cancer cells. Cancer Res. 2009, 69, 537–546. [Google Scholar] [CrossRef] [Green Version]
- Mehta, R.R.; Yamada, T.; Taylor, B.N.; Christov, K.; King, M.L.; Majumdar, D.; Lekmine, F.; Tiruppathi, C.; Shilkaitis, A.; Bratescu, L.; et al. A cell penetrating peptide derived from azurin inhibits angiogenesis and tumor growth by inhibiting phosphorylation of VEGFR-2, FAK and Akt. Angiogenesis 2011, 14, 355–369. [Google Scholar] [CrossRef]
- Hu, G.; Zheng, W.; Li, A.; Mu, Y.; Shi, M.; Li, T.; Zou, H.; Shao, H.; Qin, A.; Ye, J. A novel CAV derived cell-penetrating peptide efficiently delivers exogenous molecules through caveolae-mediated endocytosis. Vet. Res. 2018, 49, 16. [Google Scholar] [CrossRef] [Green Version]
- Tréhin, R.; Nielsen, H.M.; Jahnke, H.G.; Krauss, U.; Beck-Sickinger, A.G.; Merkle, H.P. Metabolic cleavage of cell-penetrating peptides in contact with epithelial models: Human calcitonin (hCT)-derived peptides, Tat(47–57) and penetratin(43–58). Biochem. J. 2004, 382, 945–956. [Google Scholar] [CrossRef] [Green Version]
- Elmquist, A.; Langel, Ü. In vitro uptake and stability study of pVEC and its all-D analog. Biol. Chem. 2003, 384, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Palm, C.; Jayamanne, M.; Kjellander, M.; Hällbrink, M. Peptide degradation is a critical determinant for cell-penetrating peptide uptake. Biochim. Biophys. Acta—Biomembr. 2007, 1768, 1769–1776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tugyi, R.; Uray, K.; Iván, D.; Fellinger, E.; Perkins, A.; Hudecz, F. Partial D-amino acid substitution: Improved enzymatic stability and preserved Ab recognition of a MUC2 epitope peptide. Proc. Natl. Acad. Sci. USA 2005, 102, 413–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brugidou, J.; Legrand, C.; Mery, J.; Rabie, A. The Retro-inverso Form of a Homeobox-Derived Short Peptide Is Rapidly Internalized by Cultured Neurons: A New Basis for an Efficient Intracellular Delivery System. Biochem. Biophys. Res. Commun. 1995, 214, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Derossi, D.; Calvet, S.; Trembleau, A.; Brunissen, A.; Chassaing, G.; Prochiantz, A. Cell internalization of the third helix of the antennapedia homeodomain is receptor-independent. J. Biol. Chem. 1996, 271, 18188–18193. [Google Scholar] [CrossRef] [Green Version]
- Wender, P.A.; Mitchell, D.J.; Pattabiraman, K.; Pelkey, E.T.; Steinman, L.; Rothbard, J.B. The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: Peptoid molecular transporters. Proc. Natl. Acad. Sci. USA 2000, 97, 13003–13008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, D.J.; Kim, D.T.; Steinman, L.; Fathman, C.G.; Rothbard, J.B. Polyarginine enters cells more efficiently than other polycationic homopolymers. J. Pept. Res. 2000, 56, 318–325. [Google Scholar] [CrossRef]
- Gammon, S.T.; Villalobos, V.M.; Prior, J.L.; Sharma, V.; Piwnica-Worms, D. Quantitative analysis of permeation peptide complexes labeled with technetium-99m: Chiral and sequence-specific effects on net cell uptake. Bioconjug. Chem. 2003, 14, 368–376. [Google Scholar] [CrossRef]
- Tünnemann, G.; Ter-Avetisyan, G.; Martin, R.M.; Stöckl, M.; Herrmann, A.; Cardoso, M.C. Live-cell analysis of cell penetration ability and toxicity of oligo-arginines. J. Pept. Sci. 2008, 14, 469–476. [Google Scholar] [CrossRef]
- Verdurmen, W.P.R.; Bovee-Geurts, P.H.; Wadhwani, P.; Ulrich, A.S.; Hällbrink, M.; Van Kuppevelt, T.H.; Brock, R. Preferential uptake of L-versus D-amino acid cell-penetrating peptides in a cell type-dependent manner. Chem. Biol. 2011, 18, 1000–1010. [Google Scholar] [CrossRef] [Green Version]
- Najjar, K.; Erazo-Oliveras, A.; Brock, D.J.; Wang, T.Y.; Pellois, J.P. An L- to D-amino acid conversion in an endosomolytic analog of the cell-penetrating peptide TAT influences proteolytic stability, endocytic uptake, and endosomal escape. J. Biol. Chem. 2017, 292, 847–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pujals, S.; Fernández-Carneado, J.; Ludevid, M.D.; Giralt, E. D-SAP: A New, Noncytotoxic, and Fully Protease Resistant Cell-Penetrating Peptide. ChemMedChem 2008, 3, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Fominaya, J.; Bravo, J.; Rebollo, A. Strategies to stabilize cell penetrating peptides for in vivo applications. Ther. Deliv. 2015, 6, 1171–1194. [Google Scholar] [CrossRef] [PubMed]
- Ezzat, K.; El Andaloussi, S.; Zaghloul, E.M.; Lehto, T.; Lindberg, S.; Moreno, P.M.D.; Viola, J.R.; Magdy, T.; Abdo, R.; Guterstam, P.; et al. PepFect 14, a novel cell-penetrating peptide for oligonucleotide delivery in solution and as solid formulation. Nucleic Acids Res. 2011, 39, 5284–5298. [Google Scholar] [CrossRef]
- Mäe, M.; EL Andaloussi, S.; Lundin, P.; Oskolkov, N.; Johansson, H.J.; Guterstam, P.; Langel, Ü. A stearylated CPP for delivery of splice correcting oligonucleotides using a non-covalent co-incubation strategy. J. Control. Release 2009, 134, 221–227. [Google Scholar] [CrossRef]
- Veiman, K.L.; Künnapuu, K.; Lehto, T.; Kiisholts, K.; Pärn, K.; Langel, Ü.; Kurrikoff, K. PEG shielded MMP sensitive CPPs for efficient and tumor specific gene delivery in vivo. J. Control. Release 2015, 209, 238–247. [Google Scholar] [CrossRef] [Green Version]
- Dietrich, L.; Rathmer, B.; Ewan, K.; Bange, T.; Heinrichs, S.; Dale, T.C.; Schade, D.; Grossmann, T.N. Cell Permeable Stapled Peptide Inhibitor of Wnt Signaling that Targets β-Catenin Protein-Protein Interactions. Cell Chem. Biol. 2017, 24, 958–968.e5. [Google Scholar] [CrossRef] [Green Version]
- Mandal, D.; Nasrolahi Shirazi, A.; Parang, K. Cell-penetrating homochiral cyclic peptides as nuclear-targeting molecular transporters. Angew. Chemie—Int. Ed. 2011, 50, 9633–9637. [Google Scholar] [CrossRef]
- Nischan, N.; Herce, H.D.; Natale, F.; Bohlke, N.; Budisa, N.; Cardoso, M.C.; Hackenberger, C.P.R. Covalent attachment of cyclic TAT peptides to GFP results in protein delivery into live cells with immediate bioavailability. Angew. Chemie—Int. Ed. 2015, 54, 1950–1953. [Google Scholar] [CrossRef]
- Qian, Z.; Martyna, A.; Hard, R.L.; Wang, J.; Appiah-Kubi, G.; Coss, C.; Phelps, M.A.; Rossman, J.S.; Pei, D. Discovery and Mechanism of Highly Efficient Cyclic Cell-Penetrating Peptides. Biochemistry 2016, 55, 2601–2612. [Google Scholar] [CrossRef]
- Sun, P.; Huang, W.; Kang, L.; Jin, M.; Fan, B.; Jin, H.; Wang, Q.M.; Gao, Z. SiRNA-loaded poly(Histidine-arginine)6-modified chitosan nanoparticle with enhanced cell-penetrating and endosomal escape capacities for suppressing breast tumor metastasis. Int. J. Nanomed. 2017, 12, 3221–3234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Hao, X.; Zaidi, S.S.A.; Guo, J.; Ren, X.; Shi, C.; Zhang, W.; Feng, Y. Oligohistidine and targeting peptide functionalized TAT-NLS for enhancing cellular uptake and promoting angiogenesis in vivo. J. Nanobiotechnol. 2018, 16, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arukuusk, P.; Pärnaste, L.; Oskolkov, N.; Copolovici, D.M.; Margus, H.; Padari, K.; Möll, K.; Maslovskaja, J.; Tegova, R.; Kivi, G.; et al. New generation of efficient peptide-based vectors, NickFects, for the delivery of nucleic acids. Biochim. Biophys. Acta—Biomembr. 2013, 1828, 1365–1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Andaloussi, S.; Lehto, T.; Mäger, I.; Rosenthal-Aizman, K.; Oprea, I.I.; Simonson, O.E.; Sork, H.; Ezzat, K.; Copolovici, D.M.; Kurrikoff, K.; et al. Design of a peptide-based vector, PepFect6, for efficient delivery of siRNA in cell culture and systemically in vivo. Nucleic Acids Res. 2011, 39, 3972–3987. [Google Scholar] [CrossRef] [Green Version]
- Liou, J.S.; Liu, B.R.; Martin, A.L.; Huang, Y.W.; Chiang, H.J.; Lee, H.J. Protein transduction in human cells is enhanced by cell-penetrating peptides fused with an endosomolytic HA2 sequence. Peptides 2012, 37, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Salomone, F.; Cardarelli, F.; Di Luca, M.; Boccardi, C.; Nifosì, R.; Bardi, G.; Di Bari, L.; Serresi, M.; Beltram, F. A novel chimeric cell-penetrating peptide with membrane-disruptive properties for efficient endosomal escape. J. Control. Release 2012, 163, 293–303. [Google Scholar] [CrossRef]
- Lundberg, P.; El-Andaloussi, S.; Sütlü, T.; Johansson, H.; Langel, Ü. Delivery of short interfering RNA using endosomolytic cell-penetrating peptides. FASEB J. 2007, 21, 2664–2671. [Google Scholar] [CrossRef] [Green Version]
- Lönn, P.; Kacsinta, A.D.; Cui, X.S.; Hamil, A.S.; Kaulich, M.; Gogoi, K.; Dowdy, S.F. Enhancing Endosomal Escape for Intracellular Delivery of Macromolecular Biologic Therapeutics. Sci. Rep. 2016, 6, 32301. [Google Scholar] [CrossRef]
- Ham, S.H.; Min, K.A.; Shin, M.C. Molecular tumor targeting of gelonin by fusion with F3 peptide. Acta Pharmacol. Sin. 2017, 38, 897–906. [Google Scholar] [CrossRef]
- Li, J.; Feng, L.; Fan, L.; Zha, Y.; Guo, L.; Zhang, Q.; Chen, J.; Pang, Z.; Wang, Y.; Jiang, X.; et al. Targeting the brain with PEG-PLGA nanoparticles modified with phage-displayed peptides. Biomaterials 2011, 32, 4943–4950. [Google Scholar] [CrossRef] [Green Version]
- Gao, H.; Qian, J.; Cao, S.; Yang, Z.; Pang, Z.; Pan, S.; Fan, L.; Xi, Z.; Jiang, X.; Zhang, Q. Precise glioma targeting of and penetration by aptamer and peptide dual-functioned nanoparticles. Biomaterials 2012, 33, 5115–5123. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Zhang, S.; Cao, S.; Yang, Z.; Pang, Z.; Jiang, X. Angiopep-2 and activatable cell-penetrating peptide dual-functionalized nanoparticles for systemic glioma-targeting delivery. Mol. Pharm. 2014, 11, 2755–2763. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Lee, S.H.; Na, Y.; Noh, I.; Ha, J.H.; Yoo, J.; Bang, H.B.; Park, J.H.; Jeong, K.J.; Yun, C.O.; et al. Conformation-switchable helical polypeptide eliciting selective pro-apoptotic activity for cancer therapy. J. Control. Release 2017, 264, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Ma, Y.; Zhang, W.; Li, L.; Zhang, Y.; Zhang, L.; Liu, H.; Ni, J.; Wang, R. Design of new acid-activated cellpenetrating peptides for tumor drug delivery. PeerJ 2017, 2017, e3429. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Sun, A.; Xu, R.; Tao, X.; Dong, Y.; Lv, X.; Wei, D. Cell-penetrating and endoplasmic reticulum-locating TAT-IL-24-KDEL fusion protein induces tumor apoptosis. J. Cell. Physiol. 2016, 231, 84–93. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Ise, S.; Azuma, Y.; Takeuchi, T.; Kawano, K.; Le, T.K.; Ohkanda, J.; Futaki, S. Dipicolylamine/Metal Complexes that Promote Direct Cell-Membrane Penetration of Octaarginine. Bioconjug. Chem. 2019, 30, 454–460. [Google Scholar] [CrossRef]
- Lee, H.J.; Huang, Y.W.; Chiou, S.H.; Aronstam, R.S. Polyhistidine facilitates direct membrane translocation of cell-penetrating peptides into cells. Sci. Rep. 2019, 9, 9398. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.R.; Huang, Y.; Winiarz, J.G.; Chiang, H.J.; Lee, H.J. Intracellular delivery of quantum dots mediated by a histidine- and arginine-rich HR9 cell-penetrating peptide through the direct membrane translocation mechanism. Biomaterials 2011, 32, 3520–3537. [Google Scholar] [CrossRef]
- Tsubery, H.; Mironchik, M.; Fridkin, M.; Shechter, Y. Prolonging the action of protein and peptide drugs by a novel approach of reversible polyethylene glycol modification. J. Biol. Chem. 2004, 279, 38118–38124. [Google Scholar] [CrossRef] [Green Version]
- Osman, G.; Rodriguez, J.; Chan, S.Y.; Chisholm, J.; Duncan, G.; Kim, N.; Tatler, A.L.; Shakesheff, K.M.; Hanes, J.; Suk, J.S.; et al. PEGylated enhanced cell penetrating peptide nanoparticles for lung gene therapy. J. Control. Release 2018, 285, 35–45. [Google Scholar] [CrossRef]
- Chu, Q.; Moellering, R.E.; Hilinski, G.J.; Kim, Y.W.; Grossmann, T.N.; Yeh, J.T.H.; Verdine, G.L. Towards understanding cell penetration by stapled peptides. Medchemcomm 2015, 6, 111–119. [Google Scholar] [CrossRef]
- Hilinski, G.J.; Kim, Y.W.; Hong, J.; Kutchukian, P.S.; Crenshaw, C.M.; Berkovitch, S.S.; Chang, A.; Ham, S.; Verdine, G.L. Stitched α-helical peptides via bis ring-closing metathesis. J. Am. Chem. Soc. 2014, 136, 12314–12322. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, L.A.; Ben Neriah, D.; Senecal, A.; Benard, L.; Thiruthuvanathan, V.; Yatsenko, T.; Narayanagari, S.R.; Wheat, J.C.; Todorova, T.I.; Mitchell, K.; et al. Dual inhibition of MDMX and MDM2 as a therapeutic strategy in leukemia. Sci. Transl. Med. 2018, 10, eaao3003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakagami, K.; Masuda, T.; Kawano, K.; Futaki, S. Importance of Net Hydrophobicity in the Cellular Uptake of All-Hydrocarbon Stapled Peptides. Mol. Pharm. 2018, 15, 1332–1340. [Google Scholar] [CrossRef] [PubMed]
- Park, S.E.; Sajid, M.I.; Parang, K.; Tiwari, R.K. Cyclic cell-penetrating peptides as efficient intracellular drug delivery tools. Mol. Pharm. 2019, 16, 3727–3743. [Google Scholar] [CrossRef]
- Lättig-Tünnemann, G.; Prinz, M.; Hoffmann, D.; Behlke, J.; Palm-Apergi, C.; Morano, I.; Herce, H.D.; Cardoso, M.C. Backbone rigidity and static presentation of guanidinium groups increases cellular uptake of arginine-rich cell-penetrating peptides. Nat. Commun. 2011, 2, 453. [Google Scholar] [CrossRef]
- Amoura, M.; Illien, F.; Joliot, A.; Guitot, K.; Offer, J.; Sagan, S.; Burlina, F. Head to tail cyclisation of cell-penetrating peptides: Impact on GAG-dependent internalisation and direct translocation. Chem. Commun. 2019, 55, 4566–4569. [Google Scholar] [CrossRef]
- Qian, Z.; Liu, T.; Liu, Y.Y.; Briesewitz, R.; Barrios, A.M.; Jhiang, S.M.; Pei, D. Efficient delivery of cyclic peptides into mammalian cells with short sequence motifs. ACS Chem. Biol. 2013, 8, 423–431. [Google Scholar] [CrossRef] [Green Version]
- White, T.R.; Renzelman, C.M.; Rand, A.C.; Rezai, T.; McEwen, C.M.; Gelev, V.M.; Turner, R.A.; Linington, R.G.; Leung, S.S.F.; Kalgutkar, A.S.; et al. On-resin N-methylation of cyclic peptides for discovery of orally bioavailable scaffolds. Nat. Chem. Biol. 2011, 7, 810–817. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, J.; Gilon, C.; Hoffman, A.; Kessler, H. N-methylation of peptides: A new perspective in medicinal chemistry. Acc. Chem. Res. 2008, 41, 1331–1342. [Google Scholar] [CrossRef]
- Thorén, P.E.G.; Persson, D.; Isakson, P.; Goksör, M.; Önfelt, A.; Nordén, B. Uptake of analogs of penetratin, Tat(48-60) and oligoarginine in live cells. Biochem. Biophys. Res. Commun. 2003, 307, 100–107. [Google Scholar] [CrossRef]
- Letoha, T.; Gaá, S.; Somlai, C.; Czajlik, A.; Perczel, A.; Penke, B. Membrane translocation of penetratin and its derivatives in different cell lines. J. Mol. Recognit. 2003, 16, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Derossi, D.; Chassaing, G.; Prochiantz, A. Trojan peptides: The penetratin system for intracellular delivery. Trends Cell Biol. 1998, 8, 84–87. [Google Scholar] [CrossRef]
- Delaroche, D.; Aussedat, B.; Aubry, S.; Chassaing, G.; Burlina, F.; Clodic, G.; Bolbach, G.; Lavielle, S.; Sagan, S. Tracking a new cell-penetrating (W/R) nonapeptide, through an enzyme-stable mass spectrometry reporter tag. Anal. Chem. 2007, 79, 1932–1938. [Google Scholar] [CrossRef]
- Futaki, S.; Suzuki, T.; Ohashi, W.; Yagami, T.; Tanaka, S.; Ueda, K.; Sugiura, Y. Arginine-rich peptides. An abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery. J. Biol. Chem. 2001, 276, 5836–5840. [Google Scholar] [CrossRef] [Green Version]
- Walrant, A.; Correia, I.; Jiao, C.Y.; Lequin, O.; Bent, E.H.; Goasdoué, N.; Lacombe, C.; Chassaing, G.; Sagan, S.; Alves, I.D. Different membrane behaviour and cellular uptake of three basic arginine-rich peptides. Biochim. Biophys. Acta—Biomembr. 2011, 1808, 382–393. [Google Scholar] [CrossRef]
- Jobin, M.-L.; Blanchet, M.; Henry, S.; Chaignepain, S.; Manigand, C.; Castano, S.; Lecomte, S.; Burlina, F.; Sagan, S.; Alves, I.D. The role of tryptophans on the cellular uptake and membrane interaction of arginine-rich cell penetrating peptides. Biochim. Biophys. Acta 2015, 1848, 593–602. [Google Scholar] [CrossRef]
- Song, J.; Qian, Z.; Sahni, A.; Chen, K.; Pei, D. Cyclic Cell-Penetrating Peptides with Single Hydrophobic Groups. ChemBioChem 2019, 20, 2085–2088. [Google Scholar] [CrossRef] [Green Version]
- Tompa, P.; Buzder-Lantos, P.; Tantos, A.; Farkas, A.; Szilágyi, A.; Bánóczi, Z.; Hudecz, F.; Friedrich, P. On the sequential determinants of calpain cleavage. J. Biol. Chem. 2004, 279, 20775–20785. [Google Scholar] [CrossRef] [Green Version]
- Roloff, A.; Nelles, D.A.; Thompson, M.P.; Yeo, G.W.; Gianneschi, N.C. Self-Transfecting Micellar RNA: Modulating Nanoparticle Cell Interactions via High Density Display of Small Molecule Ligands on Micelle Coronas. Bioconjug. Chem. 2018, 29, 126–135. [Google Scholar] [CrossRef]
- Mandal, S.; Mann, G.; Satish, G.; Brik, A. Enhanced Live-Cell Delivery of Synthetic Proteins Assisted by Cell-Penetrating Peptides Fused to DABCYL. Angew. Chemie—Int. Ed. 2021, 60, 7333–7343. [Google Scholar] [CrossRef]
- Alexa, A.; Ember, O.; Szabó, I.; Mo’ath, Y.; Póti, Á.L.; Reményi, A.; Bánóczi, Z. Peptide Based Inhibitors of Protein Binding to the Mitogen-Activated Protein Kinase Docking Groove. Front. Mol. Biosci. 2021, 8, 629. [Google Scholar] [CrossRef] [PubMed]
- Yousef, M.; Szabó, I.; Biri-Kovács, B.; Szeder, B.; Illien, F.; Sagan, S.; Bánóczi, Z. Modification of Short Non-Permeable Peptides to Increase Cellular Uptake and Cytostatic Activity of Their Conjugates. ChemistrySelect 2021, 6, 10111–10120. [Google Scholar] [CrossRef]
- Johnson, J.R.; Jiang, H.; Smith, B.D. Zinc(II)-coordinated oligotyrosine: A new class of cell penetrating peptide. Bioconjug. Chem. 2008, 19, 1033–1039. [Google Scholar] [CrossRef]
- Azuma, Y.; Imai, H.; Kawaguchi, Y.; Nakase, I.; Kimura, H.; Futaki, S. Modular Redesign of a Cationic Lytic Peptide To Promote the Endosomal Escape of Biomacromolecules. Angew. Chem.—Int. Ed. 2018, 57, 12771–12774. [Google Scholar] [CrossRef]
- Lécorché, P.; Walrant, A.; Burlina, F.; Dutot, L.; Sagan, S.; Mallet, J.M.; Desbat, B.; Chassaing, G.; Alves, I.D.; Lavielle, S. Cellular uptake and biophysical properties of galactose and/or tryptophan containing cell-penetrating peptides. Biochim. Biophys. Acta—Biomembr. 2012, 1818, 448–457. [Google Scholar] [CrossRef]
- Monreal, I.A.; Contreras, E.M.; Wayman, G.A.; Aguilar, H.C.; Saludes, J.P. SialoPen peptides are new cationic foldamers with remarkable cell permeability. Heliyon 2020, 6, e05780. [Google Scholar] [CrossRef]
- Erazo-Oliveras, A.; Muthukrishnan, N.; Baker, R.; Wang, T.Y.; Pellois, J.P. Improving the endosomal escape of cell-penetrating peptides and their cargos: Strategies and challenges. Pharmaceuticals 2012, 5, 1177–1209. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.T.; Zaitseva, E.; Chernomordik, L.V.; Melikov, K. Cell-penetrating peptide induces leaky fusion of liposomes containing late endosome-specific anionic lipid. Biophys. J. 2010, 99, 2525–2533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Sayed, A.; Futaki, S.; Harashima, H. Delivery of macromolecules using arginine-rich cell-penetrating peptides: Ways to overcome endosomal entrapment. AAPS J. 2009, 11, 13–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuber, G.; Dauty, E.; Nothisen, M.; Belguise, P.; Behr, J.P. Towards synthetic viruses. Adv. Drug Deliv. Rev. 2001, 52, 245–253. [Google Scholar] [CrossRef]
- Lou, D.; Saltzman, W.M. Synthetic DNA delivery systems. Nat. Biotechnol. 2000, 18, 33–37. [Google Scholar]
- Kichler, A.; Leborgne, C.; Coeytaux, E.; Danos, O. Polyethylenimine-mediated gene delivery: A mechanistic study. J. Gene Med. 2001, 3, 135–144. [Google Scholar] [CrossRef]
- Haensler, J.; Szoka, F.C. Polyamidoamine Cascade Polymers Mediate Efficient Transfection of Cells in Culture. Bioconjug. Chem. 1993, 4, 372–379. [Google Scholar] [CrossRef]
- Freeman, E.C.; Weiland, L.M.; Meng, W.S. Modeling the proton sponge hypothesis: Examining proton sponge effectiveness for enhancing intracellular gene delivery through multiscale modeling. J. Biomater. Sci. Polym. Ed. 2013, 24, 398–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonawane, N.D.; Szoka, F.C.; Verkman, A.S. Chloride Accumulation and Swelling in Endosomes Enhances DNA Transfer by Polyamine-DNA Polyplexes. J. Biol. Chem. 2003, 278, 44826–44831. [Google Scholar] [CrossRef] [Green Version]
- Lo, S.L.; Wang, S. An endosomolytic Tat peptide produced by incorporation of histidine and cysteine residues as a nonviral vector for DNA transfection. Biomaterials 2008, 29, 2408–2414. [Google Scholar] [CrossRef] [PubMed]
- Beloor, J.; Zeller, S.; Choi, C.S.; Lee, S.K.; Kumar, P. Cationic cell-penetrating peptides as vehicles for siRNA delivery. Ther. Deliv. 2015, 6, 491–507. [Google Scholar] [CrossRef]
- Khalil, I.A.; Futaki, S.; Niwa, M.; Baba, Y.; Kaji, N.; Kamiya, H.; Harashima, H. Mechanism of improved gene transfer by the N-terminal stearylation of octaarginine: Enhanced cellular association by hydrophobic core formation. Gene Ther. 2004, 11, 636–644. [Google Scholar] [CrossRef] [Green Version]
- Pei, D.; Buyanova, M. Overcoming Endosomal Entrapment in Drug Delivery. Bioconjug. Chem. 2019, 30, 273–283. [Google Scholar] [CrossRef]
- Plank, C.; Oberhauser, B.; Mechtler, K.; Koch, C.; Wagner, E. The influence of endosome-disruptive peptides on gene transfer using synthetic virus-like gene transfer systems. J. Biol. Chem. 1994, 269, 12918–12924. [Google Scholar] [CrossRef]
- Varkouhi, A.K.; Scholte, M.; Storm, G.; Haisma, H.J. Endosomal escape pathways for delivery of biologicals. J. Control. Release 2011, 151, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Morishita, T.; Aburai, K.; Sakai, K.; Abe, M.; Nakase, I.; Futaki, S.; Sakai, H.; Sakamoto, K. Key process and factors controlling the direct translocation of cell-penetrating peptide through bio-membrane. Int. J. Mol. Sci. 2020, 21, 5466. [Google Scholar] [CrossRef]
- Takeuchi, T.; Kosuge, M.; Tadokoro, A.; Sugiura, Y.; Nishi, M.; Kawata, M.; Sakai, N.; Matile, S.; Futaki, S. Direct and rapid cytosolic delivery using cell-penetrating peptides mediated by pyrenebutyrate. ACS Chem. Biol. 2006, 1, 299–303. [Google Scholar] [CrossRef]
- Guterstam, P.; Madani, F.; Hirose, H.; Takeuchi, T.; Futaki, S.; EL Andaloussi, S.; Gräslund, A.; Langel, Ü. Elucidating cell-penetrating peptide mechanisms of action for membrane interaction, cellular uptake, and translocation utilizing the hydrophobic counter-anion pyrenebutyrate. Biochim. Biophys. Acta—Biomembr. 2009, 1788, 2509–2517. [Google Scholar] [CrossRef] [Green Version]
- Katayama, S.; Hirose, H.; Takayama, K.; Nakase, I.; Futaki, S. Acylation of octaarginine: Implication to the use of intracellular delivery vectors. J. Control. Release 2011, 149, 29–35. [Google Scholar] [CrossRef]
- Marks, J.R.; Placone, J.; Hristova, K.; Wimley, W.C. Spontaneous membrane-translocating peptides by orthogonal high-throughput screening. J. Am. Chem. Soc. 2011, 133, 8995–9004. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Hristova, K.; Wimley, W.C. A highly charged voltage-sensor helix spontaneously translocates across membranes. Angew. Chem.—Int. Ed. 2012, 51, 7150–7153. [Google Scholar] [CrossRef] [Green Version]
- Fuselier, T.; Wimley, W.C. Spontaneous Membrane Translocating Peptides: The Role of Leucine-Arginine Consensus Motifs. Biophys. J. 2017, 113, 835–846. [Google Scholar] [CrossRef]
- Swiecicki, J.M.; Di Pisa, M.; Lippi, F.; Chwetzoff, S.; Mansuy, C.; Trugnan, G.; Chassaing, G.; Lavielle, S.; Burlina, F. Unsaturated acyl chains dramatically enhanced cellular uptake by direct translocation of a minimalist oligo-arginine lipopeptide. Chem. Commun. 2015, 51, 14656–14659. [Google Scholar] [CrossRef]
- Sugahara, K.N.; Teesalu, T.; Karmali, P.P.; Kotamraju, V.R.; Agemy, L.; Girard, O.M.; Hanahan, D.; Mattrey, R.F.; Ruoslahti, E. Tissue-Penetrating Delivery of Compounds and Nanoparticles into Tumors. Cancer Cell 2009, 16, 510–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laakkonen, P.; Porkka, K.; Hoffman, J.A.; Ruoslahti, E. A tumor-homing peptide with a targeting specificity related to lymphatic vessels. Nat. Med. 2002, 8, 751–755. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Lillo, A.M.; Steiniger, S.C.J.; Liu, Y.; Ballatore, C.; Anichini, A.; Mortarini, R.; Kaufmann, G.F.; Zhou, B.; Felding-Habermann, B.; et al. Targeting heat shock proteins on cancer cells: Selection, characterization, and cell-penetrating properties of a peptidic GRP78 ligand. Biochemistry 2006, 45, 9434–9444. [Google Scholar] [CrossRef] [PubMed]
- Brunner, J.; Barton, J.K. Targeting DNA mismatches with rhodium intercalators functionalized with a cell-penetrating peptide. Biochemistry 2006, 45, 12295–12302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerrato, C.P.; Pirisinu, M.; Vlachos, E.N.; Langel, Ü. Novel cell-penetrating peptide targeting mitochondria. FASEB J. 2015, 29, 4589–4599. [Google Scholar] [CrossRef] [PubMed]
- Cerrato, C.P.; Langel, Ü. Effect of a Fusion Peptide by Covalent Conjugation of a Mitochondrial Cell-Penetrating Peptide and a Glutathione Analog Peptide. Mol. Ther.—Methods Clin. Dev. 2017, 5, 221–231. [Google Scholar] [CrossRef] [Green Version]
- Szeto, H.H. Mitochondria-targeted peptide antioxidants: Novel neuroprotective agents. AAPS J. 2006, 8, 521–531. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Geisler, I.; Chmielewski, J.; Cheng, J.X. Cationic amphiphilic polyproline helix P11LRR targets intracellular mitochondria. J. Control. Release 2010, 142, 259–266. [Google Scholar] [CrossRef]
- de Jong, H.; Bonger, K.M.; Löwik, D.W.P.M. Activatable cell-penetrating peptides: 15 years of research. RSC Chem. Biol. 2020, 1, 192–203. [Google Scholar] [CrossRef]
- Jin, E.; Zhang, B.; Sun, X.; Zhou, Z.; Ma, X.; Sun, Q.; Tang, J.; Shen, Y.; Van Kirk, E.; Murdoch, W.J.; et al. Acid-active cell-penetrating peptides for in vivo tumor-targeted drug delivery. J. Am. Chem. Soc. 2013, 135, 933–940. [Google Scholar] [CrossRef]
- Svensen, N.; Walton, J.G.A.; Bradley, M. Peptides for cell-selective drug delivery. Trends Pharmacol. Sci. 2012, 33, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Sauter, M.; Strieker, M.; Kleist, C.; Wischnjow, A.; Daniel, V.; Altmann, A.; Haberkorn, U.; Mier, W. Improving antibody-based therapies by chemical engineering of antibodies with multimeric cell-penetrating peptides for elevated intracellular delivery. J. Control. Release 2020, 322, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Denkewalter, R.G.; Kolc, J.F.; Lukasavage, W.J. Macromolecular Highly Branched Homogeneous Compound Based on Lysine Units. U.S. Patent 4,289,872-A, 15 September 1983. [Google Scholar]
- Sheldon, K.; Liut, D.; Ferguson, J.; Garijepyt, J.; Baldeschwieler, J.D. Loligomers: Design of de novo peptide-based intracellular vehicles Communicated by. Proc. Natl. Acad. Sci. USA 1995, 92, 2056–2060. [Google Scholar] [CrossRef] [Green Version]
- Tung, C.H.; Mueller, S.; Weissleder, R. Novel branching membrane translocational peptide as gene delivery vector. Bioorganic Med. Chem. 2002, 10, 3609–3614. [Google Scholar] [CrossRef]
- Rudolph, C.; Plank, C.; Lausier, J.; Schillinger, U.; Müller, R.H.; Rosenecker, J. Oligomers of the arginine-rich motif of the HIV-1 TAT protein are capable of transferring plasmid DNA into cells. J. Biol. Chem. 2003, 278, 11411–11418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.M.; Ahn, M.H.; Chae, W.J.; Jung, Y.G.; Park, J.C.; Song, H.M.; Kim, Y.E.; Shin, J.A.; Park, C.S.; Park, J.W.; et al. Intranasal delivery of the cytoplasmic domain of CTLA-4 using a novel protein transduction domain prevents allergic inflammation. Nat. Med. 2006, 12, 574–579. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.I.; Park, S.H.; Jin, H.T.; Lee, C.G.; Seo, S.H.; Song, M.Y.; Lee, C.W.; Sung, Y.C. Enhanced delivery efficiency of recombinant adenovirus into tumor and mesenchymal stem cells by a novel PTD. Cancer Gene Ther. 2008, 15, 703–712. [Google Scholar] [CrossRef] [Green Version]
- Park, S.H.; Doh, J.; Park, S.I.; Lim, J.Y.; Kim, S.M.; Youn, J.I.; Jin, H.T.; Seo, S.H.; Song, M.Y.; Sung, S.Y.; et al. Branched oligomerization of cell-permeable peptides markedly enhances the transduction efficiency of adenovirus into mesenchymal stem cells. Gene Ther. 2010, 17, 1052–1061. [Google Scholar] [CrossRef] [Green Version]
- Monreal, I.A.; Liu, Q.; Tyson, K.; Bland, T.; Dalisay, D.S.; Adams, E.V.; Wayman, G.A.; Aguilar, H.C.; Saludes, J.P. Branched dimerization of Tat peptide improves permeability to HeLa and hippocampal neuronal cells. Chem. Commun. 2015, 51, 5463–5466. [Google Scholar] [CrossRef]
- Kim, H.; Kitamatsu, M.; Ohtsuki, T. Enhanced intracellular peptide delivery by multivalent cell-penetrating peptide with bioreducible linkage. Bioorganic Med. Chem. Lett. 2018, 28, 378–381. [Google Scholar] [CrossRef]
- Erazo-Oliveras, A.; Najjar, K.; Dayani, L.; Wang, T.Y.; Johnson, G.A.; Pellois, J.P. Protein delivery into live cells by incubation with an endosomolytic agent. Nat. Methods 2014, 11, 861–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eggimann, G.A.; Buschor, S.; Darbre, T.; Reymond, J.L. Convergent synthesis and cellular uptake of multivalent cell penetrating peptides derived from Tat, Antp, pVEC, TP10 and SAP. Org. Biomol. Chem. 2013, 11, 6717–6733. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, K.S.; Sung, M.; Bolewska-Pedyczak, E.; Gariépy, J. Probing the impact of valency on the routing of arginine-rich peptides into eukaryotic cells. Biochemistry 2006, 45, 1116–1127. [Google Scholar] [CrossRef] [PubMed]
- Angeles-Boza, A.M.; Erazo-Oliveras, A.; Lee, Y.J.; Pellois, J.P. Generation of endosomolytic reagents by branching of cell-penetrating peptides: Tools for the delivery of bioactive compounds to live cells in cis or trans. Bioconjug. Chem. 2010, 21, 2164–2167. [Google Scholar] [CrossRef] [Green Version]
- Neundorf, I.; Rennert, R.; Hoyer, J.; Schramm, F.; Löbner, K.; Kitanovic, I.; Wölfl, S. Fusion of a short HA2-derived peptide sequence to cell-penetrating peptides improves cytosolic uptake, but enhances cytotoxic activity. Pharmaceuticals 2009, 2, 49–65. [Google Scholar] [CrossRef] [Green Version]
- Hoyer, J.; Schatzschneider, U.; Schulz-Siegmund, M.; Neundorf, I. Dimerization of a cell-penetrating peptide leads to enhanced cellular uptake and drug delivery. Beilstein J. Org. Chem. 2012, 8, 1788–1797. [Google Scholar] [CrossRef] [Green Version]
- Gronewold, A.; Horn, M.; Ranđelović, I.; Tóvári, J.; Muñoz Vázquez, S.; Schomäcker, K.; Neundorf, I. Characterization of a Cell-Penetrating Peptide with Potential Anticancer Activity. ChemMedChem 2017, 12, 42–49. [Google Scholar] [CrossRef]
- Díaz-Perlas, C.; Oller-Salvia, B.; Sánchez-Navarro, M.; Teixidó, M.; Giralt, E. Branched BBB-shuttle peptides: Chemoselective modification of proteins to enhance blood-brain barrier transport. Chem. Sci. 2018, 9, 8409–8415. [Google Scholar] [CrossRef] [Green Version]
- Won, Y.W.; Kim, H.A.; Lee, M.; Kim, Y.H. Reducible poly(oligo-d-arginine) for enhanced gene expression in mouse lung by intratracheal injection. Mol. Ther. 2010, 18, 734–742. [Google Scholar] [CrossRef]
- Jeong, C.; Yoo, J.; Lee, D.Y.; Kim, Y.C. A branched TAT cell-penetrating peptide as a novel delivery carrier for the efficient gene transfection. Biomater. Res. 2016, 20, 28. [Google Scholar] [CrossRef] [Green Version]
- Yoo, J.; Lee, D.Y.; Gujrati, V.; Rejinold, N.S.; Lekshmi, K.M.; Uthaman, S.; Jeong, C.; Park, I.K.; Jon, S.; Kim, Y.C. Bioreducible branched poly(modified nona-arginine) cell-penetrating peptide as a novel gene delivery platform. J. Control. Release 2017, 246, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Pujals, S.; Miyamae, H.; Afonin, S.; Murayama, T.; Hirose, H.; Nakase, I.; Taniuchi, K.; Umeda, M.; Sakamoto, K.; Ulrich, A.S.; et al. Curvature engineering: Positive membrane curvature induced by epsin N-terminal peptide boosts internalization of octaarginine. ACS Chem. Biol. 2013, 8, 1894–1899. [Google Scholar] [CrossRef] [PubMed]
- Hsu, W.Y.; Masuda, T.; Afonin, S.; Sakai, T.; Arafiles, J.V.V.; Kawano, K.; Hirose, H.; Imanishi, M.; Ulrich, A.S.; Futaki, S. Enhancing the activity of membrane remodeling epsin-peptide by trimerization. Bioorganic Med. Chem. Lett. 2020, 30, 127190. [Google Scholar] [CrossRef] [PubMed]
- Futaki, S.; Nakase, I.; Suzuki, T.; Youjun, Z.; Sugiura, Y. Translocation of branched-chain arginine peptides through cell membranes: Flexibility in the spatial disposition of positive charges in membrane-permeable peptides. Biochemistry 2002, 41, 7925–7930. [Google Scholar] [CrossRef]
- Chua, B.Y.; Zeng, W.; Jackson, D.C. Simple branched arginine-based structures can enhance the cellular uptake of peptide cargos. Int. J. Pept. Res. Ther. 2007, 13, 431–437. [Google Scholar] [CrossRef]
- Huang, C.W.; Li, Z.; Conti, P.S. In vivo near-infrared fluorescence imaging of integrin α 2β 1 in prostate cancer with cell-penetrating-peptide- conjugated DGEA probe. J. Nucl. Med. 2011, 52, 1979–1986. [Google Scholar] [CrossRef] [Green Version]
- Rewatkar, P.V.; Parekh, H.S.; Parat, M.O. Molecular determinants of the cellular entry of asymmetric peptide dendrimers and role of caveolae. PLoS ONE 2016, 11, e0147491. [Google Scholar] [CrossRef]
- Sommer, P.; Fluxa, V.S.; Darbre, T.; Reymond, J.L. Proteolysis of peptide dendrimers. ChemBioChem 2009, 10, 1527–1536. [Google Scholar] [CrossRef]
- Eggimann, G.A.; Blattes, E.; Buschor, S.; Biswas, R.; Kammer, S.M.; Darbre, T.; Reymond, J.L. Designed cell penetrating peptide dendrimers efficiently internalize cargo into cells. Chem. Commun. 2014, 50, 7254–7257. [Google Scholar] [CrossRef] [Green Version]
- Bryson, D.I.; Zhang, W.; McLendon, P.M.; Reineke, T.M.; Santos, W.L. Toward targeting RNA structure: Branched peptides as cell-permeable ligands to TAR RNA. ACS Chem. Biol. 2012, 7, 210–217. [Google Scholar] [CrossRef] [Green Version]
- Kozhikhova, K.V.; Andreev, S.M.; Shilovskiy, I.P.; Timofeeva, A.V.; Gaisina, A.R.; Shatilov, A.A.; Turetskiy, E.A.; Andreev, I.M.; Smirnov, V.V.; Dvornikov, A.S.; et al. A novel peptide dendrimer LTP efficiently facilitates transfection of mammalian cells. Org. Biomol. Chem. 2018, 16, 8181–8190. [Google Scholar] [CrossRef]
- Kwok, A.; Eggimann, G.A.; Reymond, J.L.; Darbre, T.; Hollfelder, F. Peptide dendrimer/lipid hybrid systems are efficient DNA transfection reagents: Structure-activity relationships highlight the role of charge distribution across dendrimer generations. ACS Nano 2013, 7, 4668–4682. [Google Scholar] [CrossRef] [PubMed]
- Plank, C.; Tang, M.X.; Wolfe, A.R.; Szoka, F.C. Branched cationic peptides for gene delivery: Role of type and number of cationic residues in formation and in vitro activity of DNA polyplexes. Hum. Gene Ther. 1999, 10, 319–332. [Google Scholar] [CrossRef]
- Jiang, H.; Hu, X.Y.; Mosel, S.; Knauer, S.K.; Hirschhäuser, C.; Schmuck, C. A Branched Tripeptide with an Anion-Binding Motif as a New Delivery Carrier for Efficient Gene Transfection. ChemBioChem 2019, 20, 1410–1416. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Bi, Y.; Zhang, H.; Dong, S.; Teng, L.; Lee, R.J.; Yang, Z. Cell-Penetrating Peptides in Diagnosis and Treatment of Human Diseases: From Preclinical Research to Clinical Application. Front. Pharmacol. 2020, 11, 697. [Google Scholar] [CrossRef] [PubMed]
- Staecker, H.; Jokovic, G.; Karpishchenko, S.; Kienle-Gogolok, A.; Krzyzaniak, A.; Der Lin, C.; Navratil, P.; Tzvetkov, V.; Wright, N.; Meyer, T. Efficacy and safety of AM-111 in the treatment of acute unilateral sudden deafness—A double-blind, randomized, placebo-controlled phase 3 study. Otol. Neurotol. 2019, 40, 584–594. [Google Scholar] [CrossRef] [PubMed]
- Chiquet, C.; Aptel, F.; Creuzot-Garcher, C.; Berrod, J.P.; Kodjikian, L.; Massin, P.; Deloche, C.; Perino, J.; Kirwan, B.A.; de Brouwer, S.; et al. Postoperative Ocular Inflammation: A Single Subconjunctival Injection of XG-102 Compared to Dexamethasone Drops in a Randomized Trial. Am. J. Ophthalmol. 2017, 174, 76–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miampamba, M.; Liu, J.; Harootunian, A.; Gale, A.J.; Baird, S.; Chen, S.L.; Nguyen, Q.T.; Tsien, R.Y.; González, J.E. Sensitive in vivo Visualization of Breast Cancer Using Ratiometric Protease-activatable Fluorescent Imaging Agent, AVB-620. Theranostics 2017, 7, 3369. [Google Scholar] [CrossRef] [PubMed]
- Flynn, C.R.; Cheung-flynn, J.; Smoke, C.C.; Lowry, D.; Roberson, R.; Sheller, M.R.; Brophy, C.M. Internalization and Intracellular Trafficking of a PTD-Conjugated Anti-Fibrotic Peptide, AZX100, in Human Dermal Keloid Fibroblasts. J. Pharm. Sci. 2010, 99, 3100–3121. [Google Scholar] [CrossRef]
- Kinali, M.; Arechavala-Gomeza, V.; Feng, L.; Cirak, S.; Hunt, D.; Adkin, C.; Guglieri, M.; Ashton, E.; Abbs, S.; Nihoyannopoulos, P.; et al. Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: A single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 2009, 8, 918. [Google Scholar] [CrossRef] [Green Version]
- Klein, A.F.; Varela, M.A.; Arandel, L.; Holland, A.; Naouar, N.; Arzumanov, A.; Seoane, D.; Revillod, L.; Bassez, G.; Ferry, A.; et al. Peptide-conjugated oligonucleotides evoke long-lasting myotonic dystrophy correction in patient-derived cells and mice. J. Clin. Investig. 2019, 129, 4739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coriat, R.; Faivre, S.J.; Mir, O.; Dreyer, C.; Ropert, S.; Bouattour, M.; Desjardins, R.; Goldwasser, F.; Raymond, E. Pharmacokinetics and safety of DTS-108, a human oligopeptide bound to SN-38 with an esterase-sensitive cross-linker in patients with advanced malignancies: A Phase I study. Int. J. Nanomed. 2016, 11, 6207–6216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyaji, Y.; Walter, S.; Chen, L.; Kurihara, A.; Ishizuka, T.; Saito, M.; Kawai, K.; Okazaki, O. Distribution of KAI-9803, a Novel δ-Protein Kinase C Inhibitor, after Intravenous Administration to Rats. Drug Metab. Dispos. 2011, 39, 1946–1953. [Google Scholar] [CrossRef]
- Müller, R.; Misund, K.; Holien, T.; Bachke, S.; Gilljam, K.M.; Våtsveen, T.K.; Rø, T.B.; Bellacchio, E.; Sundan, A.; Otterlei, M. Targeting proliferating cell nuclear antigen and its protein interactions induces apoptosis in multiple myeloma cells. PLoS ONE 2013, 8, e70430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guergnon, J.; Dessauge, F.; Dominguez, V.; Viallet, J.; Bonnefoy, S.; Yuste, V.J.; Mercereau-Puijalon, O.; Cayla, X.; Rebollo, A.; Susin, S.A.; et al. Use of Penetrating Peptides Interacting with PP1/PP2A Proteins As a General Approach for a Drug Phosphatase Technology. Mol. Pharmacol. 2006, 69, 1115–1124. [Google Scholar] [CrossRef] [Green Version]
- Kumthekar, P.; Tang, S.C.; Brenner, A.J.; Kesari, S.; Piccioni, D.E.; Anders, C.; Carrillo, J.; Chalasani, P.; Kabos, P.; Puhalla, S.; et al. ANG1005, a Brain-Penetrating Peptide-Drug Conjugate, Shows Activity in Patients with Breast Cancer with Leptomeningeal Carcinomatosis and Recurrent Brain Metastases. Clin. Cancer Res. 2020, 26, 2789–2799. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide or Conjugate | Sequence | Effect of Modification on a | Ref. | |
|---|---|---|---|---|
| Uptake | Stability | |||
| R9 | RRRRRRRRR | [103,108] | ||
| r9 | rrrrrrrrr | +/− | + | [103] |
| hLF | KCFQWQRNMRKVRGPPVSCIKR | [103] | ||
| penetratin | RQIKIWFQNRRKWKK | [103,108] | ||
| L-dfTat | 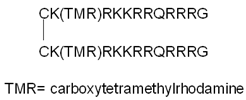 | + | − (compared to D-dfTat) | [104] |
| D-dfTat |  | − (compared to L-dfTat) | + | [104] |
| PepFect14 | Stearyl–AGYLLGKLLOOLAAAALOOLL | + | + | [107] |
| PF144 | 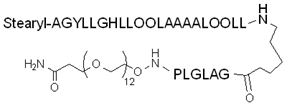 | (different tissue distribution) | N.D. | [109] |
| NLS–StAx–h |  | + (relative to StAx) | N.D. | [110] |
| [WR]5 |  | + | + | [111] |
| cyclic TAT (for conjugation) | 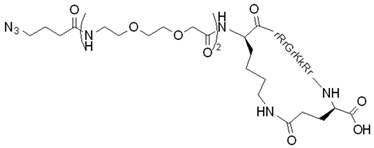 | + | + | [112] |
| CPP12 | 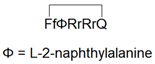 | + | + | [113] |
| H6R6 | HHHHHHRRRRRR | N.D. | N.D. | [114] |
| REDV–TAT–NLS–H12 | REDVYGRKKRRQRRRPKKKRKVHHHHHHHHHHHH | + | N.D. | [115] |
| Stearyl–TP10 | Stearyl–AGYLLGKINLKALAALAKKIL | + | N.D. | [108] |
| NickFect51 | 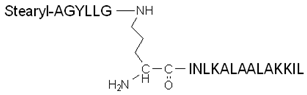 | + | + | [116] |
| PepFect6 |  | + | + | [117] |
| R9–INF7–RFP | RRRRRRRRRGLFEAIEGFIENGWEGMIDGWYG-mCherry | + | N.D. | [118] |
| TAT–CM18 | KWKLFKKIGAVLKVLTTGYGRKKRRQRRRC-atto633 | + | N.D. | [119] |
| EB1 | LIRLWSHLIHIWFQNRRLKWKKK | + | N.D. | [120] |
| GFPβ11–TAT–PEG(6)–GWWG/GFWFG |  | + | N.D. | [121] |
| F3 | KDEPQRRSARLSAKPAPPKPEPKPKKAPAKK | [122] | ||
| TGN | TGNYKALHPHNG | [123,124] | ||
| Angiopep-2 | TFFYGGSRGKRNNFKTEEY | [125] | ||
| ACPP (MMP-2-activated) | 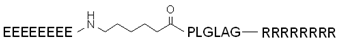 | [125] | ||
| RP4F | 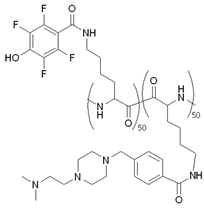 | [126] | ||
| TH | AGYLLGHINLHHLAHL(Aib)HHIL | N.D. | N.D. | [127] |
| TAT–IL-24–KDEL | YGRKKRRQRRR-IL24-KDEL | [128] | ||
| DPA–R8 | 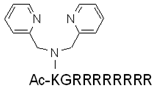 | + | N.D. | [129] |
| HR9 | CHHHHHRRRRRRRRRHHHHHC | + | N.D. | [130,131] |
| HL6 | CHHHHHRRWQWRHHHHHC | + | N.D. | [130] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szabó, I.; Yousef, M.; Soltész, D.; Bató, C.; Mező, G.; Bánóczi, Z. Redesigning of Cell-Penetrating Peptides to Improve Their Efficacy as a Drug Delivery System. Pharmaceutics 2022, 14, 907. https://doi.org/10.3390/pharmaceutics14050907
Szabó I, Yousef M, Soltész D, Bató C, Mező G, Bánóczi Z. Redesigning of Cell-Penetrating Peptides to Improve Their Efficacy as a Drug Delivery System. Pharmaceutics. 2022; 14(5):907. https://doi.org/10.3390/pharmaceutics14050907
Chicago/Turabian StyleSzabó, Ildikó, Mo’ath Yousef, Dóra Soltész, Csaba Bató, Gábor Mező, and Zoltán Bánóczi. 2022. "Redesigning of Cell-Penetrating Peptides to Improve Their Efficacy as a Drug Delivery System" Pharmaceutics 14, no. 5: 907. https://doi.org/10.3390/pharmaceutics14050907
APA StyleSzabó, I., Yousef, M., Soltész, D., Bató, C., Mező, G., & Bánóczi, Z. (2022). Redesigning of Cell-Penetrating Peptides to Improve Their Efficacy as a Drug Delivery System. Pharmaceutics, 14(5), 907. https://doi.org/10.3390/pharmaceutics14050907