
Activation of NLRP3 Is Required for a Functional and Beneficial Microglia Response after Brain Trauma

 ,
,  ,
, 
Abstract
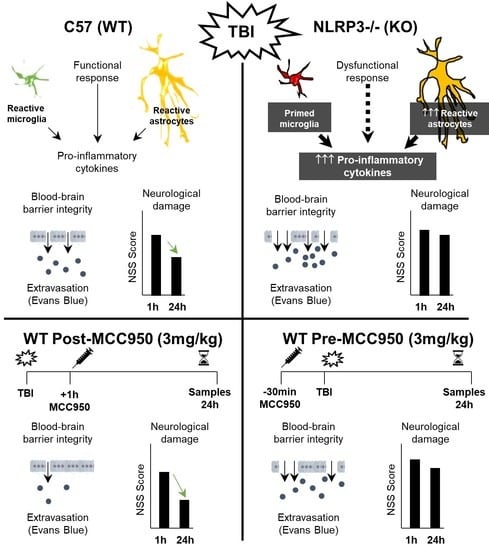
:
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Traumatic Brain Injury Model
2.3. Neurological Severity Score (NSS) Test
2.4. Treatments
2.5. Blood–Brain Barrier Integrity Assessment
2.6. Tissue Preparation
2.7. RNA and Protein Extraction
2.8. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
2.9. Western Blot
2.10. Statistical Analyses
3. Results
3.1. Inflammatory Profile in WT Mice 24 h after TBI
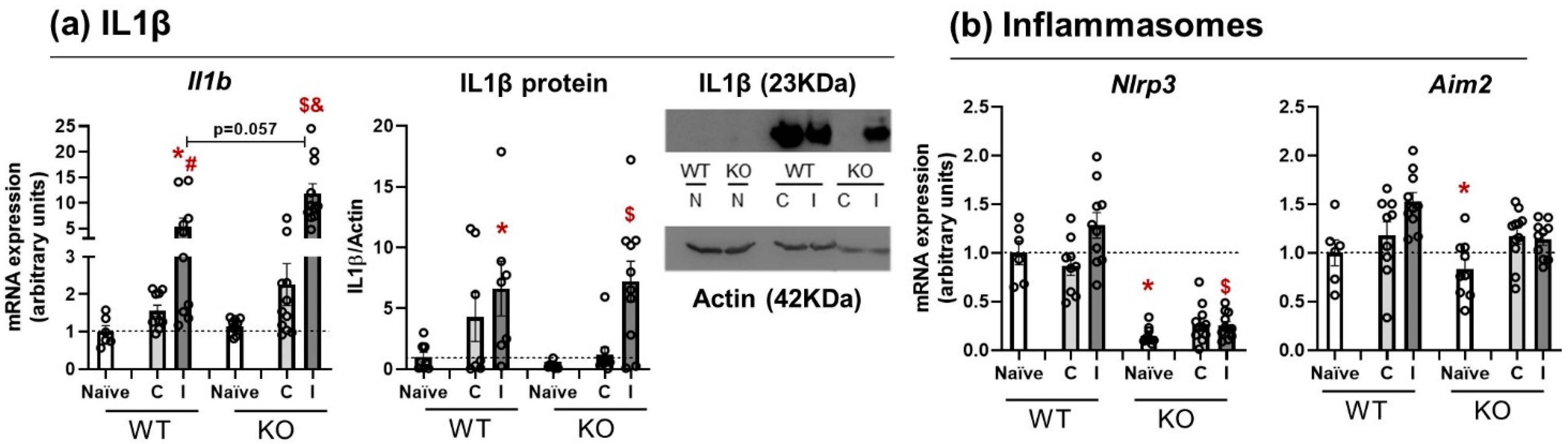
3.2. Il1β mRNA Expression and Protein Release Are Equal in WT and NLRP3 KO Mice
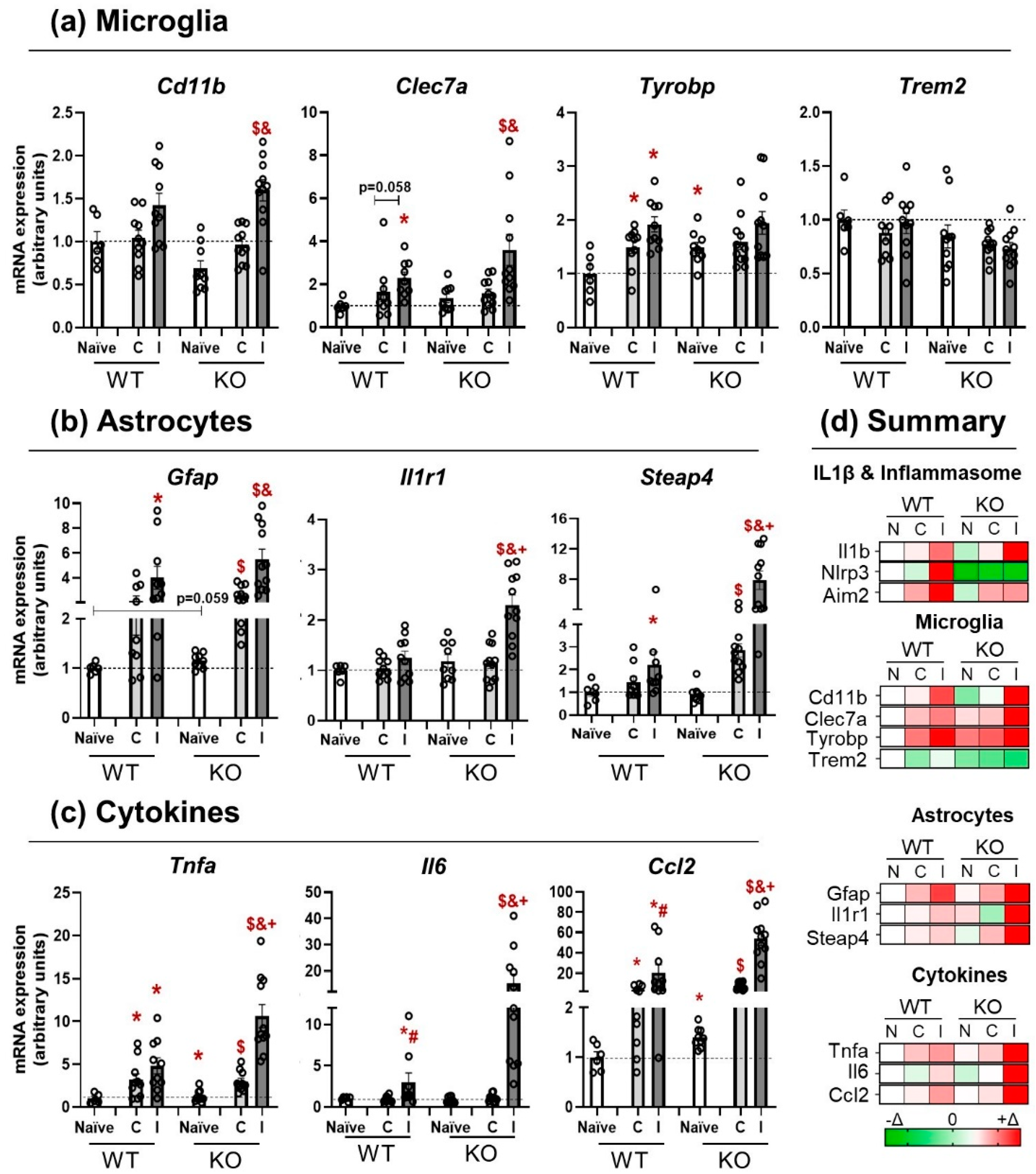
3.3. NLRP3 KO Mice Showed an Exaggerated Inflammatory Response after TBI
3.4. Activation of NLRP3 Is Critical for Blood–Brain Barrier Integrity and Neurological Damage
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- James, S.L.; Bannick, M.S.; Montjoy-Venning, W.C.; Lucchesi, L.R.; Dandona, L.; Dandona, R.; Hawley, C.; Hay, S.I.; Jakovljevic, M.; Khalil, I.; et al. Global, Regional, and National Burden of Traumatic Brain Injury and Spinal Cord Injury, 1990–2016: A Systematic Analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 56–87. [Google Scholar] [CrossRef] [Green Version]
- Hiskens, M.I. Targets of Neuroprotection and Review of Pharmacological Interventions in Traumatic Brain Injury. J. Pharmacol. Exp. Ther. 2022, 382, 149–166. [Google Scholar] [CrossRef]
- Capizzi, A.; Woo, J.; Verduzco-Gutierrez, M. Traumatic Brain Injury: An Overview of Epidemiology, Pathophysiology, and Medical Management. Med. Clin. N. Am. 2020, 104, 213–238. [Google Scholar] [CrossRef]
- Yang, Q.; Zhou, J. Neuroinflammation in the Central Nervous System: Symphony of Glial Cells. Glia 2019, 67, 1017–1035. [Google Scholar] [CrossRef] [PubMed]
- Hickey, W.F.; Kimura, H. Perivascular Microglial Cells of the CNS Are Bone Marrow-Derived and Present Antigen In Vivo. Science 1988, 239, 290–292. [Google Scholar] [CrossRef] [PubMed]
- Jha, M.K.; Jo, M.; Kim, J.H.; Suk, K. Microglia-Astrocyte Crosstalk: An Intimate Molecular Conversation. Neuroscientist 2019, 25, 227–240. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.; Shi, L.; Wang, Y.; Chen, S.; Zhang, J. Recent Advances of the NLRP3 Inflammasome in Central Nervous System Disorders. J. Immunol. Res. 2016, 2016, 9238290. [Google Scholar] [CrossRef]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and Regulation of the Inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef]
- Liu, H.D.; Li, W.; Chen, Z.R.; Hu, Y.C.; Zhang, D.D.; Shen, W.; Zhou, M.L.; Zhu, L.; Hang, C.H. Expression of the NLRP3 Inflammasome in Cerebral Cortex after Traumatic Brain Injury in a Rat Model. Neurochem. Res. 2013, 38, 2072–2083. [Google Scholar] [CrossRef]
- Johann, S.; Heitzer, M.; Kanagaratnam, M.; Goswami, A.; Rizo, T.; Weis, J.; Troost, D.; Beyer, C. NLRP3 Inflammasome Is Expressed by Astrocytes in the SOD1 Mouse Model of ALS and in Human Sporadic ALS Patients. Glia 2015, 63, 2260–2273. [Google Scholar] [CrossRef]
- De Rivero Vaccari, J.P.; Lotocki, G.; Alonso, O.F.; Bramlett, H.M.; Dietrich, W.D.; Keane, R.W. Therapeutic Neutralization of the NLRP1 Inflammasome Reduces the Innate Immune Response and Improves Histopathology after Traumatic Brain Injury. J. Cereb. Blood Flow Metab. 2009, 29, 1251–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuwar, R.; Rolfe, A.; Di, L.; Xu, H.; He, L.; Jiang, Y.; Zhang, S.; Sun, D. A Novel Small Molecular NLRP3 Inflammasome Inhibitor Alleviates Neuroinflammatory Response Following Traumatic Brain Injury. J. Neuroinflamm. 2019, 16, 81. [Google Scholar] [CrossRef] [PubMed]
- Ismael, S.; Nasoohi, S.; Ishrat, T. MCC950, the Selective Inhibitor of Nucleotide Oligomerization Domain-Like Receptor Protein-3 Inflammasome, Protects Mice against Traumatic Brain Injury. J. Neurotrauma 2018, 35, 1294–1303. [Google Scholar] [CrossRef] [Green Version]
- Palomino-Antolin, A.; Narros-Fernández, P.; Farré-Alins, V.; Sevilla-Montero, J.; Decouty-Pérez, C.; Lopez-Rodriguez, A.B.; Fernández, N.; Monge, L.; Casas, A.I.; Calzada, M.J.; et al. Time-Dependent Dual Effect of NLRP3 Inflammasome in Brain Ischaemia. Br. J. Pharmacol. 2022, 179, 1395–1410. [Google Scholar] [CrossRef]
- A Flierl, M.; Stahel, P.F.; Beauchamp, K.M.; Morgan, S.J.; Smith, W.R.; Shohami, E. Mouse Closed Head Injury Model Induced by a Weight-Drop Device. Nat. Protoc. 2009, 4, 1328–1337. [Google Scholar] [CrossRef] [PubMed]
- Homsi, S.; Federico, F.; Croci, N.; Palmier, B.; Plotkine, M.; Marchand-Leroux, C.; Jafarian-Tehrani, M. Minocycline Effects on Cerebral Edema: Relations with Inflammatory and Oxidative Stress Markers Following Traumatic Brain Injury in Mice. Brain Res. 2009, 1291, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Homsi, S.; Piaggio, T.; Croci, N.; Noble, F.; Plotkine, M.; Marchand-Leroux, C.; Jafarian-Tehrani, M. Blockade of Acute Microglial Activation by Minocycline Promotes Neuroprotection and Reduces Locomotor Hyperactivity after Closed Head Injury in Mice: A Twelve-Week Follow-up Study. J. Neurotrauma 2010, 27, 911–921. [Google Scholar] [CrossRef]
- Siopi, E.; Llufriu-Dabén, G.; Fanucchi, F.; Plotkine, M.; Marchand-Leroux, C.; Jafarian-Tehrani, M. Evaluation of Late Cognitive Impairment and Anxiety States Following Traumatic Brain Injury in Mice: The Effect of Minocycline. Neurosci. Lett. 2012, 511, 110–115. [Google Scholar] [CrossRef]
- Lopez-Rodriguez, A.B.; Siopi, E.; Finn, D.P.; Marchand-Leroux, C.; Garcia-Segura, L.M.; Jafarian-Tehrani, M.; Viveros, M.-P.M.-P. CB1 and CB2 Cannabinoid Receptor Antagonists Prevent Minocycline-Induced Neuroprotection Following Traumatic Brain Injury in Mice. Cereb. Cortex 2015, 25, 35–45. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Yin, D.; Ren, H.; Gao, W.; Li, F.; Sun, D.; Wu, Y.; Zhou, S.; Lyu, L.; Yang, M.; et al. Selective NLRP3 Inflammasome Inhibitor Reduces Neuroinflammation and Improves Long-Term Neurological Outcomes in a Murine Model of Traumatic Brain Injury. Neurobiol. Dis. 2018, 117, 15–27. [Google Scholar] [CrossRef]
- Witcher, K.G.; Bray, C.E.; Dziabis, J.E.; McKim, D.B.; Benner, B.N.; Rowe, R.K.; Kokiko-Cochran, O.N.; Popovich, P.G.; Lifshitz, J.; Eiferman, D.S.; et al. Traumatic Brain Injury-Induced Neuronal Damage in the Somatosensory Cortex Causes Formation of Rod-Shaped Microglia That Promote Astrogliosis and Persistent Neuroinflammation. Glia 2018, 66, 2719. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.W.; McGeachy, M.J.; Bayır, H.; Clark, R.S.B.; Loane, D.J.; Kochanek, P.M. The Far-Reaching Scope of Neuroinflammation after Traumatic Brain Injury. Nat. Rev. Neurol. 2017, 13, 572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, W.T.; Pham, L.; Symons, G.F.; Monif, M.; Shultz, S.R.; McDonald, S.J. The NLRP3 Inflammasome in Traumatic Brain Injury: Potential as a Biomarker and Therapeutic Target. J. Neuroinflamm. 2020, 17, 104. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Li, W.; Huang, S.; Yin, Z.; Xu, X.; Chen, F.; Kong, X.; Wang, H.; Zhang, J.; Lei, P. The Pathological Role of NLRs and AIM2 Inflammasome-Mediated Pyroptosis in Damaged Blood-Brain Barrier after Traumatic Brain Injury. Brain Res. 2018, 1697, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Mahmood, A.; Chopp, M. Animal Models of Traumatic Brain Injury. Nat. Rev. Neurosci. 2013, 14, 128–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaidt, M.M.; Hornung, V. Alternative Inflammasome Activation Enables IL-1β Release from Living Cells. Curr. Opin. Immunol. 2017, 44, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Afonina, I.S.; Tynan, G.A.; Logue, S.E.; Cullen, S.P.; Bots, M.; Lüthi, A.U.; Reeves, E.P.; McElvaney, N.G.; Medema, J.P.; Lavelle, E.C.; et al. Granzyme B-Dependent Proteolysis Acts as a Switch to Enhance the Proinflammatory Activity of IL-1α. Mol. Cell 2011, 44, 265–278. [Google Scholar] [CrossRef] [Green Version]
- Feltham, R.; Vince, J.E.; Lawlor, K.E. Caspase-8: Not so Silently Deadly. Clin. Transl. Immunol. 2017, 6, e124. [Google Scholar] [CrossRef]
- Maelfait, J.; Beyaert, R. Non-Apoptotic Functions of Caspase-8. Biochem. Pharmacol. 2008, 76, 1365–1373. [Google Scholar] [CrossRef]
- Gringhuis, S.I.; Kaptein, T.M.; Wevers, B.A.; Theelen, B.; Van Der Vlist, M.; Boekhout, T.; Geijtenbeek, T.B.H. Dectin-1 Is an Extracellular Pathogen Sensor for the Induction and Processing of IL-1β via a Noncanonical Caspase-8 Inflammasome. Nat. Immunol. 2012, 13, 246–254. [Google Scholar] [CrossRef]
- Krajewska, M.; You, Z.; Rong, J.; Kress, C.; Huang, X.; Yang, J.; Kyoda, T.; Leyva, R.; Banares, S.; Hu, Y.; et al. Neuronal Deletion of Caspase 8 Protects against Brain Injury in Mouse Models of Controlled Cortical Impact and Kainic Acid-Induced Excitotoxicity. PLoS ONE 2011, 6, e24341. [Google Scholar] [CrossRef] [PubMed]
- Beer, R.; Franz, G.; Krajewski, S.; Pike, B.R.; Hayes, R.L.; Reed, J.C.; Wang, K.K.; Klimmer, C.; Schmutzhard, E.; Poewe, W.; et al. Temporal and Spatial Profile of Caspase 8 Expression and Proteolysis after Experimental Traumatic Brain Injury. J. Neurochem. 2001, 78, 862–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorente, L.; Martín, M.M.; Pérez-Cejas, A.; González-Rivero, A.F.; Ramos-Gómez, L.; Solé-Violán, J.; Cáceres, J.J.; Ferrer-Moure, C.; Jiménez, A. Low Blood Caspase-8 Levels in Survivor Patients of Traumatic Brain Injury. Neurol. Sci. 2021, 42, 5065–5070. [Google Scholar] [CrossRef] [PubMed]
- Pyrillou, K.; Burzynski, L.C.; Clarke, M.C.H. Alternative Pathways of IL-1 Activation, and Its Role in Health and Disease. Front. Immunol. 2020, 11, 3288. [Google Scholar] [CrossRef] [PubMed]
- Jurga, A.M.; Paleczna, M.; Kuter, K.Z. Overview of General and Discriminating Markers of Differential Microglia Phenotypes. Front. Cell. Neurosci. 2020, 14, 198. [Google Scholar] [CrossRef]
- Hickman, S.E.; Kingery, N.D.; Ohsumi, T.K.; Borowsky, M.L.; Wang, L.C.; Means, T.K.; El Khoury, J. The Microglial Sensome Revealed by Direct RNA Sequencing. Nat. Neurosci. 2013, 16, 1896–1905. [Google Scholar] [CrossRef] [Green Version]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef]
- Audrain, M.; Haure-Mirande, J.V.; Mleczko, J.; Wang, M.; Griffin, J.K.; St George-Hyslop, P.H.; Fraser, P.; Zhang, B.; Gandy, S.; Ehrlich, M.E. Reactive or Transgenic Increase in Microglial TYROBP Reveals a TREM2-Independent TYROBP–APOE Link in Wild-Type and Alzheimer’s-Related Mice. Alzheimer’s Dement. 2021, 17, 149–163. [Google Scholar] [CrossRef]
- Zhang, B.; Gaiteri, C.; Bodea, L.G.; Wang, Z.; McElwee, J.; Podtelezhnikov, A.A.; Zhang, C.; Xie, T.; Tran, L.; Dobrin, R.; et al. Integrated Systems Approach Identifies Genetic Nodes and Networks in Late-Onset Alzheimer’s Disease. Cell 2013, 153, 707–720. [Google Scholar] [CrossRef] [Green Version]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic Reactive Astrocytes Are Induced by Activated Microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Barres, B.A. Not Everything Is Scary about a Glial Scar. Nature 2016, 532, 182–183. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.; Dunne, A.; Lopez-Rodriguez, A.B. Astrocytes: Heterogeneous and Dynamic Phenotypes in Neurodegeneration and Innate Immunity. Neuroscientist 2019, 25, 455–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, M.A.; Burda, J.E.; Ren, Y.; Ao, Y.; O’Shea, T.M.; Kawaguchi, R.; Coppola, G.; Khakh, B.S.; Deming, T.J.; Sofroniew, M.V. Astrocyte Scar Formation Aids Central Nervous System Axon Regeneration. Nature 2016, 532, 195–200. [Google Scholar] [CrossRef] [Green Version]
- Clarke, L.E.; Liddelow, S.A.; Chakraborty, C.; Münch, A.E.; Heiman, M.; Barres, B.A. Normal Aging Induces A1-like Astrocyte Reactivity. Proc. Natl. Acad. Sci. USA 2018, 115, E1896–E1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Béchade, C.; Cantaut-Belarif, Y.; Bessis, A. Microglial Control of Neuronal Activity. Front. Cell. Neurosci. 2013, 7, 32. [Google Scholar] [CrossRef] [Green Version]
- Clark, A.K.; Gruber-Schoffnegger, D.; Drdla-Schutting, R.; Gerhold, K.J.; Malcangio, M.; Sandkühler, J. Selective Activation of Microglia Facilitates Synaptic Strength. J. Neurosci. 2015, 35, 4552–4570. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.K.; Hayashi, H.; Ishikawa, T.; Shibata, K.; Shigetomi, E.; Shinozaki, Y.; Inada, H.; Roh, S.E.; Kim, S.J.; Lee, G.; et al. Cortical Astrocytes Rewire Somatosensory Cortical Circuits for Peripheral Neuropathic Pain. J. Clin. Investig. 2016, 126, 1983–1997. [Google Scholar] [CrossRef]
- Nikolakopoulou, A.M.; Koeppen, J.; Garcia, M.; Leish, J.; Obenaus, A.; Ethell, I.M. Astrocytic Ephrin-B1 Regulates Synapse Remodeling Following Traumatic Brain Injury. ASN Neurol. 2016, 8, 1759091416630220. [Google Scholar] [CrossRef] [Green Version]
- Pascual, O.; Ben Achour, S.; Rostaing, P.; Triller, A.; Bessis, A. Microglia Activation Triggers Astrocyte-Mediated Modulation of Excitatory Neurotransmission. Proc. Natl. Acad. Sci. USA 2012, 109, E197–E205. [Google Scholar] [CrossRef] [Green Version]
- Perez, E.J.; Tapanes, S.A.; Loris, Z.B.; Balu, D.T.; Sick, T.J.; Coyle, J.T.; Liebl, D.J. Enhanced Astrocytic D-Serine Underlies Synaptic Damage after Traumatic Brain Injury. J. Clin. Investig. 2017, 127, 3114–3125. [Google Scholar] [CrossRef]
- Villapol, S.; Byrnes, K.R.; Symes, A.J. Temporal Dynamics of Cerebral Blood Flow, Cortical Damage, Apoptosis, Astrocyte-Vasculature Interaction and Astrogliosis in the Pericontusional Region after Traumatic Brain Injury. Front. Neurol. 2014, 5, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosa, J.M.; Farré-Alins, V.; Ortega, M.C.; Navarrete, M.; Lopez-Rodriguez, A.B.; Palomino-Antolín, A.; Fernández-López, E.; Vila-del Sol, V.; Decouty, C.; Narros-Fernández, P.; et al. TLR4 Pathway Impairs Synaptic Number and Cerebrovascular Functions through Astrocyte Activation Following Traumatic Brain Injury. Br. J. Pharmacol. 2021, 178, 3395–3413. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wise, L.; Fukuchi, K.I. TLR4 Cross-Talk With NLRP3 Inflammasome and Complement Signaling Pathways in Alzheimer’s Disease. Front. Immunol. 2020, 11, 724. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.V.; Deng, M.; Ting, J.P.Y. The NLRP3 Inflammasome: Molecular Activation and Regulation to Therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Quintas, C.; Vale, N.; Gonçalves, J.; Queiroz, G. Microglia P2Y13 Receptors Prevent Astrocyte Proliferation Mediated by P2Y1 Receptors. Front. Pharmacol. 2018, 9, 418. [Google Scholar] [CrossRef]
- Sun, H.; Liang, R.; Yang, B.; Zhou, Y.; Liu, M.; Fang, F.; Ding, J.; Fan, Y.; Hu, G. Aquaporin-4 Mediates Communication between Astrocyte and Microglia: Implications of Neuroinflammation in Experimental Parkinson’s Disease. Neuroscience 2016, 317, 65–75. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Bergamini, G.; Rajendran, L. Cell-to-Cell Communication by Extracellular Vesicles: Focus on Microglia. Neuroscience 2019, 405, 148–157. [Google Scholar] [CrossRef]
- De Vries, H.E.; Blom-Roosemalen, M.C.M.; Van Oosten, M.; De Boer, A.G.; Van Berkel, T.J.C.; Breimer, D.D.; Kuiper, J. The Influence of Cytokines on the Integrity of the Blood-Brain Barrier in Vitro. J. Neuroimmunol. 1996, 64, 37–43. [Google Scholar] [CrossRef]
- Zhang, J.; Sadowska, G.B.; Chen, X.; Park, S.Y.; Kim, J.E.; Bodge, C.A.; Cummings, E.; Lim, Y.P.; Makeyev, O.; Besio, W.G.; et al. Anti–IL-6 Neutralizing Antibody Modulates Blood-Brain Barrier Function in the Ovine Fetus. FASEB J. 2015, 29, 1739. [Google Scholar] [CrossRef] [Green Version]
- Dimitrijevic, O.B.; Stamatovic, S.M.; Keep, R.F.; Andjelkovic, A.V. Effects of the Chemokine CCL2 on Blood-Brain Barrier Permeability during Ischemia-Reperfusion Injury. J. Cereb. Blood Flow Metab. 2006, 26, 797–810. [Google Scholar] [CrossRef] [Green Version]
- Chodobski, A.; Zink, B.J.; Szmydynger-Chodobska, J. Blood-Brain Barrier Pathophysiology in Traumatic Brain Injury. Transl. Stroke Res. 2011, 2, 492–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Task | Description |
|---|---|
| Exit circle | Exit a circle of 30 cm od diameter in 2 min |
| Seeking behavior | Interest on the environment and movement |
| Motor skills | Straight walk Monoparesis or hemiparesis |
| Startle reflex | Response to a loud hand clap |
| Beam balancing | Ability to balance on a surface of 10 × 10 mm |
| Beam walk | Cross a 30 cm-long beam of 3 cm-width Cross a 30 cm-long beam of 2 cm-width Cross a 30 cm-long beam of 1 cm-width |
| Round stick balancing | Ability to balance on a 5 mm round stick |
| Gen | Primer Sequences |
|---|---|
| 18S | Fw: 5′-CGCCGCTAGAGGTGAAATTCT-3′ Rv: 5′-CATTCTTGGCAAATGTCTTTCG-3′ |
| Aim2 | Fw: 5′-AAGAGAGCCAGGGAAACTCC-3′ Rv: 5′-CACCTCCATTGTCCCTGTTT-3′ |
| ASC | Fw: 5′-GAAGCTGCTGACAGTGCAAC-3′ Rv: 5′-GAAGAGTCTGGAGCTGTGGC-3′ |
| Casp1 | Fw: 5′-AGATGGCACATTTCCAGGAC-3′ Rv: 5′-GATCCTCCAGCAGCAACTTC-3′ |
| Casp11 | Fw: 5′-ACGATGTGGTGGTGAAAGAGGAGC-3′ Rv: 5′-TGTCTCGGTAGGACAAGTGATGTGG-3′ |
| Ccl2 | Fw: 5′-ACAAGAGGATCACCAGCAGC-3′ Rv: 5′-GGACCCATTCCTTCTTGGGG-3′ |
| Ccl5 | Fw: 5′-GCAGTCGTGTTTGTCACTCGAA-3′ Rv: 5′-GATGTATTCTTGAACCCACTTCTTCTC-3′ |
| Cd11b | Fw: 5′-CCTTGTTCTCTTTGATGCAG-3′ Rv: 5′-GTGATGACAACTAGGATCTT-3′ |
| Cd11c | Fw: 5′-CTGGATAGCCTTTCTTCTGCTG-3′ Rv: 5′-GCACACTGTGTCCGAACTC-3′ |
| Clec7a | Fw: 5′-CCCAACTCGTTTCAAGTCAG-3′ Rv: 5′-AGACCTCTGATCCATGAATCC-3′ |
| Gfap | Fw: 5′-CTCCAACCTCCAGATCCGAG-3′ Rv: 5′-TCCACAGTCTTTACCAGATGT-3′ |
| Iba1 | Fw: 5′-CCGAGGAGACGTTCAGCTAC-3′ Rv: 5′-GACATCCACCTCCAATCAGG-3′ |
| Il18 | Fw: 5′-GACTCTTGCGTCAACTTCAAGG-3′ Rv: 5′-CAGGCTGTCTTTTGTCAACGA-3′ |
| Il1b | Fw: 5′-AACCTGCTGGTGTGTGACGTTC-3′ Rv: 5′-CAGCACGAGGCTTTTTTGTTGT-3′ |
| Il1r1 | Fw: 5′-GCAATATCCGGTCACACGAGTA-3′ Rv: 5′-ATCATTGATCCTGGGTCAGCTT-3′ Probe: 5′-TCCTGAGCCCTCGGAATGAGACGATC-3′ |
| Il6 | Fw: 5′-TTCTCTGGGAAATCGTGGAAA-3′ Rv: 5′-CTGCAAGTGCATCATCGTTGT-3′ |
| Nlrc4 | Fw: 5′-TGGTGACAATAGGGCTCCTC-3′ Rv: 5′-CTGTTCCCTTTGCTCACCTC-3′ |
| Nlrp3 | Fw: 5′-GCCCAAGGAGGAAGAAGAAG-3′ Rv: 5′-TCCGGTTGGTGCTTAGACTT-3′ |
| Steap4 | Fw: 5′-TGCAAGCCGGCAGGTGTTTGT-3′ Rv: 5′-TCCAGTGGGGTGAGCCCAAGA-3′ |
| Tnfa | Fw: 5′-GCCTCTTCTCATTCCTGCTTG-3′ Rv: 5′-CTGATGAGAGGGAGGCCATT-3′ |
| Tyrobp | Fw: 5′-CGTACAGGCCCAGAGTGAC-3′ Rv: 5′-CACCAAGTCACCCAGAACAA-3′ |
| Vimentin | Fw: 5′-GCTGCAGGCCCAGATTCA-3′ Rv: 5′-TTCATACTGCTGGCGCACAT-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lopez-Rodriguez, A.B.; Decouty-Perez, C.; Farré-Alins, V.; Palomino-Antolín, A.; Narros-Fernández, P.; Egea, J. Activation of NLRP3 Is Required for a Functional and Beneficial Microglia Response after Brain Trauma. Pharmaceutics 2022, 14, 1550. https://doi.org/10.3390/pharmaceutics14081550
Lopez-Rodriguez AB, Decouty-Perez C, Farré-Alins V, Palomino-Antolín A, Narros-Fernández P, Egea J. Activation of NLRP3 Is Required for a Functional and Beneficial Microglia Response after Brain Trauma. Pharmaceutics. 2022; 14(8):1550. https://doi.org/10.3390/pharmaceutics14081550
Chicago/Turabian StyleLopez-Rodriguez, Ana Belen, Céline Decouty-Perez, Victor Farré-Alins, Alejandra Palomino-Antolín, Paloma Narros-Fernández, and Javier Egea. 2022. "Activation of NLRP3 Is Required for a Functional and Beneficial Microglia Response after Brain Trauma" Pharmaceutics 14, no. 8: 1550. https://doi.org/10.3390/pharmaceutics14081550
APA StyleLopez-Rodriguez, A. B., Decouty-Perez, C., Farré-Alins, V., Palomino-Antolín, A., Narros-Fernández, P., & Egea, J. (2022). Activation of NLRP3 Is Required for a Functional and Beneficial Microglia Response after Brain Trauma. Pharmaceutics, 14(8), 1550. https://doi.org/10.3390/pharmaceutics14081550