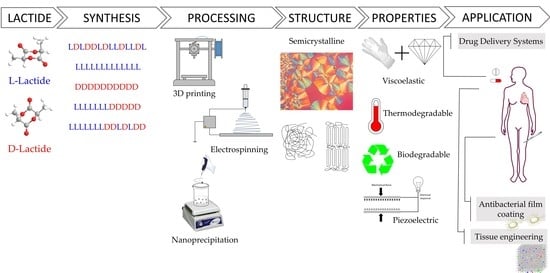
1. Introduction
Biomaterials are natural or engineered substances that interact with components of living systems that can be exploited for a medical purpose, either as therapeutic or diagnostic agents [
1]. The development of novel, customised solutions urged by society to minimise detrimental invasive side effects involves complex multifunctional compounds that feature ambivalent properties. Likewise, the molecular engineered design should consider the processing steps required to generate the final material as well as the mechanism of application to attain a high performance of the targeted activities. Particularly, metals are typically used when mechanical strength or electrical conductivity is required, whilst ceramics exhibit a high compressive strength and relative chemical inertness and polymers possess a great potential due to the chemical flexibility that endows them with a wide range of physical and mechanical properties, as well as activities through their functionalisation [
2]. The use of biopolymers as agents for medical applications dates back thousands of years, when, in ancient India and Egypt, wounds were sutured by using natural polymers such as catgut and silk. Likewise, naturally occurring polymers, such as polysaccharides and proteins, are abundantly available and have been widely used in biomedicine ranging from wound dressing to arterial and skin grafts. However, its application in the field of medicine is limited due to the risk of infections, antigenicity, and batch-to-batch variability [
3]. In contrast, the application of synthetic polymers in medicine was reported more recently during the Second World War, when Nicholas Harold used poly(methyl methacrylate) (PMMA) as an artificial corneal substitute [
4]. Subsequently, several biostable (non-degradable under physiological conditions) synthetic polymers were employed in the biomedical field, such as polyethylene terephthalate (PET) for vascular grafts, polydimethylsiloxane (PDMS) for breast implants, and polyethylene (PE) for hip joint replacements [
3], due to their mass production at an industrial scale that endows them with a low cost, batch-to-batch reproducibility, and flexibility for performing function-specific applications. In addition, hydrolysable polymers such as polylactic acid (PLA) were considered valuable for degradable surgical implants to avoid a subsequent clinical intervention for removing the medical implant [
5]. Since then, the development of biodegradable (under physiological conditions) polymers, such as polyesters, poly-ester-urethanes, polycarbonates, etc., for biomedical applications, such as bone grafts, sutures, and 3D scaffolds, and for pharmaceutical applications such as drug delivery systems or polymer therapeutics has increased exponentially due to their tunable degradation properties, the ease of their processing and administration, as well as their chemical and biological properties that resemble numerous biological tissues [
6,
7].
The design of novel polymeric materials must fulfil the environmental and societal demands to diminish the carbon footprint required to adopt manufacturing strategies that meet the European policies such as “A European Strategy for Plastics in a Circular Economy”, which was launched in 2018 to address how plastics are designed, used, and recycled [
8]. Likewise, the European Parliament has recently recognised the potential role of bioplastics and compostable plastics in the circular economy and sustainability [
9,
10]. Consequently, bio-based plastics synthesised from renewable resources such as PLA are promising environmentally friendly candidates for the development of biomedical and pharmaceutical applications whilst contributing to the circular economy.
PLA is a compostable polymer derived from corn sugar, potato, and sugar cane [
11,
12] whose promising physicochemical properties are comparable to those of petroleum-based polymers, such as PE, polypropylene, polystyrene, polycarbonate, and PET [
13]. PLA is a semicrystalline polymer that hydrolyses in physiological media, yielding lactic acid, a non-toxic component that is eliminated via the Krebs cycle as water and carbon dioxide [
14]. The biocompatibility, biodegradability, and resorbability characteristics of PLA have promoted its use in the biomedical field for a wide range of applications (suture threads, bone fixation screws, drug delivery systems, etc.), offering an alternative to conventional biocompatible materials such as metals and ceramics [
15]. In addition, the ability to tailor the mechanical, thermal, and degradation properties of PLA derivatives due to the range of afforded nanostructures depending on the chemical architecture and processing conditions allows for designing personalised medical solutions. Indeed, the novel available synthetic PLA approaches to generate multiblock copolymers as well as the advent of current processing technologies broaden its suitability to advance into the customisation of the generated end-products to minimise the adverse side effects. In particular, several highly cutting-edge PLA-based therapeutic applications have recently attained the clinical phase, such as drug-eluting stents [
16,
17,
18,
19] as well as personalised pharmaceutical agents that were designed from an interdisciplinary approach to avoid the serendipitous progress, emphasising the beneficial interaction between the materials and the biomedical fields. Likewise, the frequent dismissed PLA nanostructure of the designed biomedical solutions hamper the systematic advance by correlating the structure–property relationship of the system with the application performance. Several reviews about PLA have been published covering a wide range of topics, such as the PLA synthesis [
20,
21], the physicochemical and mechanical properties of PLA [
22], the crystallisation and structure–properties relationship of PLA [
23], the characteristics of the promising stereocomplex PLA phase [
24,
25], and PLA applications in widespread fields [
11,
15,
26,
27,
28,
29,
30]. Herein, the state-of-the-art of PLA-based biomedical applications is reviewed from a global approach by highlighting the interconnections among the architectural designing parameters with the desired applications, with an emphasis on the stereocomplex phase of PLA.
2. Poly-(lactic Acid) (PLA)
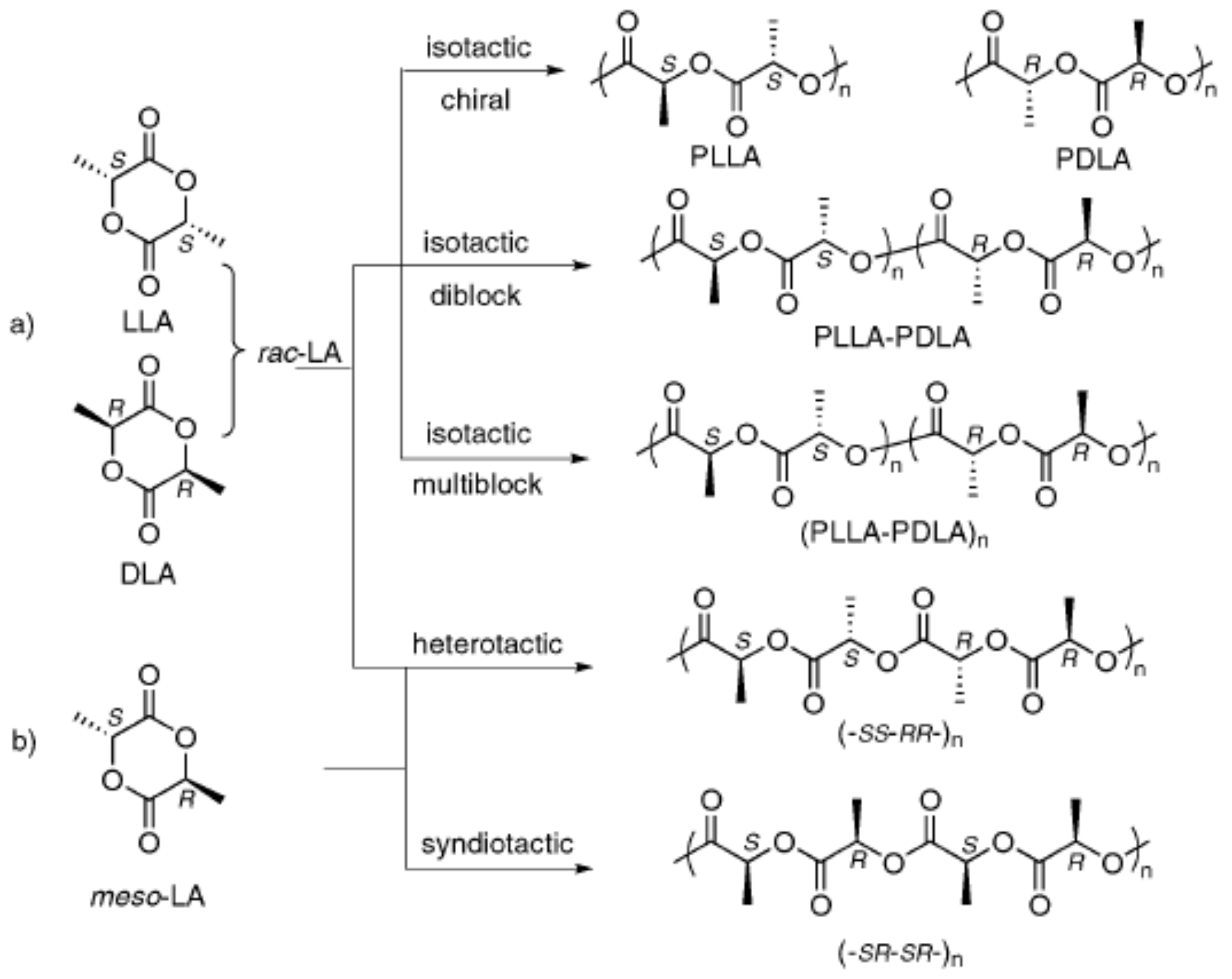
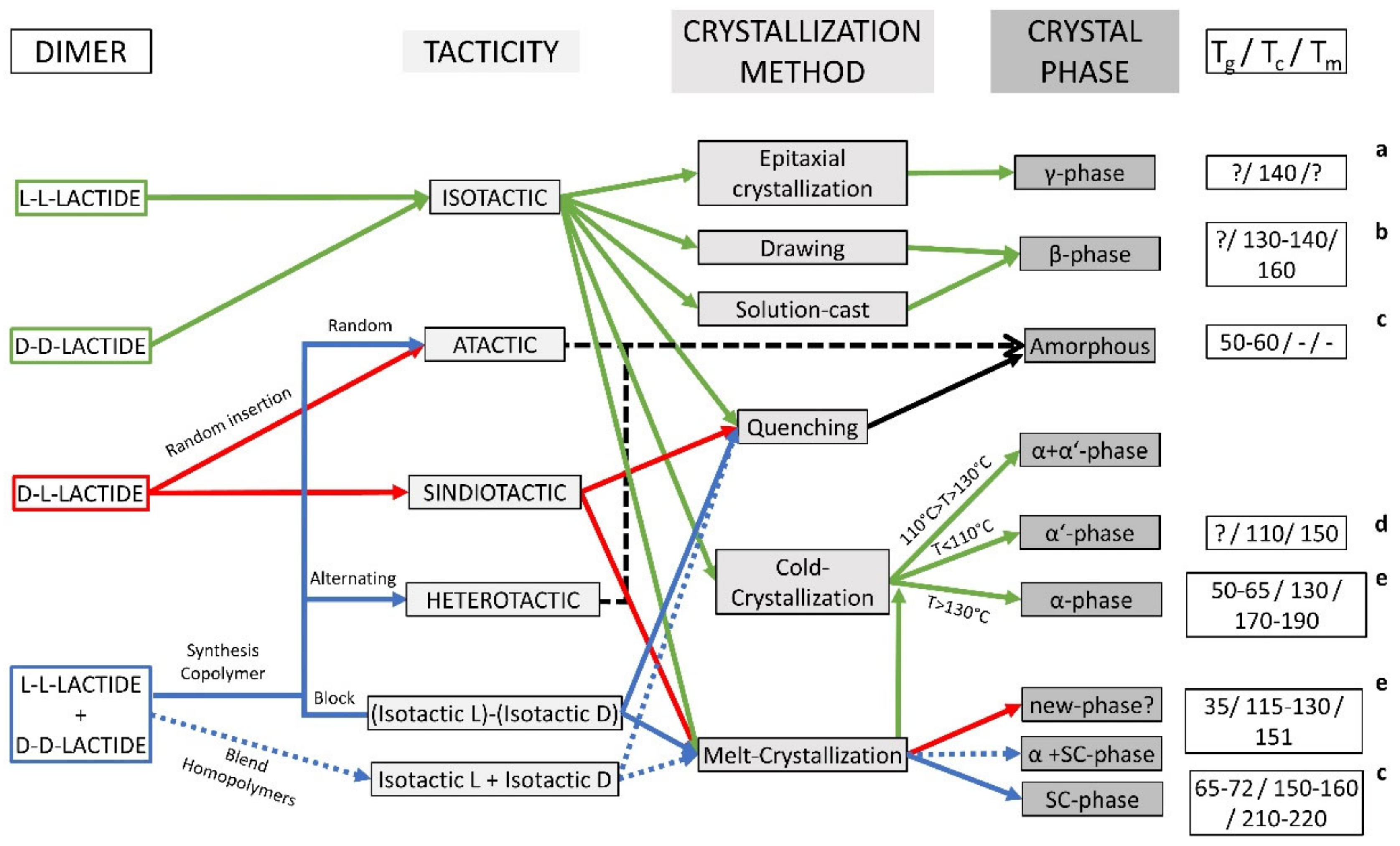
Polylactic acid is a poly-α-hydroxy acid synthesised from lactic acid (LA; 2-hydroxypropanoic acid) which exists in two optically active stereoisomers, namely, L-LA and D-LA (S and R in absolute configuration, respectively) [
31]. Approximately 90% of the total lactic acid produced worldwide is made by bacterial fermentation, which offers advantages in both the utilisation of a renewable source and the production of optically pure L- and D-lactic acid, depending on the strain selected (the chemical synthesis of lactic acid always results in a racemic mixture). The dehydrated cyclic dimer of lactic acid is commonly called lactide (3,6-dimethyl-1,4-dioxane-2,5-dione). Lactide exists in three different forms due to the presence of two asymmetric carbon atoms in the molecule: L-lactide, D-lactide, and meso-lactide. In addition, a racemate of D-lactide and L-lactide exists as rac-lactide [
31]. The chirality of PLA adds new functionality to PLA applications such as the specific recognition and interaction with chiral molecules (drugs, proteins, DNA, etc.) [
32].
The polymerisation of optically pure L- and D-lactide yields isotactic homopolymers of Poly–(L-lactide) (PLLA) and Poly–(D-Lactide) (PDLA), respectively (
Figure 1). Both PLLA and PDLA are semicrystalline polymers, showing a melting temperature (T
m) around 170 °C [
20,
33] and a thermal degradation temperature around 200 °C [
34]. The PLA derivative crystallinity as well as their melting and glass transition temperatures (T
g) usually decrease with the diminishing optical purity of the lactate units [
22,
35]. PLLA polymers with a D-lactide content lower than 10% tend to be crystalline (or PDLA with L-lactide content), whilst homopolymers with a lower optical purity are amorphous [
36]. The random insertion during the polymerisation of D- and L-LA units of both rac- and meso-lactide monomers generates an atactic polymer, Rac-PLA, which is completely amorphous. Moreover, syndiotactic PLA is obtained when D- and L-lactic acid units are placed alternatively along the chain, whereas a heterotactic chain architecture is attained when D- and L-lactide units are inserted alternatively on the polymer chain [
37]. Syndiotactic PLA is a semicrystalline polymer exhibiting higher T
c than isotactic PLA but lower T
m, whilst heterotactic PLA is amorphous [
38]. Furthermore, the one-pot sequential addition polymerisation method [
39] of D- and L-lactide monomers yields stereo-block copolymers with blocks of opposite chirality, featuring melting temperatures 50 °C higher than those of isotactic homopolymers (220 °C) [
25].
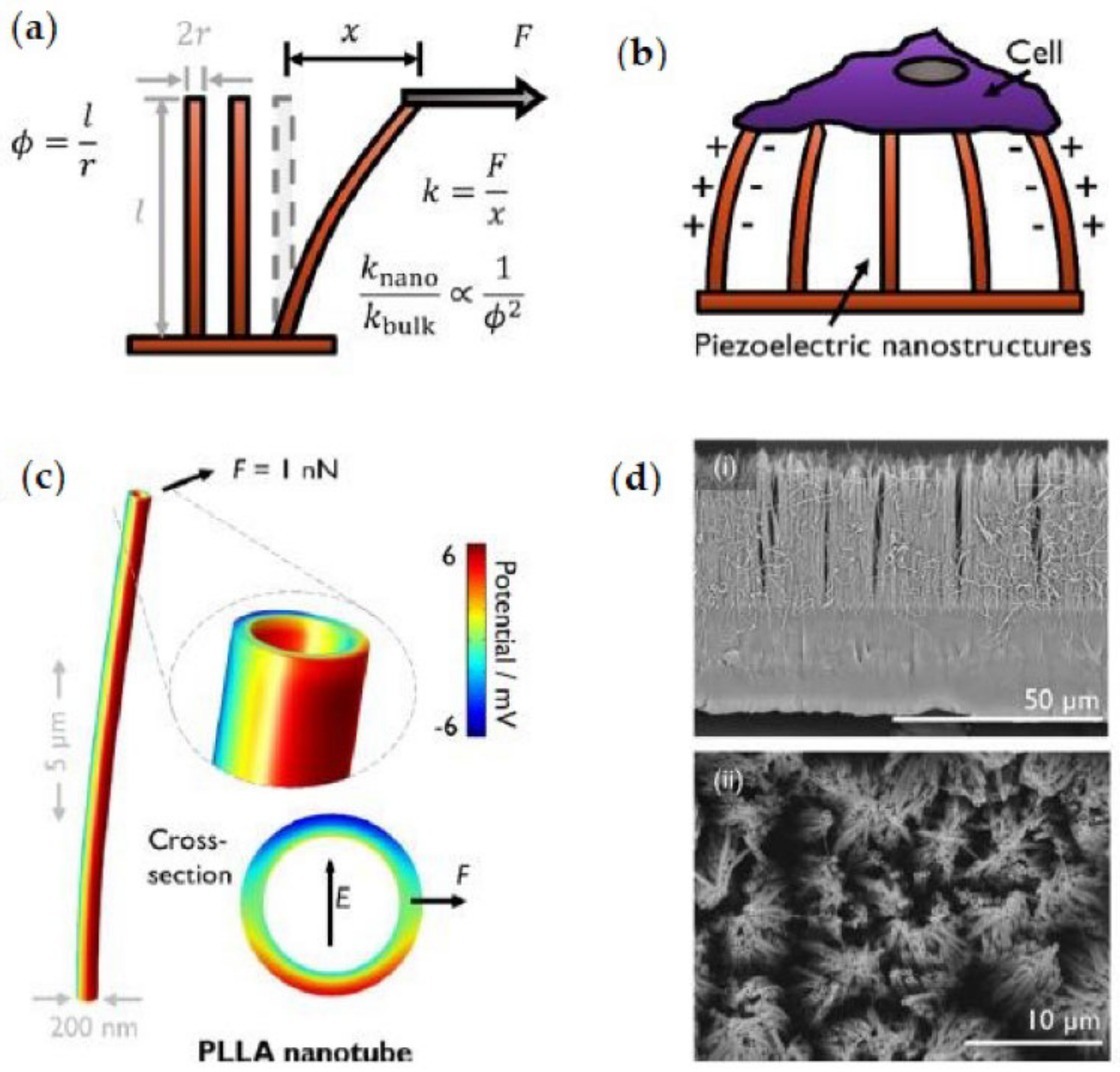
PLA displays different crystalline phases (α, α′, β, γ) established by the chain architecture and the specific crystallisation mechanism or thermo-mechanical history imposed during its processing, which define the properties of the final product [
40]. The α-phase is the more stable PLA homocrystal structure that corresponds to an orthorhombic unit cell in which the helices are packed in a hexagonal fashion, containing two antiparallel chains per unit cell. The α-phase is normally obtained when isotactic PLLA or PDLA are crystallised from the melt above 130 °C or by crystallisation from the solution, characterised by a melting temperature of around 170 °C [
41,
42]. The α′-phase (or δ-phase) is the disordered form of the α-phase that is generated either from crystallisation from the melt at temperatures below 110 °C or by cold-crystallisation after quenching PLA to the glassy state. The α’-phase is also organised in the orthorhombic crystallographic unit cell; however, it contains two parallel helices per unit cell, which increase the lattice parameters when compared to the ordered α phase. A mixture of α- and α’-phases is obtained when PLA is crystallised between 110 °C and 130 °C, although the α’-phase recrystallises into the α-phase when heated near the T
m (150–160 °C) [
41]. Moreover, the β-phase is developed by stretching PLA fibres in the α-phase at a high temperature (130–140 °C) and high draw ratios as well as by casting thin films from the solution [
42,
43]. The chain conformation of the β-phase is a threefold helix in a trigonal unit cell containing three chains per unit cell, and its T
m is ca. 10 °C lower than α-phase T
m [
44,
45]. In addition, the β-phase exhibits piezoelectricity that allows for the interchange of mechanical and electrical energy [
45], broadening its potential applications. Finally, the γ-phase is produced by the epitaxial crystallisation of PLA on hexamethylbenzene, forming two antiparallel threefold helices in an orthorhombic unit cell [
46].
Furthermore, a new crystal structure, the stereocomplex (SC) phase, is formed from the co-crystallisation of the two stereoisomers of PLA (PLLA and PDLA) that feature a trigonal unit cell comprised of six threefold helices per unit cell. The structural peculiarity of the SC phase, with the nearest neighbours of any stem being of a different polymeric chain, provides them easy access to the growth front for both enantiomeric species. In addition, the specific C-H···O-H hydrogen bonds within the crystal lattice that stabilise the structure [
47] endow stereocomplexes with a higher melting point (220 °C) and degradation temperature (220–260 °C) [
34]. The SC phase was first found by casting a mixed solution of both enantiomers [
48], and since then, the SC phase has typically been obtained intentionally from the blend of both enantiomers in the solution (in an appropriate solvent such as dichloromethane or chloroform at room temperature or acetonitrile around boiling temperature [
49]) or in the solid-state from the melt [
24]. However, the SC crystallisation of the blended enantiomers diminishes for high-molecular-weight (HM
w) PLA, and enantiomeric homocrystals (HC) in the α-phase are obtained instead [
50]. Additionally, the critical M
w to exclusively obtain SC crystallisation is lower for blends obtained from the melt than those obtained from the solution [
51], which hampers its industrial application. The optical purities of the polymers and the mixing ratio of the isomeric chains also affect the obtained ratio of SC-to-HC crystallites, and thus, the preparation of pure SC-PLA requires meticulous specific conditions [
52]. SC crystallites can also be generated through the synthesis of block copolymers by the one-pot sequential monomer addition to a truly living polymerisation catalyst, which allows for the retention of the SC crystallisation in HM
w polymers [
53,
54].
The new synthetic approach affords a wide range of chain architectures that can be generated through different ratios of L- and D-Lactide monomers that offer the possibility to tailor the properties of the final polymeric product depending on the intended application. Furthermore, understanding the advantages and drawbacks of the different synthetic processing methods to obtain PLA is crucial to tailoring the foreseen applications. Moreover, since PLA still exhibits performance drawbacks such as low mechanical properties, a low thermal resistance, and a low hydrophobicity, which limit its applications in some biomedical fields, novel materials with unique properties can be obtained through the blend or copolymerisation of PLA with other biodegradable or non-biodegradable polymers, such as polyethylene (PE), polypropylene (PP), Polyhydroxhyalkanoates (PHA), PMMA, Poly(ethylene-
co-vinyl acetate) (PEVA), etc. [
13,
55,
56]. In addition, nanocomposites can be fabricated by mixing PLA with other complementary compounds such as silk [
57], gelatin [
58], collagen [
59], tungsten disulfide [
60,
61,
62], natural fibres (flax, jute, hemp,) [
63], ceramics (ZnO, TiO
2) [
64,
65], etc. to enhance their performance.
3. Synthesis of PLA
PLA was first synthesised by polycondensation by Théophile-Jules Pelouze in 1845. In 1932, Wallace Hume Carothers developed a novel synthetic method based on the ring-opening polymerisation (ROP) of the cycle lactide monomer to synthesise PLLA. ROP was later patented by Du Pont in 1954 to synthesise vinyl fluoride (U.S. Patent No. 2674632, 1954). However, HM
w PLA by ROP on an industrial scale was only attained by the mid-1990s [
31].
The lactic acid monomer can be converted to PLA through a polycondensation process by the reaction of the hydroxyl (–OH) and carboxylic acid (–COOH) groups with the removal of the detrimental byproducts such as water. Generally, catalysts are added to polymerisation to increase the reaction rate. The removal of water, enhanced under vacuum pressure, is critical to producing HM
w polymers due to the increased viscosity of the reaction mixture as the reaction proceeds. However, side reactions, such as transesterification, can also occur during the polycondensation of lactic acid, resulting in the formation of ring structures of different sizes, such as lactides. Transesterification reactions lower the overall M
w and the stereocontrol over the chain architecture, decreasing the physical properties of the PLA afforded as well as reducing the reaction yield [
35]. The HM
w PLA is mainly synthesised by ROP due to the accurate chemical control in terms of molecular weight, polydispersity, polymer chain-ends, and tacticity. Moreover, ROP requires relatively mild conditions (130 °C) when compared to polycondensation (180–200 °C) [
35,
66]. Three reaction mechanisms have been proposed for ROP of lactide: anionic, cationic, and coordination-insertion mechanisms. In both anionic and cationic polymerisations, a monomer-activation mechanism occurs first, which permits the catalyst to be independent of the propagating polymer and can thus be easily removed as the polymerisation finishes. However, undesirable side and racemisation reactions are likely to occur due to the highly activated monomers. On the contrary, coordination-insertion polymerisation attains HM
w PLA with higher control over the M
w distribution [
14,
35]. Metal complexes of several metals have been widely employed as the catalysts for the ROP of lactides [
35], of which the most studied are stannous 2-ethylhexanoate [Sn(Oct)
2], aluminium isopropoxide [Al(O i-Pr)
3], and zinc(II) lactate [Zn(Lact)
2] [
66]. Sn(Oct)
2, is the catalyst utilised for the industrial synthesis of PLA, largely due to its approval by the FDA for use in medical (<20 ppm [
67]) and food applications. Moreover, lauryl alcohol (1-dodecanol) is usually added as an initiator [
35].
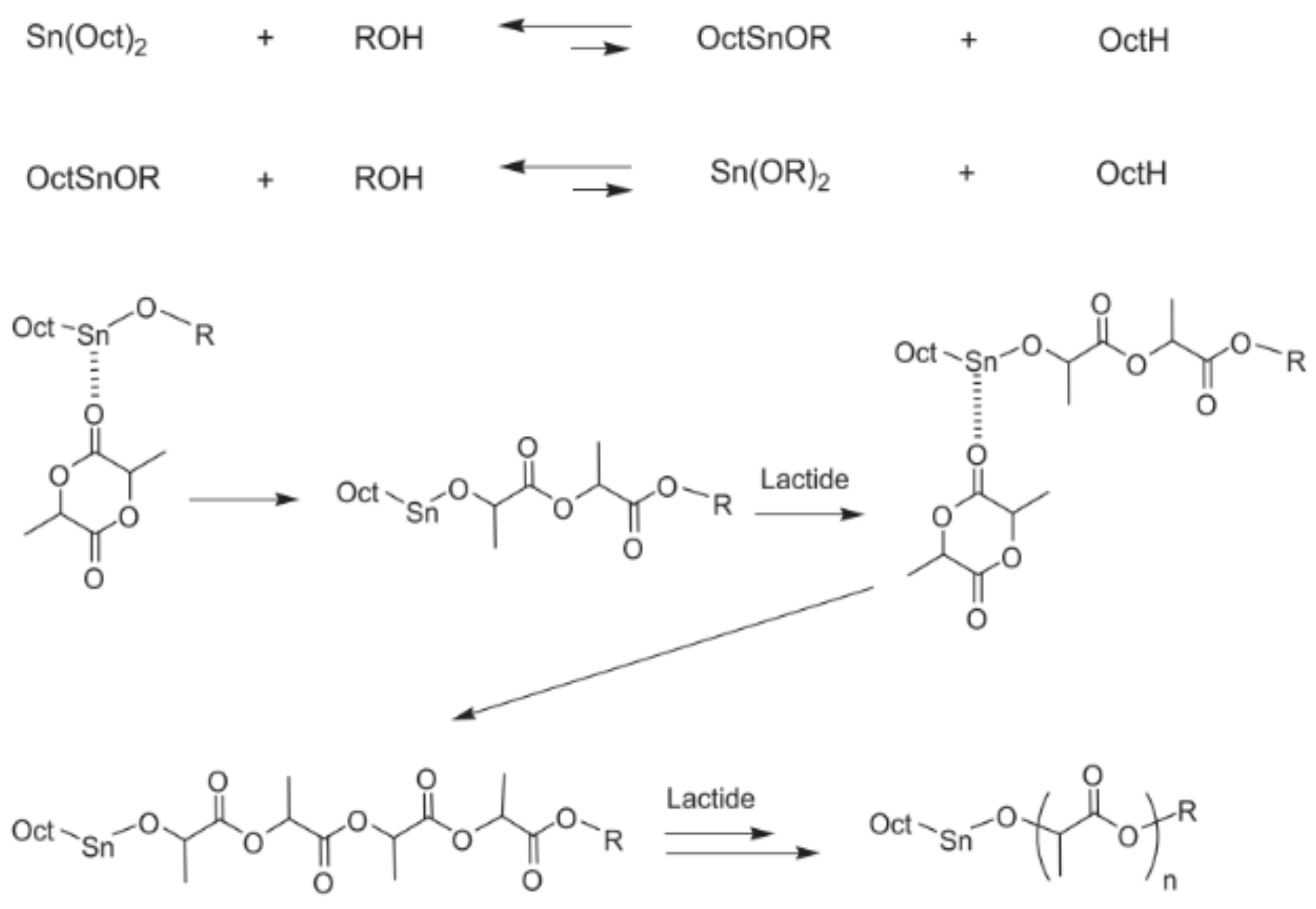
The polymerisation is induced by a coordination-insertion three-step mechanism supported by the catalyst [
66], which was first formulated in 1971 by Dittrich and Schulz [
14] (
Figure 2). Firstly, Sn(Oct)
2 reacts with the lauryl alcohol to form a tin alkoxide. Subsequently, the exocyclic carbonyl oxygen of lactide temporarily coordinates with the tin atom of the catalyst in the alkoxide form. The formed coordination system enhances the nucleophilicity of the alkoxide part of the initiator as well as the electrophilicity of the lactide carbonyl group that enables the reaction to each other. Finally, the acyl-oxygen bond of lactide is disrupted, and the generated linear chain of the lactide turns into the alkoxide part of the catalyst, promoting the coordination with a new lactide molecule and, thus, the polymerisation propagation [
35]. Finally, the active metal-alkoxide bond is hydrolysed as the monomer is entirely consumed, and the formation of a hydroxyl end-group occurs [
66]. In the last stage of the propagation step, as the monomer concentration becomes significantly lower (~80%), both intra- and inter-molecular transesterification reactions occur, and the molecular weight distribution increases. However, the degrees of racemisation and chain scrambling achieved by metal complexes that follow coordination-insertion mechanisms are notably lower than those obtained by anionic or cationic catalysis [
35]. The microstructure of the final polymers depends both on the initial monomers added to the reaction mixture (
Scheme 1) and the catalyst stereocontrol. The control exerted by the catalyst over the nanostructure of the PLA, particularly to synthesise the PLA stereo block copolymers of HM
w, is essential to tailor the properties of the final product, and the synthesis of novel catalysts, particularly metal-based catalysts, for polymerisation by the coordination mechanism has attracted much attention since the pioneering work of Kleine et al. in the 1950s [
66,
68,
69]. However, several drawbacks to controlling the synthesis of stereoblock HM
w PLA have emerged since then, such as the decrease in the living character of the catalyst due to the increase in the reaction heterogeneity [
70], the detrimental side reactions due to the multiple nuclearities exhibited by the catalysts [
71], as well as the long reaction time required to achieve the desired architectures and molecular weight [
14]. Recently, novel catalysts that fulfil the synthetic requirements whilst exhibiting low toxicity for the application of the PLA derivatives in the biomedical and pharmaceutical fields were just attained [
54,
72,
73], which offer the possibility to design multiblock copolymers simultaneously featuring the PLA stereoblock to attain higher physicochemical properties with other complementary blocks to tackle the PLA limitations.
4. PLA Processing
Once synthesised, PLA is usually manipulated into its final shape by the use of different processing techniques that apply diverse thermomechanical histories. Melt processing is a three-step process generally used to transform PLA into different commodity products at an industrial scale [
75]. Firstly, the polymer is melted to subsequently be moulded into the desired shape, which is then generally cooled to stabilise its dimensions. Widespread industrial techniques such as extrusion and injection moulding are the two most common melt-based processes for manufacturing thermoplastic polymers. Novel common additive manufacturing technologies with promising personalised biomedical applications such as fused deposition modelling follow a melt-based process [
76]. Likewise, the tailoring of T
m of PLA is crucial for the performance of applications employing melt-based processing techniques, as the process temperature must be above T
m to form a homogeneous melt but low enough to minimise thermal degradation [
77]. In addition, the cooling rate during the third step will influence the properties of the final product, which determine the crystallisation conditions that dictate the crystallinity degree and crystalline phase. Particularly, quenching PLA from the melt at a high cooling rate (>500 °C/min, such as during injection moulding) will result in a highly amorphous polymer [
78], whilst semicrystalline PLA is obtained when the cooling rate is reduced (>30 °C/min [
79]). The α-form is usually developed from PLLA or PDLA during typical melt processing; however, a mixture of the α and α′ phases is obtained when the cooling rate is higher than 2 °C/min since the α-form has very slow crystallisation kinetics (pure α-phase is obtained at slower rates) [
80]. Furthermore, post-production treatments such as annealing can be implemented to increase the thermal stability and mechanical properties of the final product. The α-phase develops completely from a melt-crystallised material exhibiting a mixture of α and α’when annealing at 140 °C for 1 h [
80]. Additionally, obtaining only the SC phase from the melt from the equimolar blend of PLLA and PDLA enantiomers can be achieved by restricted thermodynamic conditions such as relatively low cooling rates (20 °C/min) to avoid phase separation or from low-molecular-weight enantiomers (~20 kDa) [
50] and isothermal crystallisation at temperatures above the T
m of homocrystals (~175 °C) [
51], as well as by melt-spinning under high-tensile-stress conditions [
81] (
Scheme 1). However, PLA materials crystallised in the stereocomplex phase have not reached the market yet [
82].
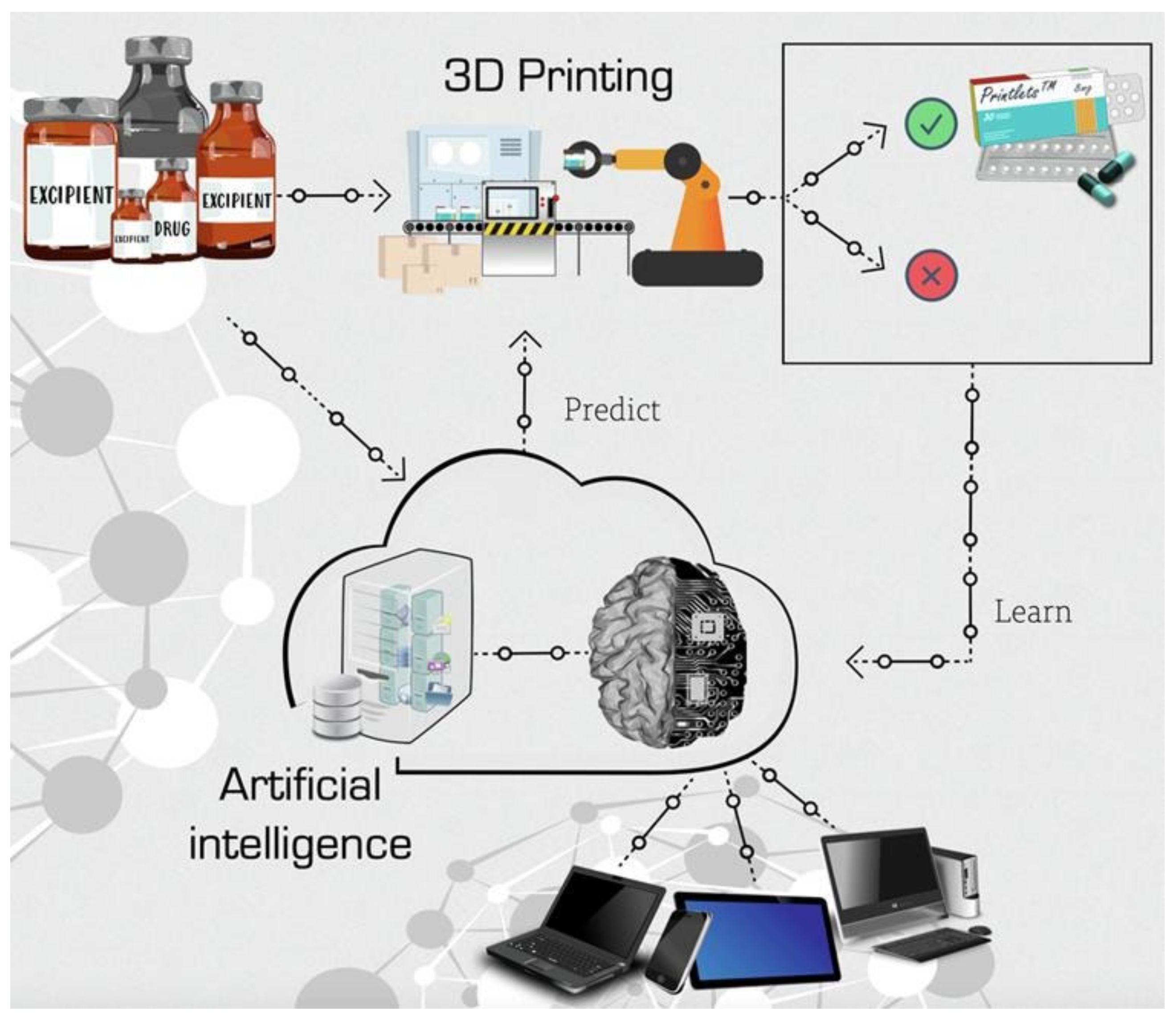
Three-dimensional (3D) printing is an additive manufacturing technology with the unique ability to produce personalised objects with complex designs at reduced costs and a high-resolution precision [
83,
84]. They have already reached the market in the biomedical field such as the manufacturing of 3D scaffolds for studying the response of particular tissues to different stimuli [
85]. In particular, PLA is the most-used polymer for 3D printing since the slow crystallisation rate compared to that of polyolefins (i-PP or PET) avoids warping between layers [
86]. Classical processing methods such as particulate leaching, gas foaming, or solvent-casting were used for the tuning of the internal architecture of 3D scaffolds due to their adequacy for replacing tissues with a high regenerative capacity. However, 3D printing enables the required control over the scaffold architecture for fewer regenerative tissues, such as tendons or nerves [
87]. Likewise, PLA 3D-printed scaffolds have already been investigated for bone [
88,
89,
90,
91,
92,
93], neural [
94], and musculoskeletal soft [
95] tissue engineering. In addition, PLA nanofibres have also been used as part of a fibrous bioink for the 3D printing of a meniscus construct to study the proliferation of human adipose-derived stem cells that provide a higher cell proliferation and metabolic activity [
95]. Recently, the assessment of the PLA scaffold geometry effect on the orthopaedic applications [
96] revealed that the presence of hydroxyapatite (HA) in the scaffold efficiently enables mineralisation as well as induces the crystallisation of PLA after being 3D printed, whilst PLA without HA remained amorphous. The presence or absence of crystalline domains within the 3D-printed PLA scaffold will invariably influence the hydrolysis degradation rate, which is crucial to controlling the optimum performance of the biomaterial. However, the usual lack of structural study for most of the reported 3D-printed PLA scaffolds for biomedical applications impedes the determination of the relationship between the processing conditions, the crystalline structure, and the biomedical performance.
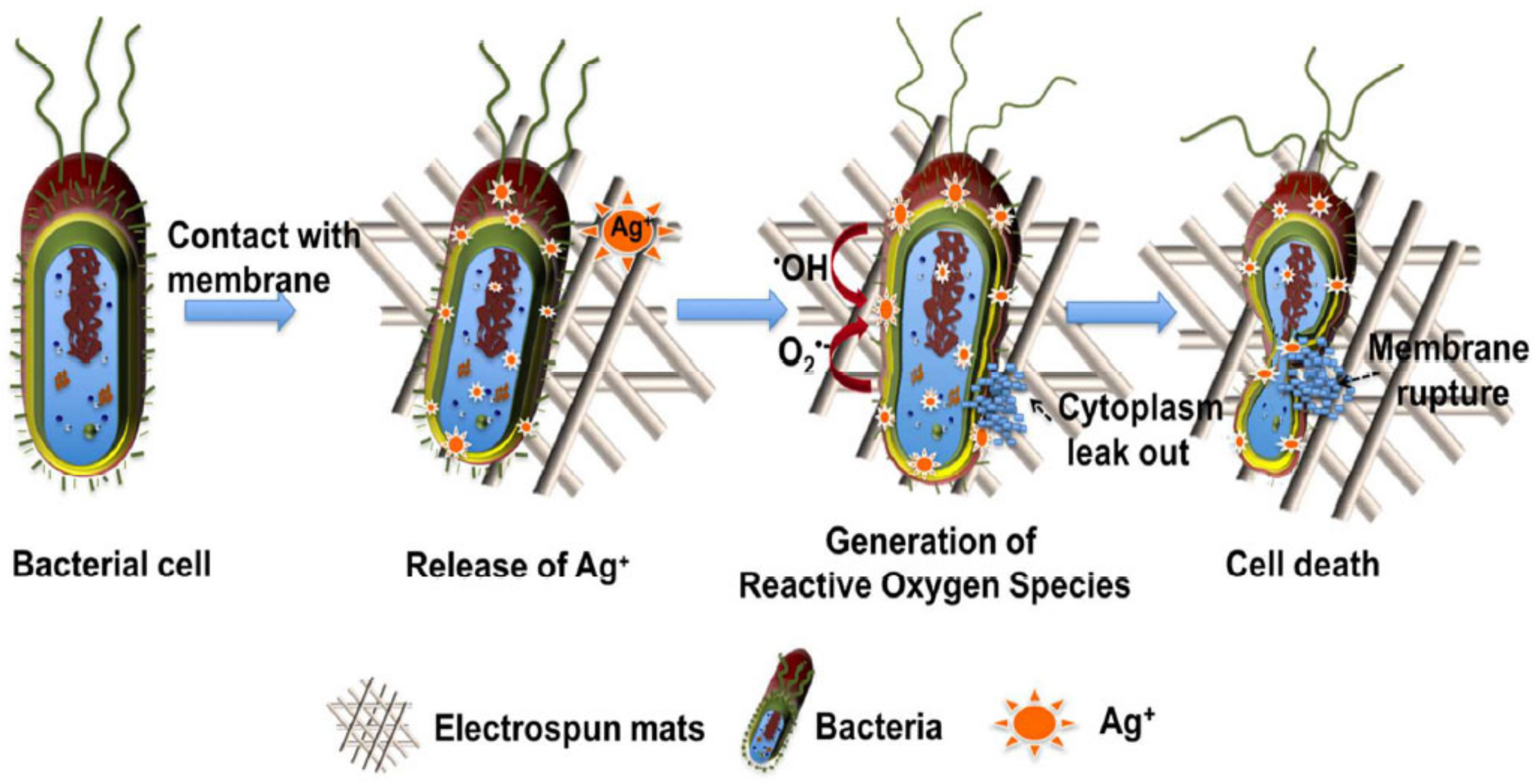
Furthermore, in the biomedical industry, electrospinning has been considered a promising method to fabricate polymer nanofibres due to its simplicity and the cost-effectiveness of the technique. Electrospinning, unlike drying spinning, which relies on mechanical extrusion, uses the electrostatic force to spin the solution into fibres [
78]. The fibres thus obtained have a nanometric diameter, producing materials with a high area/volume ratio, a high flexibility, and superior mechanical properties compared with other material formats [
97]. Electrospun PLA usually exhibits either an amorphous structure or a semicrystalline structure, although with a very low crystallinity (~10%), due to the rapid solidification of the fibres during the process, which entails post-processing thermal treatment between the T
g and the T
m to enhance the crystallinity by cold-crystallisation in the α-phase [
98,
99]. Recently, the straightforward fabrication of PLA electrospun fibres exhibiting the β-phase without further post-processing treatment was achieved [
100], facilitating the development of PLA-based devices with piezoelectric properties for potential biomedical applications, as will be further discussed [
101]. Furthermore, PLA fibres in the SC phase were also generated from the electrospinning of PLLA/PDLA blends, resulting in more uniform fibres [
102]; however, an annealing post-processing step is usually required to obtain crystalline structures [
103,
104]. In addition, the tensile strength and Young’s modulus was found to be modulated by varying the spinning method, i.e., either by melt-spun, solution-spun, and/or as-spun [
103]. Moreover, plasma protein adsorption was also investigated on solution cast films of PEG-PLA and compared to the SC phase formed by the PEG-PLLA/PEG-PDLA blend. The absorption of both albumin and fibrinogen was higher on the SC films than on the homopolymer counterparts. The SC crystallisation was revealed to hamper the migration of the PEG to the surface of the film, prolonging protein adsorption and cell attachment over a longer period [
105]. The large efforts dedicated to electrospun nanofibres in the biomedical field raise the possibility to mimic the extracellular matrix, since the human tissues and organs are formed by nanofibrous scaffolds [
106,
107]. However, industrial-scale production of PLA nano-fibres has not been achieved yet due to the low throughput of the technique and the requirement of specific solvents [
78]. Nevertheless, PLA nanofibres are a topic in continuous research due to their crucial role in several biomedical applications such as bone regeneration [
108,
109], drug delivery systems [
110], and wound dressing [
111].
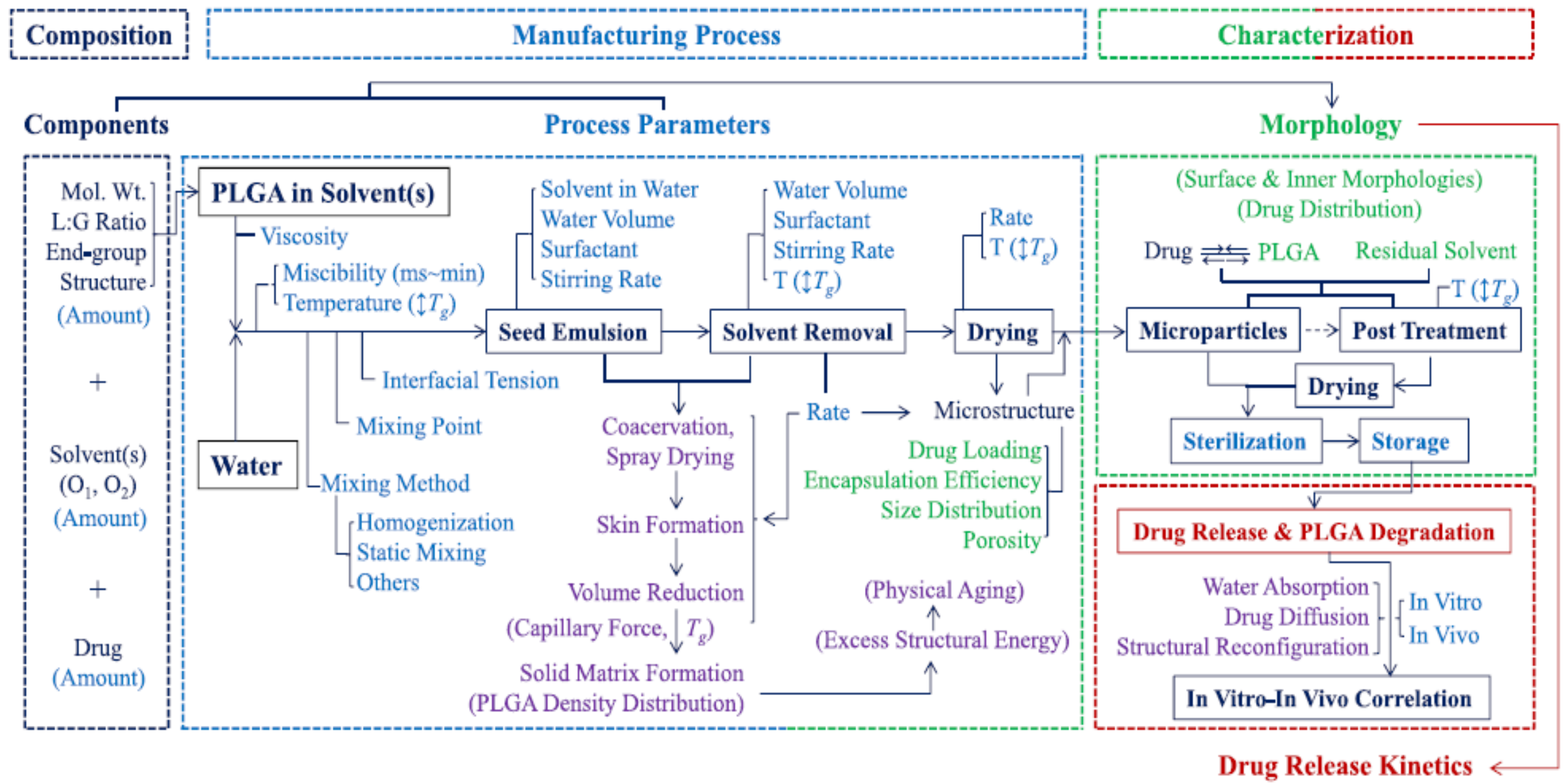
Furthermore, PLA can also be processed into nanoparticles to generate drug delivery systems in which the drug release rate can be controlled by varying parameters such as the processing method, the microstructure of the starting polymer, or its concentration in the organic solvent [
112]. However, contradictory results are usually found when relating certain processing parameters to the final nanoparticle release kinetics [
113,
114,
115], and hence, further studies based on the structure–property relationship are required to understand the mechanistic process occurring during nanoparticles formation to design drug nanocarriers with tailored drug release profiles [
112] (
Figure 3). Furthermore, the methods for preparing nanoformulations most commonly utilised at the laboratory scale, such as nanoprecipitation or emulsification-solvent evaporation, lack reproducibility between batches. However, novel approaches such as supercritical technology, electrospraying, or premix membrane emulsification have emerged as methods that are better adapted for industrial production, enabling the scale-up process of nanoparticles [
116].
In addition, PLA is generally employed to afford hydrogels, as is capable of absorbing a large amount of water that can be programmed to be expanded or shrunk due to external condition changes [
117]. Hydrogels are aqueous dispersions that solidify by the decrease in polymer solubility in response to different physical and/or chemical stimuli—typically, pH or temperature—that are used to control the drug delivery systems [
118]. PLA is usually copolymerised with a hydrophilic polymer to form associative micelles that constitute the gel in which the nanostructure and rheological properties can be tuned by varying the stereochemistry of the PLA [
119,
120]. Moreover, PLA can also be part of hydrogel materials as a mechanically reinforcing and/or drug-eluting component [
58,
121,
122]. PLA-based hydrogel studies are less common than PLA-based nanoparticle studies. However, a deeper structural characterisation is usually accomplished.
7. Conclusions
Polymers were envisaged as an alternative to conventional materials as well as to realise novel highly functionalised applications in the pharmaceutical and biomedical fields due to their ease in being shaped, lightness, industrial scalability, and chemical and biological inert nature. The current status of the cutting-edge applications in the pharmaceutical and biomedical fields based on PLA has been revised, covering the updates on the synthetic routes to attain multiblock PLA architectures and their structure–property relationships as well as the processing conditions impacting the performance of the final material. PLA is a bio-based polymer that has attracted significant interest due to its biodegradation capability and low price. PLA derivatives attaining different architectures are obtained depending on both the starting lactide derivative and the polymerisation method. The recently employed living polymerisation assisted by a single-site catalyst has enabled the attainment of PLA derivatives with high stereocontrol, high Mw, and narrow polydispersity, including stereo-block copolymers, which expands the availability of multiblock polymers on demand. In particular, high-Mw PLA stereo-block copolymers and their derivatives feature enhanced physicochemical properties that enlarge the potential biomedical and pharmaceutical applications. In addition, the rich PLA nanostructure, with diverse crystalline phases featuring different thermo-mechanical properties, as well as particular physicochemical properties such as piezoelectricity, are obtained through several processing techniques such as electro-spinning, 3D printing, or nanoparticle formulation. Likewise, the PLA crystalline domains degrade in physiological media at different rates depending on the crystalline PLA phase as well as the physicochemical and morphological parameters related to the surrounding medium, which must be considered when designing a biomedical device for a specific application.
Remarkably, PLA-based materials have already reached the clinical level in both pharmaceutical and biomedical applications such as sutures, stents, microparticles, and dermal fillers. In addition, PLA-based nanoparticles and 3D scaffolds are currently being developed for drug delivery and tissue regeneration applications, respectively. The piezoelectricity and chirality exhibited by PLA enlarge the functionalities and, in particular, the molecular recognition for potential biomedical devices. Likewise, the generation of bionanocomposites based on PLA affords the customisation of therapeutic materials with multifaceted capabilities. Furthermore, personalised therapeutic approaches have become feasible with the advent of 3D printing technology designing specific pharmaceutical formulations for specific patients, pointing toward personalised medicine.
However, the current gap between the PLA structural characterisation and the performance of PLA in biomedical applications hinders the systematic conception of personalised biomedical solutions based on the correlation of the structure–property relationship with the required functionalities. The end-user requirements should be considered from an interdisciplinary approach from the genesis of PLA-based material to realistically reach the pharmaceutical/biomedical market. Likewise, the PLA-nanoparticles formulation has greatly advanced to attain drug carriers down to nanoparticles with a fair control; however, the nanostructure development during the polymer processing is frequently dismissed, which is key to tuning the drug release profile as well as diminishing the batch-to-batch variability usually obtained through conventional techniques that hamper the scalability of nanoparticle formulation to the industrial scale. Moreover, the comprehensive nanostructure control achieved for a wide range of smart PLA-based hydrogels with specific functionalities must be followed by in vitro and in vivo studies to correlate the physicochemical properties with its performance under physiological conditions. In addition, the screening focus to develop new synthetic pathways to efficiently conjugate drugs to PLA should be also associated with a targeted PLA conjugate nanostructure in the applied medium in order to be translated reliably to both the release mechanism and the molecular recognition activity required to efficiently target the damaged tissue. Moreover, the tissue regeneration progress achieved from the materials approach requires a further understanding of the particular physicochemical and mechanical properties of each tissue, the alterations occurring under pathological conditions, and its evolution upon healing for designing materials according to the real requirements. Furthermore, the performance of the PLA-based stents suffers from structural stability due to the exhibited lack of radial strength that could be tacked by iterative structure-properties studies of novel multiblock copolymers. The mastering of novel polymer processing techniques, especially 3D techniques, should be achieved to generate customised materials with performing mechanical properties to attain consistently demanding applications.


 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}