
A Physiologically Based Pharmacokinetic and Pharmacodynamic Model of the CYP3A4 Substrate Felodipine for Drug–Drug Interaction Modeling

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Software
2.2. Clinical Data
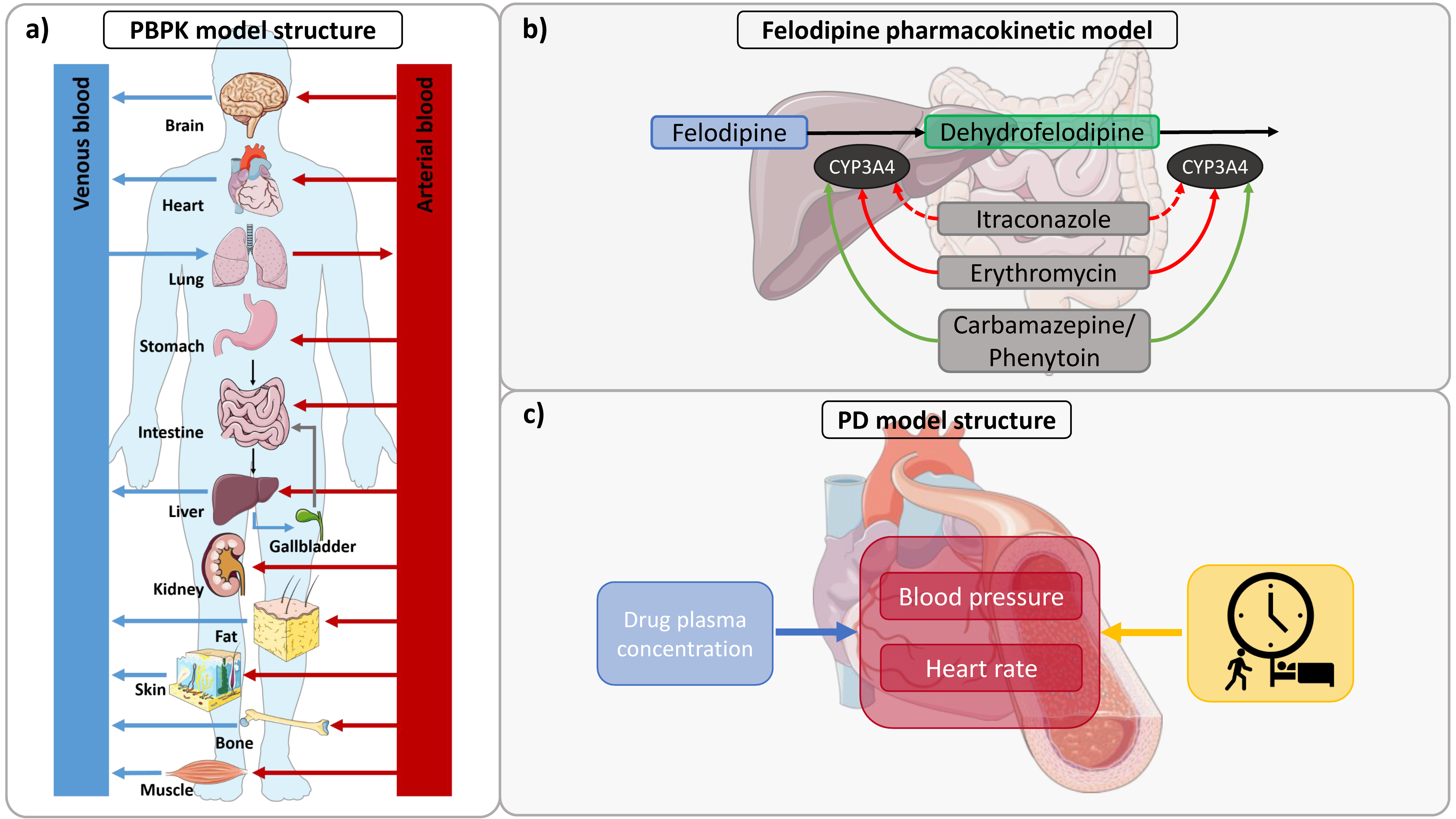
2.3. PBPK Model Building
2.4. PD Model Building
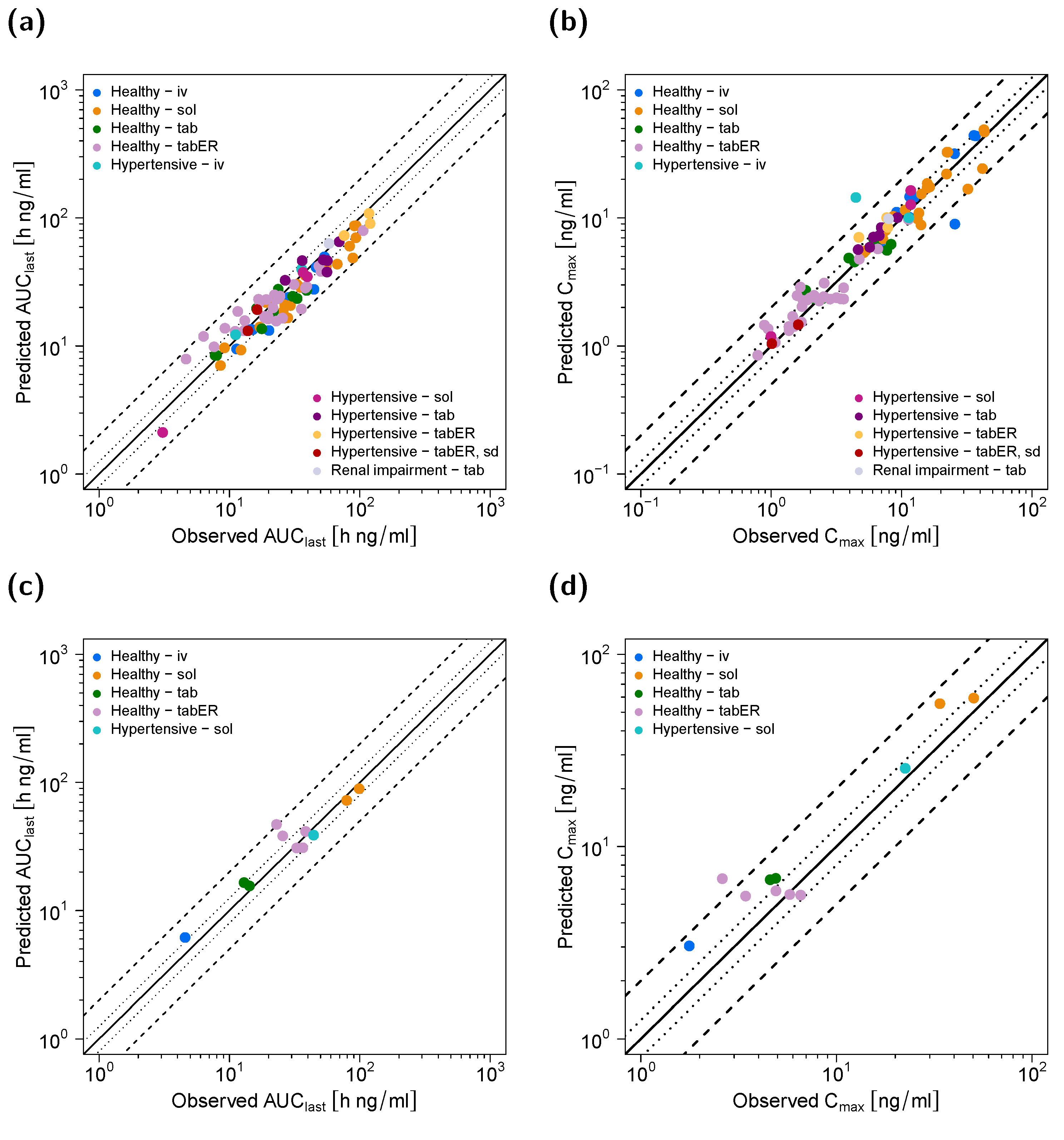
2.5. Model Evaluation
2.6. DDI Modeling
3. Results
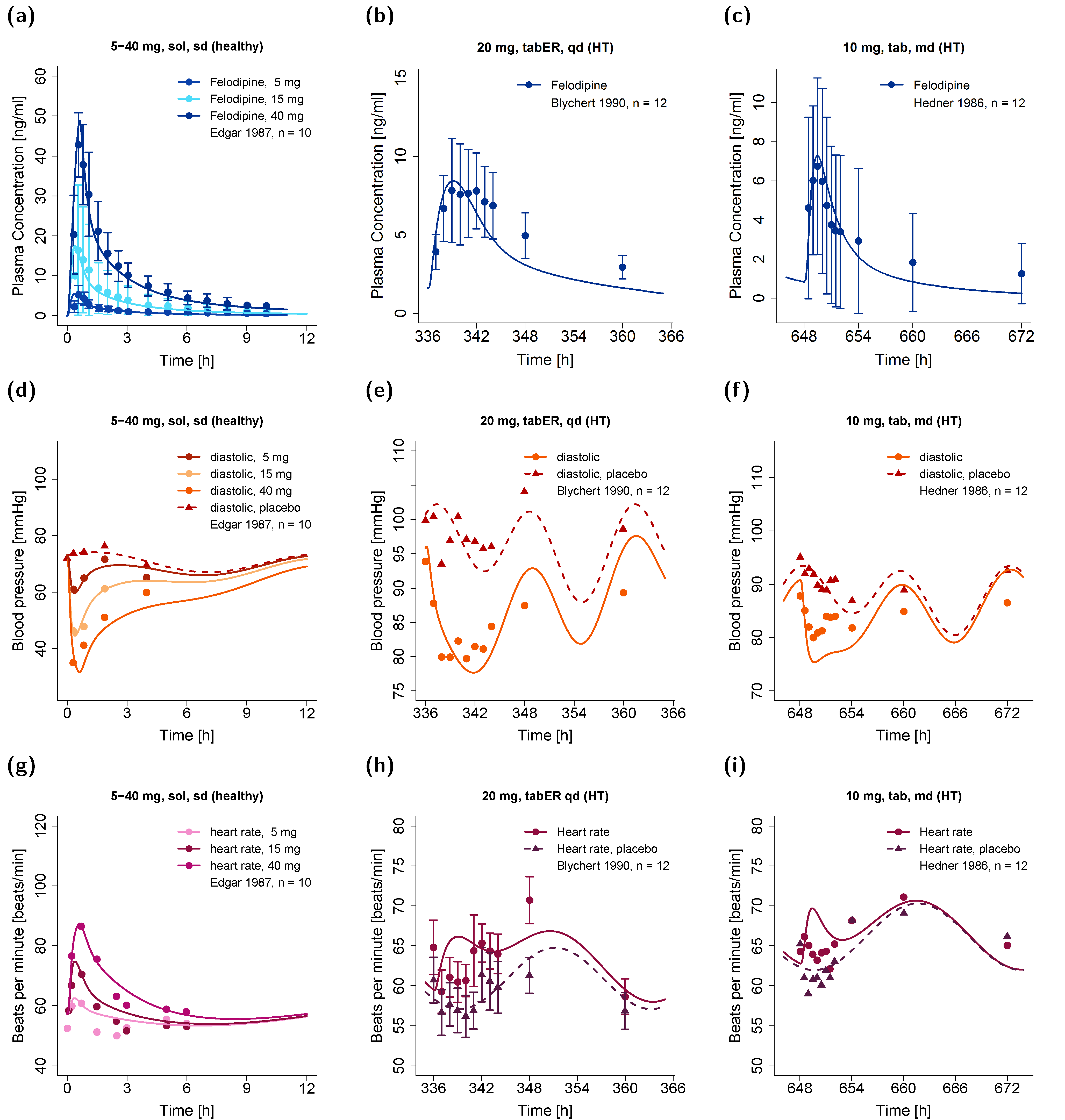
3.1. Pharmacokinetic Model
3.2. Pharmacodynamic Model
3.3. DDI Modeling
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- AstraZeneca LP. Plendil (Felodipine). Extended-Release Tablets. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/019834s025lbl.pdf (accessed on 26 October 2021).
- Heumann Pharma. Felodipin Retard Heumann. Available online: https://www.heumann.de/fileadmin/user_upload/produkte/infos/Fachinformation-Felodipin-retard-Heumann.pdf (accessed on 26 October 2021).
- Williams, B.; Mancia, G.; Spiering, W.; Agabiti Rosei, E.; Azizi, M.; Burnier, M.; Clement, D.L.; Coca, A.; de Simone, G.; Dominiczak, A.; et al. 2018 ESC/ESH Guidelines for the management of arterial hypertension. Eur. Heart J. 2018, 39, 3021–3104. [Google Scholar] [CrossRef] [PubMed]
- Blychert, E.; Edgar, B.; Elmfeldt, D.; Hedner, T. Plasma concentration—Effect relationships for felodipine: A meta analysis. Clin. Pharmacol. Ther. 1992, 52, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson-Hasselgren, B.; Edgar, B.; Rönn, O. Dose-dependent effects of felodipine on diuresis and natriuresis in healthy subjects. J. Cardiovasc. Pharmacol. 1988, 12, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Benet, L.Z.; Broccatelli, F.; Oprea, T.I. BDDCS applied to over 900 drugs. AAPS J. 2011, 13, 519–547. [Google Scholar] [CrossRef] [Green Version]
- Edgar, B.; Lundborg, P.; Regårdh, C.G. Clinical pharmacokinetics of felodipine. A summary. Drugs 1987, 34, 16–27. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. Drug Development and Drug Interactions. Table of Substrates, Inhibitors and Inducers. Available online: https://www.fda.gov/drugs/drug-interactions-labeling/drug-development-and-drug-interactions-table-substrates-inhibitors-and-inducers (accessed on 31 March 2022).
- Walsky, R.L.; Obach, R.S. Validated assays for human cytochrome P450 activities. Drug Metab. Dispos. 2004, 32, 647–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritz, A.; Busch, D.; Lapczuk, J.; Ostrowski, M.; Drozdzik, M.; Oswald, S. Expression of clinically relevant drug-metabolizing enzymes along the human intestine and their correlation to drug transporters and nuclear receptors: An intra-subject analysis. Basic Clin. Pharmacol. Toxicol. 2019, 124, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Lundahl, J.; Regårdh, C.G.; Edgar, B.; Johnsson, G. Effects of grapefruit juice ingestion-pharmacokinetics and haemodynamics of intravenously and orally administered felodipine in healthy men. Eur. J. Clin. Pharmacol. 1997, 52, 139–145. [Google Scholar] [CrossRef]
- Neuvonen, P.J.; Suhonen, R. Itraconazole interacts with felodipine. J. Am. Acad. Dermatol. 1995, 33, 134–135. [Google Scholar] [CrossRef]
- Jalava, K.M.; Olkkola, K.T.; Neuvonen, P.J. Itraconazole greatly increases plasma concentrations and effects of felodipine. Clin. Pharmacol. Ther. 1997, 61, 410–415. [Google Scholar] [CrossRef]
- Bailey, D.G.; Bend, J.R.; Arnold, J.M.; Tran, L.T.; Spence, J.D. Erythromycin-felodipine interaction: Magnitude, mechanism, and comparison with grapefruit juice. Clin. Pharmacol. Ther. 1996, 60, 25–33. [Google Scholar] [CrossRef]
- Capewell, S.; Freestone, S.; Critchley, J.A.J.H.; Pottage, A.; Prescott, L.F. Reduced felodipine bioavailability in patients taking anticonvulsants. Lancet 1988, 2, 480–482. [Google Scholar] [CrossRef]
- European Medicines Agency. Guideline on the Investigation of Drug Interactions. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-drug-interactions-revision-1_en.pdf (accessed on 12 July 2022).
- European Medicines Agency. Guideline on the Reporting of Physiologically Based Pharmacokinetic (PBPK) Modelling and Simulation. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-reporting-physiologically-based-pharmacokinetic-pbpk-modelling-simulation_en.pdf (accessed on 12 July 2022).
- Zhang, X.; Yang, Y.; Grimstein, M.; Fan, J.; Grillo, J.A.; Huang, S.-M.; Zhu, H.; Wang, Y. Application of PBPK Modeling and Simulation for Regulatory Decision Making and Its Impact on US Prescribing Information: An Update on the 2018-2019 Submissions to the US FDA’s Office of Clinical Pharmacology. J. Clin. Pharmacol. 2020, 60 (Suppl. 1), S160–S178. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. Physiologically Based Pharmacokinetic Analyses—Format and Content. Available online: https://www.fda.gov/media/101469/download (accessed on 12 July 2022).
- Mitchell, M.; Muftakhidinov, B.; Winchen, T. Engauge Digitizer Software. Available online: http://markummitchell.github.io/engauge-digitizer (accessed on 22 March 2022).
- Wojtyniak, J.; Britz, H.; Selzer, D.; Schwab, M.; Lehr, T. Data Digitizing: Accurate and Precise Data Extraction for Quantitative Systems Pharmacology and Physiologically-Based Pharmacokinetic Modeling. CPT Pharmacomet. Syst. Pharmacol. 2020, 9, 322–331. [Google Scholar] [CrossRef] [PubMed]
- National Center for Health Statistics Hyattsville MD 20782. Third National Health and Nutrition Examination Survey, (NHANES III). Available online: http://www.cdc.gov/nchs/nhanes.htm (accessed on 12 July 2022).
- Valentin, J. Basic anatomical and physiological data for use in radiological protection: Reference values. A report of age- and gender-related differences in the anatomical and physiological characteristics of reference individuals. ICRP Publication 89. Ann. ICRP 2002, 32, 5–265. [Google Scholar] [CrossRef]
- Tanaka, G.; Kawamura, H. Anatomical and Physiological Characteristics for Asian Reference Man: Male and Female of Different Ages: Tanaka Model; Division of Radioecology; Report Number: NIRS-M-115; National Institute of Radiological Sciences: Hitachinaka, Japan, 1996. [Google Scholar]
- Schlender, J.F.; Meyer, M.; Thelen, K.; Krauss, M.; Willmann, S.; Eissing, T.; Jaehde, U. Development of a Whole-Body Physiologically Based Pharmacokinetic Approach to Assess the Pharmacokinetics of Drugs in Elderly Individuals. Clin. Pharm. 2016, 55, 1573–1589. [Google Scholar] [CrossRef] [Green Version]
- Open Systems Pharmacology Suite Community PK-Sim® Ontogeny Database Documentation, Version 7.3. Available online: https://github.com/Open-Systems-Pharmacology/OSPSuite.Documentation/blob/master/PK-Sim Ontogeny Database Version 7.3.pdf (accessed on 12 July 2022).
- Weitschies, W.; Wedemeyer, R.-S.; Kosch, O.; Fach, K.; Nagel, S.; Söderlind, E.; Trahms, L.; Abrahamsson, B.; Mönnikes, H. Impact of the intragastric location of extended release tablets on food interactions. J. Control. Release 2005, 108, 375–385. [Google Scholar] [CrossRef]
- Chae, D.; Kim, Y.; Park, K. Characterization of circadian blood pressure patterns using non-linear mixed effects modeling. Transl Clin. Pharmacol. 2019, 27, 24–32. [Google Scholar] [CrossRef] [Green Version]
- Lott, D.; Lehr, T.; Dingemanse, J.; Krause, A. Modeling Tolerance Development for the Effect on Heart Rate of the Selective S1P1 Receptor Modulator Ponesimod. Clin. Pharmacol. Ther. 2018, 103, 1083–1092. [Google Scholar] [CrossRef]
- Frechen, S.; Dallmann, A.; Solodenko, J. Building and Evaluation of a PBPK Model for Erythromycin in Healthy Adults. Available online: https://github.com/Open-Systems-Pharmacology/OSP-PBPK-Model-Library/blob/master/Erythromycin/Erythromycin_evaluation_report.md (accessed on 14 March 2022).
- Hanke, N.; Frechen, S.; Moj, D.; Britz, H.; Eissing, T.; Wendl, T.; Lehr, T. PBPK Models for CYP3A4 and P-gp DDI prediction: A modeling network of rifampicin, itraconazole, clarithromycin, midazolam, alfentanil, and digoxin. CPT Pharmacomet. Syst. Pharmacol. 2018, 7, 647–659. [Google Scholar] [CrossRef] [Green Version]
- Fuhr, L.M.; Marok, F.Z.; Hanke, N.; Selzer, D.; Lehr, T. Pharmacokinetics of the CYP3A4 and CYP2B6 Inducer Carbamazepine and Its Drug-Drug Interaction Potential: A Physiologically Based Pharmacokinetic Modeling Approach. Pharmaceutics 2021, 13, 270. [Google Scholar] [CrossRef] [PubMed]
- Guest, E.J.; Aarons, L.; Houston, J.B.; Rostami-Hodjegan, A.; Galetin, A. Critique of the two-fold measure of prediction success for ratios: Application for the assessment of drug-drug interactions. Drug Metab. Dispos. 2011, 39, 170–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, B.; Bengtsson, B.; Elmfeldt, D.; Lundborg, P.; Nyberg, G.; Raner, S.; Rönn, O. Acute diuretic/natriuretic properties of felodipine in man. Drugs 1985, 29, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Edgar, B.; Regårdh, C.G.; Johnsson, G.; Johansson, L.; Lundborg, P.; Löfberg, I.; Rönn, O. Felodipine kinetics in healthy men. Clin. Pharmacol. Ther. 1985, 38, 205–211. [Google Scholar] [CrossRef]
- Edgar, B.; Regårdh, C.G.; Lundborg, P.; Romare, S.; Nyberg, G.; Rönn, O. Pharmacokinetic and pharmacodynamic studies of felodipine in healthy subjects after various single, oral and intravenous doses. Biopharm. Drug Dispos. 1987, 8, 235–248. [Google Scholar] [CrossRef]
- Sluiter, H.E.; Huysmans, F.T.; Thien, T.A.; Koene, R.A.P. Haemodynamic effects of intravenous felodipine in normotensive and hypertensive subjects. Drugs 1985, 29, 144–153. [Google Scholar] [CrossRef]
- Sutfin, T.A.; Lind, T.; Gabrielsson, M.; Regårdh, C.G. Biliary secretion of felodipine metabolites in man after intravenous [14C]felodipine. Eur. J. Clin. Pharmacol. 1990, 38, 421–424. [Google Scholar] [CrossRef]
- Soons, P.A.; Mulders, T.M.T.; Uchida, E.; Schoemaker, H.C.; Cohen, A.F.; Breimer, D.D. Stereoselective pharmacokinetics of oral felodipine and nitrendipine in healthy subjects: Correlation with nifedipine pharmacokinetics. Eur. J. Clin. Pharmacol. 1993, 44, 163–169. [Google Scholar] [CrossRef]
- Abrahamsson, B.; Johansson, D.; Torstensson, A.; Wingstrand, K. Evaluation of solubilizers in the drug release testing of hydrophilic matrix extended-release tablets of felodipine. Pharm. Res. 1994, 11, 1093–1097. [Google Scholar] [CrossRef]
- Bengtsson-Hasselgren, B.; Rönn, O.; Blychert, L.O.; Edgar, B.; Raner, S. Acute effects of felodipine and nifedipine on hepatic and forearm blood flow in healthy men. Eur. J. Clin. Pharmacol. 1990, 38, 529–533. [Google Scholar] [CrossRef]
- Johnsson, G.; Murray, G.; Tweddel, A.; Hutton, I. Haemodynamic effects of a new vasodilator drug, felodipine, in healthy subjects. Eur. J. Clin. Pharmacol. 1983, 24, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Wingstrand, K.; Abrahamsson, B.; Edgar, B. Bioavailability from felodipine extended-release tablets with different dissolution properties. Int. J. Pharm. 1990, 60, 151–156. [Google Scholar] [CrossRef]
- Soons, P.A.; Roosemalen, M.C.M.; Breimer, D.D. Enantioselective determination of felodipine and other chiral dihydropyridine calcium entry blockers in human plasma. J. Chromatogr. 1990, 528, 343–356. [Google Scholar] [CrossRef]
- Blychert, E.; Wingstrand, K.; Edgar, B.; Lidman, K. Plasma concentration profiles and antihypertensive effect of conventional and extended-release felodipine tablets. Br. J. Clin. Pharmacol 1990, 29, 39–45. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.-Q.; Chen, Q.-Y.; Wang, X.; Liu, Y.-X.; Chu, X.-M.; Cao, X.-M.; Li, J.-H.; Yamazoe, Y. Different roles of pummelo furanocoumarin and cytochrome P450 3A5*3 polymorphism in the fate and action of felodipine. Curr. Drug Metab. 2007, 8, 623–630. [Google Scholar] [CrossRef]
- Hardy, B.G.; Bartle, W.R.; Myers, M.; Bailey, D.G.; Edgar, B. Effect of indomethacin on the pharmacokinetics and pharmacodynamics of felodipine. Br. J. Clin. Pharmacol. 1988, 26, 557–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, D.G.; Arnold, J.M.; Munoz, C.; Spence, J.D. Grapefruit juice-felodipine interaction: Mechanism, predictability, and effect of naringin. Clin. Pharmacol. Ther. 1993, 53, 637–642. [Google Scholar] [CrossRef]
- Edgar, B.; Bailey, D.; Bergstrand, R.; Johnsson, G.; Regårdh, C.G. Acute effects of drinking grapefruit juice on the pharmacokinetics and dynamics of felodipine-and its potential clinical relevance. Eur. J. Clin. Pharmacol. 1992, 42, 313–317. [Google Scholar] [CrossRef]
- Sakamoto, T.; Ohtake, Y.; Itoh, M.; Tabata, S.; Kuriki, T.; Uno, K. Determination of felodipine enantiomers using chiral stationary phase liquid chromatography and gas chromatography/mass spectrometry, and the study of their pharmacokinetic profiles in human and dog. Biomed. Chromatogr. 1993, 7, 99–103. [Google Scholar] [CrossRef]
- Landahl, S.; Edgar, B.; Gabrielsson, M.; Larsson, M.; Lernfelt, B.; Lundborg, P.; Regårdh, C.G. Pharmacokinetics and blood pressure effects of felodipine in elderly hypertensive patients. A comparison with young healthy subjects. Clin. Pharm. 1988, 14, 374–383. [Google Scholar] [CrossRef]
- Smith, S.R.; Wilkins, M.R.; Jack, D.B.; Kendall, M.J.; Laugher, S. Pharmacokinetic interactions between felodipine and metoprolol. Eur. J. Clin. Pharmacol. 1987, 31, 575–578. [Google Scholar] [CrossRef] [PubMed]
- Goosen, T.C.; Cillié, D.; Bailey, D.G.; Yu, C.; He, K.; Hollenberg, P.F.; Woster, P.M.; Cohen, L.; Williams, J.A.; Rheeders, M.; et al. Bergamottin contribution to the grapefruit juice-felodipine interaction and disposition in humans. Clin. Pharmacol. Ther. 2004, 76, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Hasselgren, B.; Rönn, O.; Edgar, B.; Johansson, P.; Wall, B. Pharmacokinetics and hemodynamic and diuretic/natriuretic effects of felodipine administered as an extended-release tablet. Cardiovasc. Drugs Ther. 1990, 4, 1495–1500. [Google Scholar] [CrossRef]
- Lown, K.S.; Bailey, D.G.; Fontana, R.J.; Janardan, S.K.; Adair, C.H.; Fortlage, L.A.; Brown, M.B.; Guo, W.; Watkins, P.B. Grapefruit juice increases felodipine oral availability in humans by decreasing intestinal CYP3A protein expression. J. Clin. Investig. 1997, 99, 2545–2553. [Google Scholar] [CrossRef]
- Lundahl, J.; Regårdh, C.G.; Edgar, B.; Johnsson, G. Relationship between time of intake of grapefruit juice and its effect on pharmacokinetics and pharmacodynamics of felodipine in healthy subjects. Eur. J. Clin. Pharmacol. 1995, 49, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Lundahl, J.U.E.; Regårdh, C.G.; Edgar, B.; Johnsson, G. The interaction effect of grapefruit juice is maximal after the first glass. Eur. J. Clin. Pharmacol. 1998, 54, 75–81. [Google Scholar] [CrossRef]
- Patel Devang, S.; Shanker, N.; Shah Sweety, K.; Thakkar Vaishali, K.; Mehta Nirali, N.; Srivstava Ambrish, K.; Singh, S.; Patel Chitrang, G. Bioequivalence study of two oral extended release formulations of felodipine 10 mg tablets in healthy volunteers under fed condition. Pharma Sci. Monit. Int. J. Pharm. Sci. 2011, 2, 9–20. [Google Scholar]
- Pop, A.; Vlase, L. Leucuta Pharmacokinetic study of felodipine after single oral dose of slow release formulations in healthy volunteers. Farmacia 2008, 56, 474–482. [Google Scholar]
- Xiang, Q.; Li, C.; Zhao, X.; Cui, Y.M. The influence of CYP3A5*3 and BCRPC421A genetic polymorphisms on the pharmacokinetics of felodipine in healthy Chinese volunteers. J. Clin. Pharm. Ther. 2017, 42, 345–349. [Google Scholar] [CrossRef]
- Bailey, D.G.; Kreeft, J.H.; Munoz, C.; Freeman, D.J.; Bend, J.R. Grapefruit juice-felodipine interaction: Effect of naringin and 6’,7’-dihydroxybergamottin in humans. Clin. Pharmacol. Ther. 1998, 64, 248–256. [Google Scholar] [CrossRef]
- Gelal, A.; Balkan, D.; Ozzeybek, D.; Kaplan, Y.C.; Gurler, S.; Guven, H.; Benowitz, N.L. Effect of menthol on the pharmacokinetics and pharmacodynamics of felodipine in healthy subjects. Eur. J. Clin. Pharmacol. 2005, 60, 785–790. [Google Scholar] [CrossRef] [PubMed]
- Madsen, J.K.; Jensen, J.D.; Jensen, L.W.; Pedersen, E.B. Pharmacokinetic interaction between cyclosporine and the dihydropyridine calcium antagonist felodipine. Eur. J. Clin. Pharmacol. 1996, 50, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Carrasco, J.C.; Carrasco-Portugal, M.d.C.; Flores-Murrieta, F.J.; Canizales-Quinteros, S. Oral pharmacokinetics of felodipine in mexican healthy volunteers: Evidence for interethnic differences. Int. J. Pharmacol. 2015, 11, 382–386. [Google Scholar] [CrossRef] [Green Version]
- Bailey, D.G.; Arnold, J.M.; Bend, J.R.; Tran, L.T.; Spence, J.D. Grapefruit juice-felodipine interaction: Reproducibility and characterization with the extended release drug formulation. Br. J. Clin. Pharmacol. 1995, 40, 135–140. [Google Scholar]
- Bailey, D.G.; Dresser, G.K.; Kreeft, J.H.; Munoz, C.; Freeman, D.J.; Bend, J.R. Grapefruit-felodipine interaction: Effect of unprocessed fruit and probable active ingredients. Clin. Pharmacol. Ther. 2000, 68, 468–477. [Google Scholar] [CrossRef]
- Bailey, D.G.; Dresser, G.K.; Bend, J.R. Bergamottin, lime juice, and red wine as inhibitors of cytochrome P450 3A4 activity: Comparison with grapefruit juice. Clin. Pharmacol. Ther. 2003, 73, 529–537. [Google Scholar] [CrossRef]
- Dresser, G.K.; Bailey, D.G.; Carruthers, S.G. Grapefruit juice-felodipine interaction in the elderly. Clin. Pharmacol. Ther. 2000, 68, 28–34. [Google Scholar] [CrossRef]
- Dresser, G.K.; Urquhart, B.L.; Proniuk, J.; Tieu, A.; Freeman, D.J.; Arnold, J.M.; Bailey, D.G. Coffee inhibition of CYP3A4 in vitro was not translated to a grapefruit-like pharmacokinetic interaction clinically. Pharmacol. Res. Perspect. 2017, 5, e00346. [Google Scholar] [CrossRef]
- A Comparative Study on the Relative Bioavailability of 2.5 and 5mg ER Tablets of Felodipine. 19834. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/pre96/19834-S002_PLENDIL TABLETS_BIOEQR.PDF (accessed on 22 April 2022).
- Blychert, E.; Hedner, T.; Dahlöf, C.; Elmfeldt, D. Plasma concentration-effect relationships of intravenous and extended-release oral felodipine in hypertensive patients. J. Cardiovasc. Pharmacol. 1990, 15, 428–435. [Google Scholar] [CrossRef]
- Edgar, B.; Regårdh, C.G.; Attman, P.O.; Aurell, M.; Herlitz, H.; Johnsson, G. Pharmacokinetics of felodipine in patients with impaired renal function. Br. J. Clin. Pharmacol. 1989, 27, 67–74. [Google Scholar] [CrossRef] [Green Version]
- Larsson, R.; Karlberg, B.E.; Gelin, A.; Aberg, J.; Regårdh, C.-G. Acute and steady-state pharmacokinetics and antihypertensive effects of felodipine in patients with normal and impaired renal function. J. Clin. Pharmacol. 1990, 30, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Hedner, T.; Samuelsson, O.; Sjögren, E.; Elmfeldt, D. Treatment of essential hypertension with felodipine in combination with a diuretic. Eur. J. Clin. Pharmacol. 1986, 30, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Hedner, T.; Elmfeldt, D.; Dahlöf, C.; Sjögren, E. Comparison of antihypertensive effect and pharmacokinetics of conventional and extended release felodipine tablets in patients with arterial hypertension. Drugs 1987, 34, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Leenen, F.H.H.; Coletta, E. Pharmacokinetic and antihypertensive profile of amlodipine and felodipine-ER in younger versus older patients with hypertension. J. Cardiovasc. Pharmacol. 2010, 56, 669–675. [Google Scholar] [CrossRef]
- Les Laboratoires Servier. Servier Medical Art. Available online: https://smart.servier.com/ (accessed on 14 June 2022).
- Human Metabolome Database. Metabocard for Felodipine (HMDB0015158). Available online: https://hmdb.ca/metabolites/HMDB0015158 (accessed on 28 March 2022).
- Regårdh, C.G.; Edgar, B.; Olsson, R.; Kendall, M.; Collste, P.; Shansky, C. Pharmacokinetics of felodipine in patients with liver disease. Eur. J. Clin. Pharmacol. 1989, 36, 473–479. [Google Scholar] [CrossRef]
- Felle, K.; Persson, B.; Vessman, J. Dissolution test for felodipine tablets using chemical oxidation in situ to maintain ‘sink conditions’. J. Pharm Biomed. Anal. 1984, 2, 527–536. [Google Scholar] [CrossRef]
- Takano, J.; Maeda, K.; Bolger, M.B.; Sugiyama, Y. The Prediction of the Relative Importance of CYP3A/P-glycoprotein to the Nonlinear Intestinal Absorption of Drugs by Advanced Compartmental Absorption and Transit Model. Drug Metab. Dispos. 2016, 44, 1808–1818. [Google Scholar] [CrossRef]
- DRUGBANK Online-Felodipine. Available online: https://go.drugbank.com/drugs/DB01023 (accessed on 28 March 2022).
- Van der Lee, R.; Pfaffendorf, M.; Koopmans, R.P.; van Lieshout, J.J.; van Montfrans, G.A.; van Zwieten, P.A. Comparison of the time courses and potencies of the vasodilator effects of nifedipine and felodipine in the human forearm. Blood Press 2001, 10, 217–222. [Google Scholar] [CrossRef]
- Berben, P.; Brouwers, J.; Augustijns, P. Assessment of Passive Intestinal Permeability Using an Artificial Membrane Insert System. J. Pharm. Sci. 2018, 107, 250–256. [Google Scholar] [CrossRef] [Green Version]
- Galetin, A.; Clarke, S.E.; Houston, J.B. Quinidine and haloperidol as modifiers of CYP3A4 activity: Multisite kinetic model approach. Drug Metab. Dispos. 2002, 30, 1512–1522. [Google Scholar] [CrossRef] [Green Version]
- Chemaxon. Chemicalize-Dehydrofelodipine. Available online: https://chemicalize.com (accessed on 28 March 2022).
- Gertz, M.; Houston, J.B.; Galetin, A. Physiologically based pharmacokinetic modeling of intestinal first-pass metabolism of CYP3A substrates with high intestinal extraction. Drug Metab. Dispos. 2011, 39, 1633–1642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heikkinen, A.T.; Baneyx, G.; Caruso, A.; Parrott, N. Application of PBPK modeling to predict human intestinal metabolism of CYP3A substrates-An evaluation and case study using GastroPlusTM. Eur. J. Pharm. Sci. 2012, 47, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Jamei, M.; Abrahamsson, B.; Brown, J.; Bevernage, J.; Bolger, M.B.; Heimbach, T.; Karlsson, E.; Kotzagiorgis, E.; Lindahl, A.; McAllister, M.; et al. Current status and future opportunities for incorporation of dissolution data in PBPK modeling for pharmaceutical development and regulatory applications: OrBiTo consortium commentary. Eur. J. Pharm. Biopharm. 2020, 155, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Wade, J.R.; Sambol, N.C. Felodipine population dose-response and concentration-response relationships in patients with essential hypertension. Clin. Pharmacol. Ther. 1995, 57, 569–581. [Google Scholar] [CrossRef]
- Blychert, E.; Edgar, B.; Elmfeldt, D.; Hedner, T. A population study of the pharmacokinetics of felodipine. Br. J. Clin. Pharmacol. 1991, 31, 15–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oparil, S.; Acelajado, M.C.; Bakris, G.L.; Berlowitz, D.R.; Cífková, R.; Dominiczak, A.F.; Grassi, G.; Jordan, J.; Poulter, N.R.; Rodgers, A.; et al. Hypertension. Nat. Rev. Dis. Prim. 2018, 4, 18014. [Google Scholar] [CrossRef] [Green Version]
- Muller, D.N.; Kvakan, H.; Luft, F.C. Immune-related effects in hypertension and target-organ damage. Curr. Opin. Nephrol. Hypertens. 2011, 20, 113–117. [Google Scholar] [CrossRef]
- De Jong, L.M.; Jiskoot, W.; Swen, J.J.; Manson, M.L. Distinct Effects of Inflammation on Cytochrome P450 Regulation and Drug Metabolism: Lessons from Experimental Models and a Potential Role for Pharmacogenetics. Genes 2020, 11, 1509. [Google Scholar] [CrossRef]
- Zhao, P.; Vieira, M.D.L.T.; Grillo, J.A.; Song, P.; Wu, T.-C.; Zheng, J.H.; Arya, V.; Berglund, E.G.; Atkinson, A.J.; Sugiyama, Y.; et al. Evaluation of Exposure Change of Nonrenally Eliminated Drugs in Patients With Chronic Kidney Disease Using Physiologically Based Pharmacokinetic Modeling and Simulation. J. Clin. Pharmacol 2012, 52, 91–108. [Google Scholar] [CrossRef]
- Elmfeldt, D.; Hedner, T. Antihypertensive effects of felodipine compared with placebo. Drugs 1985, 29, 109–116. [Google Scholar] [CrossRef]
- Beck, K.R.; Thompson, G.R.; Odermatt, A. Drug-induced endocrine blood pressure elevation. Pharmacol Res. 2020, 154, 104311. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, W.J.; McHardy, I.; Thompson, G.R. Itraconazole induced hypertension and hypokalemia: Mechanistic evaluation. Mycoses 2018, 61, 337–339. [Google Scholar] [CrossRef] [PubMed]
- Liedholm, H.; Nordin, G. Erythromycin-Felodipine Interaction. DICP 1991, 25, 1007–1008. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Route | Dose (mg) | n (PK) | n (DBP) | n (HR) | Reference |
|---|---|---|---|---|---|
| Healthy Individuals | |||||
| iv | 1–3 | 9 | 1 | 1 | [5,11,34,35,36,37,38] |
| sol, sd | 5–40 | 15 | 8 | 8 | [5,7,34,35,36,39,40,41,42,43,44] |
| sol, md | 10 | 1 | 0 | 0 | [45] |
| tab, sd | 5–10 | 4 | 1 | 2 | [7,46,47,48,49,50] |
| tab, md | 5–10 | 4 | 0 | 0 | [45,51,52] |
| tabER, sd | 5–40 | 24 | 7 | 6 | [7,11,13,14,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69] |
| tabER, md | 5–10 | 6 | 1 | 1 | [45,57,68,70] |
| Profiles (healthy) | 63 | 18 | 18 | ||
| Hypertensive Individuals | |||||
| iv | 1–2.25 | 2 | 1 | 1 | [71,72] |
| sol, sd | 0.83–10 | 3 | 2 | 0 | [34,72] |
| tab, sd | 10 | 2 | 2 | 0 | [73] |
| tab, md | 5–10 | 5 | 5 | 1 | [51,73,74,75] |
| tabER, sd | 20 | 3 | 1 | 1 | [71,76] |
| tabER, md | 20 | 4 | 1 | 1 | [71,75,76] |
| Profiles (hypertensive) | 19 | 12 | 4 | ||
| Profiles (total) | 82 | 30 | 22 | ||
| Parameter | Unit | Model | Literature | Reference | Description |
|---|---|---|---|---|---|
| Felodipine | |||||
| MW | g/mol | 384.25 | 384.25 | [78] | Molecular weight |
| fu, plasma | % | 0.36 | 0.36 | [79] | fraction unbound in plasma |
| Solubility (pH) | mg/L | 7.15 (6.5) | 1.2 (7) 7.15 (6.5) 14.3 (7.1) 19.7 (7) | [80,81,82] | Solubility at reference pH |
| Solubility-tabER (pH) | mg/L | 0.89 (7) | - | - | Solubility at reference pH used for extended-release tablets |
| logP | - | 4.36 | 3.44 3.80 4.36 4.46 4.64 | [78,81,82,83] | Lipophilicity |
| Intestinal permeability | cm/min | 2.76 × 10−4 | 4.42 × 10−4 3.06 × 10−4 2.64 × 10−4 | [81,84] | Transcellular intestinal permeability |
| GFR fraction | - | 1 | - | - | Fraction of filtered drug in the urine |
| Km-CYP3A4 | µmol/L | 2.81 | 0.94 2.81 26.4 | [9,81,85] | CYP3A4 Michaelis–Menten constant |
| kcat-CYP3A4 | 1/min | 250.44 | - | - | CYP3A4 catalytic rate constant |
| Weibull time-tab | min | 54.86 | - | - | Tablet dissolution profile shape |
| Weibull shape-tab | - | 1.32 | - | - | Tablet dissolution time (50% dissolved) |
| Weibull time-tabER | min | 173.04 | - | - | Extended-release tablet dissolution profile shape |
| Weibull shape-tabER | - | 1.30 | - | - | Extended-release tablet dissolution time (50% dissolved) |
| Partition coefficient | - | Diverse | RR | Cell to plasma partition coefficients | |
| Cellular permeability | cm/min | 0.42 | PK-Sim | Permeability into the cellular space | |
| Dehydrofelodipine | |||||
| MW | g/mol | 382.24 | 382.24 | [86] | Molecular weight |
| pKa (base) | - | 4.06 | 4.06 | [86] | Acid dissociation constant |
| fu, plasma | % | 0.68 | - | - | fraction unbound in plasma |
| Solubility (pH) | mg/L | 2.93 | 2.93 | [86] | Solubility at reference pH |
| logP | - | 3.32 | 4.24 | [86] | Lipophilicity |
| Intestinal permeability | cm/min | 1.38 × 10−4 | estimated via PK-Sim® | Transcellular intestinal permeability | |
| GFR fraction | - | 1 | - | - | Fraction of filtered drug in the urine |
| CL-CYP3A4 | 1/min | 35.74 | - | - | CYP3A4-mediated clearance |
| CL-hepatic | 1/min | 2.76 | - | - | Unspecific hepatic clearance |
| Partition coefficient | - | Diverse | S | Cell to plasma partition coefficients | |
| Cellular permeability | cm/min | 0.04 | CDS | Permeability into the cellular space | |
| Parameter | Unit | Model | Literature | Reference | Description |
|---|---|---|---|---|---|
| Diastolic Blood Pressure Model | |||||
| Emax | mmHg | 56.18 | - | - | Maximum effect on diastolic blood pressure |
| EC50 | µmol/L | 0.04 | - | - | Concentration for half-maximal effect |
| amp24 | % | 2.14 | 2.14 | [28] | Amplitude for 24 h period |
| amp12 | % | 5.93 | 5.93 | [28] | Amplitude for 12 h period |
| phase24 | h | 5.28 (2.10) a | - | - | Phase for 24 h period |
| phase12 | h | 0.16 (2.10) a | - | - | Phase for 12 h period |
| BPmean | mmHg | 70.4 (6.76) a; 91.9 (6.60) a,b | 69.5 | [28] | Mean diastolic blood pressure over 24 h |
| Heart Rate Model | |||||
| Emax | bpm | 39.71 | - | - | Maximum effect on heart rate |
| EC50 | µmol/L | 0.05 | - | - | Concentration for half-maximal effect |
| h | - | 1.40 | - | - | Hill coefficient |
| amp | % | 6.3 | 6.3 | [29] | Amplitude |
| phase | h | 11.8 (5.13) a | 9.2 | [29] | Phase |
| HRmean | bpm | 62.2 (4.21) a; 66.4 (3.32) a,b | 66.2 | [29] | Mean heart rate over 24 h |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fuhr, L.M.; Marok, F.Z.; Mees, M.; Mahfoud, F.; Selzer, D.; Lehr, T. A Physiologically Based Pharmacokinetic and Pharmacodynamic Model of the CYP3A4 Substrate Felodipine for Drug–Drug Interaction Modeling. Pharmaceutics 2022, 14, 1474. https://doi.org/10.3390/pharmaceutics14071474
Fuhr LM, Marok FZ, Mees M, Mahfoud F, Selzer D, Lehr T. A Physiologically Based Pharmacokinetic and Pharmacodynamic Model of the CYP3A4 Substrate Felodipine for Drug–Drug Interaction Modeling. Pharmaceutics. 2022; 14(7):1474. https://doi.org/10.3390/pharmaceutics14071474
Chicago/Turabian StyleFuhr, Laura Maria, Fatima Zahra Marok, Maximilian Mees, Felix Mahfoud, Dominik Selzer, and Thorsten Lehr. 2022. "A Physiologically Based Pharmacokinetic and Pharmacodynamic Model of the CYP3A4 Substrate Felodipine for Drug–Drug Interaction Modeling" Pharmaceutics 14, no. 7: 1474. https://doi.org/10.3390/pharmaceutics14071474
APA StyleFuhr, L. M., Marok, F. Z., Mees, M., Mahfoud, F., Selzer, D., & Lehr, T. (2022). A Physiologically Based Pharmacokinetic and Pharmacodynamic Model of the CYP3A4 Substrate Felodipine for Drug–Drug Interaction Modeling. Pharmaceutics, 14(7), 1474. https://doi.org/10.3390/pharmaceutics14071474