
Cutting-Edge Delivery Systems and Adjuvants in Tolerogenic Vaccines: A Review

 , , , , and
, , , , and
Abstract
:1. Introduction
2. The Concept of Immune Tolerance
3. Strategies Used in Tolerogenic Vaccines to Induce Antigen-Specific Tolerance
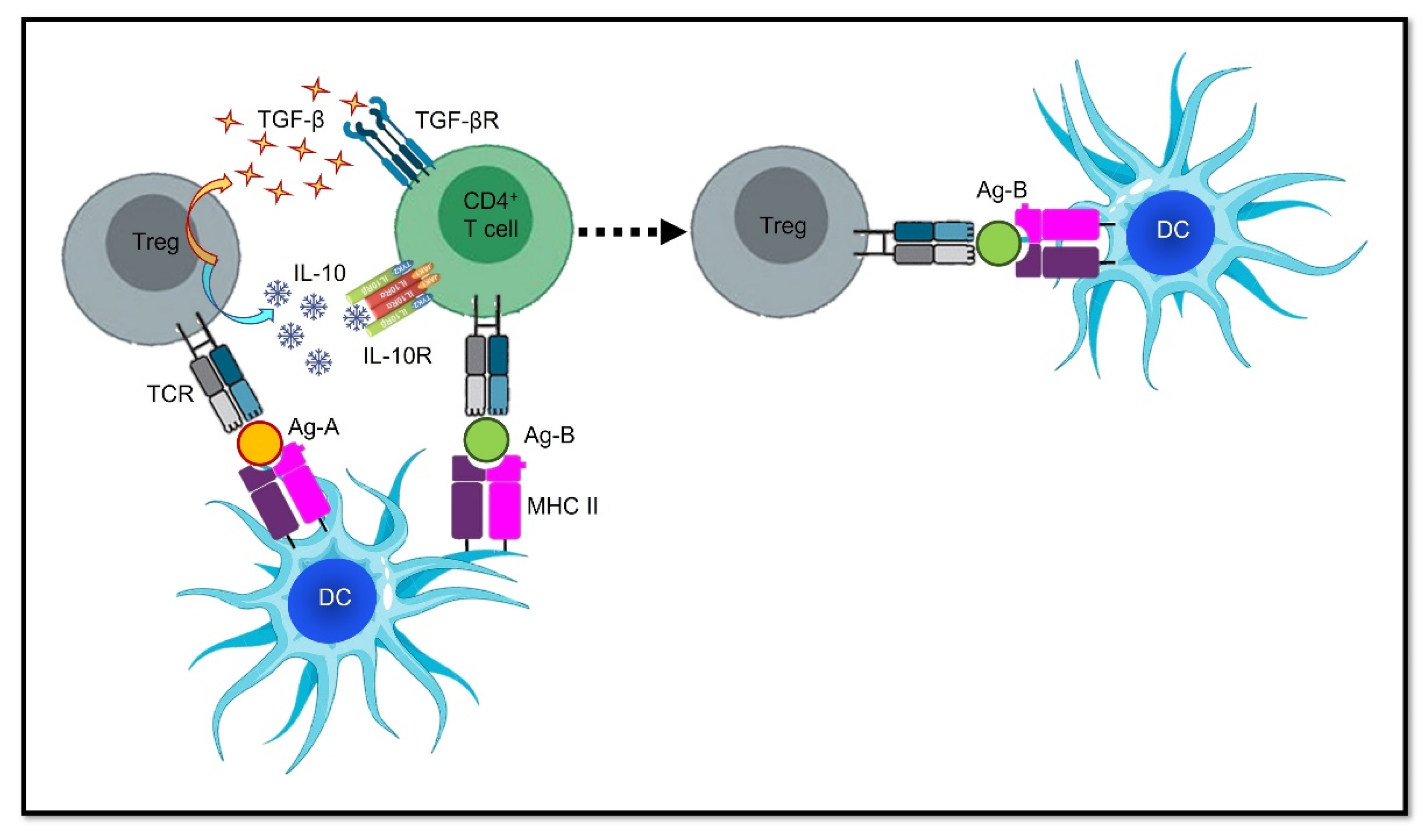
3.1. Deprivation of Co-Stimulatory Signals
3.2. Inhibition of Pro-Inflammatory Stimuli
3.3. Harnessing Tolerogenic Physiological Mechanisms
3.4. Induction of a Tolerogenic Phenotype
3.5. Dendritic Cell-Based Vaccines
3.5.1. Ex Vivo DC Education
3.5.2. In Vivo DC Targeting Strategies
3.6. Nucleic Acid-Based Tolerogenic Vaccines
4. Epitope Spreading: Hurdle or Advantage?
5. Focus on the Revolutionary Role of Micro- and Nanoparticle-Based Vaccines
5.1. Protein-Based Nanoparticles
5.2. Extracellular Vesicles as Particle-Based Vaccines
5.3. Technical Considerations in Particulate Vaccine Manufacturing
6. Concluding Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Baker, K.; Isaacs, J. Novel therapies for immune-mediated inflammatory diseases: What can we learn from their use in rheumatoid arthritis, spondyloarthritis, systemic lupus erythematosus, psoriasis, Crohn’s disease and ulcerative colitis? Ann. Rheum. Dis. 2017, 77, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Cauwels, A.; Tavernier, J. Tolerizing Strategies for the Treatment of Autoimmune Diseases: From ex vivo to in vivo Strategies. Front. Immunol. 2020, 11, 674. [Google Scholar] [CrossRef] [PubMed]
- Galli, E.; Fortina, A.B.; Ricci, G.; Maiello, N.; Neri, I.; Baldo, E.; Berti, I.; Bonamonte, D.; Capra, L.; Carboni, E.; et al. Narrative review on the management of moderate-severe atopic dermatitis in pediatric age of the Italian Society of Pediatric Allergology and Immunology (SIAIP), of the Italian Society of Pediatric Dermatology (SIDerP) and of the Italian Society of Pediatrics (SIP). Ital. J. Pediatr. 2022, 48, 95. [Google Scholar] [CrossRef] [PubMed]
- Cappellano, G.; Abreu, H.; Casale, C.; Dianzani, U.; Chiocchetti, A. Nano-Microparticle Platforms in Developing Next-Generation Vaccines. Vaccines 2021, 9, 606. [Google Scholar] [CrossRef]
- Jensen-Jarolim, E.; Bachmann, M.F.; Bonini, S.; Jacobsen, L.; Jutel, M.; Klimek, L.; Mahler, V.; Mösges, R.; Moingeon, P.; O’hehir, R.E.; et al. State-of-the-art in marketed adjuvants and formulations in Allergen Immunotherapy: A position paper of the European Academy of Allergy and Clinical Immunology (EAACI). Allergy 2020, 75, 746–760. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Principles of Vaccination. In Centers for Disease Control and Prevention; Pink Book Webinar Series; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2020; pp. 1–7. [Google Scholar]
- Cappellano, G.; Comi, C.; Chiocchetti, A.; Dianzani, U. Exploiting PLGA-Based Biocompatible Nanoparticles for Next-Generation Tolerogenic Vaccines against Autoimmune Disease. Int. J. Mol. Sci. 2019, 20, 204. [Google Scholar] [CrossRef]
- Hanabuchi, S.; Ito, T.; Park, W.-R.; Watanabe, N.; Shaw, J.L.; Roman, E.; Arima, K.; Wang, Y.-H.; Voo, K.S.; Cao, W.; et al. Thymic Stromal Lymphopoietin-Activated Plasmacytoid Dendritic Cells Induce the Generation of FOXP3+ Regulatory T Cells in Human Thymus. J. Immunol. 2010, 184, 2999–3007. [Google Scholar] [CrossRef]
- Xing, Y.; Hogquist, K. T-Cell Tolerance: Central and Peripheral. Cold Spring Harb. Perspect. Biol. 2012, 4, a006957. [Google Scholar] [CrossRef]
- Nemazee, D. Mechanisms of central tolerance for B cells. Nat. Rev. Immunol. 2017, 17, 281–294. [Google Scholar] [CrossRef]
- Lutz, M.B.; Backer, R.A.; Clausen, B.E. Revisiting Current Concepts on the Tolerogenicity of Steady-State Dendritic Cell Subsets and Their Maturation Stages. J. Immunol. 2021, 206, 1681–1689. [Google Scholar] [CrossRef]
- Oh, J.; Shin, J.-S. The Role of Dendritic Cells in Central Tolerance. Immune Netw. 2015, 15, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, H.; Matsumoto, T. Mechanisms of Tolerance Induction by Dendritic Cells In Vivo. Front. Immunol. 2018, 9, 350. [Google Scholar] [CrossRef]
- Zhu, J.; Paul, W.E. Heterogeneity and plasticity of T helper cells. Cell Res. 2010, 20, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Boggio, E.; Gigliotti, C.L.; Rossi, D.; Toffoletti, E.; Cappellano, G.; Clemente, N.; Puglisi, S.; Lunghi, M.; Cerri, M.; Vianelli, N.; et al. Decreased function of Fas and variations of the perforin gene in adult patients with primary immune thrombocytopenia. Br. J. Haematol. 2016, 176, 258–267. [Google Scholar] [CrossRef]
- Wu, H.; Gong, J.; Liu, Y. Indoleamine 2,3-dioxygenase regulation of immune response (Review). Mol. Med. Rep. 2018, 17, 4867–4873. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Mellor, A.L. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol. 2012, 34, 137–143. [Google Scholar] [CrossRef]
- Metz, R.; Rust, S.; DuHadaway, J.B.; Mautino, M.R.; Munn, D.H.; Vahanian, N.N.; Link, C.J.; Prendergast, G.C. IDO inhibits a tryptophan sufficiency signal that stimulates mTOR: A novel IDO effector pathway targeted by D-1-methyl-tryptophan. OncoImmunology 2012, 1, 1460–1468. [Google Scholar] [CrossRef] [PubMed]
- Badawy, A.A.-B. Tryptophan metabolism, disposition and utilization in pregnancy. Biosci. Rep. 2015, 35, e00261. [Google Scholar] [CrossRef]
- Antonioli, L.; Pacher, P.; Vizi, E.S.; Haskó, G. CD39 and CD73 in immunity and inflammation. Trends Mol. Med. 2013, 19, 355–367. [Google Scholar] [CrossRef]
- Mandapathil, M.; Hilldorfer, B.; Szczepanski, M.J.; Czystowska, M.; Szajnik, M.; Ren, J.; Lang, S.; Jackson, E.K.; Gorelik, E.; Whiteside, T.L. Generation and Accumulation of Immunosuppressive Adenosine by Human CD4+CD25highFOXP3+ Regulatory T Cells. J. Biol. Chem. 2010, 285, 7176–7186. [Google Scholar] [CrossRef] [Green Version]
- Bono, M.R.; Tejon, G.; Flores-Santibañez, F.; Fernandez, D.; Rosemblatt, M.; Sauma, D. Retinoic Acid as a Modulator of T Cell Immunity. Nutrients 2016, 8, 349. [Google Scholar] [CrossRef] [PubMed]
- Clemente-Casares, X.; Blanco, J.; Ambalavanan, P.; Yamanouchi, J.; Singha, S.; Fandos, C.; Tsai, S.; Wang, J.; Garabatos, N.; Izquierdo, C.; et al. Expanding antigen-specific regulatory networks to treat autoimmunity. Nature 2016, 530, 434–440. [Google Scholar] [CrossRef]
- Tsai, S.; Shameli, A.; Yamanouchi, J.; Clemente-Casares, X.; Wang, J.; Serra, P.; Yang, Y.; Medarova, Z.; Moore, A.; Santamaria, P. Reversal of Autoimmunity by Boosting Memory-like Autoregulatory T Cells. Immunity 2010, 32, 568–580. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, T.K.; Maldonado, R.A. Nanoparticles for the Induction of Antigen-Specific Immunological Tolerance. Front. Immunol. 2018, 9, 230. [Google Scholar] [CrossRef]
- Capini, C.; Jaturanpinyo, M.; Chang, H.-I.; Mutalik, S.; McNally, A.; Street, S.; Steptoe, R.; O’Sullivan, B.; Davies, N.; Thomas, R. Antigen-Specific Suppression of Inflammatory Arthritis Using Liposomes. J. Immunol. 2009, 182, 3556–3565. [Google Scholar] [CrossRef] [PubMed]
- Linke, M.; Fritsch, S.D.; Sukhbaatar, N.; Hengstschläger, M.; Weichhart, T. mTORC1 and mTORC2 as regulators of cell metabolism in immunity. FEBS Lett. 2017, 591, 3089–3103. [Google Scholar] [CrossRef]
- Tostanoski, L.H.; Chiu, Y.-C.; Gammon, J.M.; Simon, T.; Andorko, J.I.; Bromberg, J.S.; Jewell, C.M. Reprogramming the Local Lymph Node Microenvironment Promotes Tolerance that Is Systemic and Antigen Specific. Cell Rep. 2016, 16, 2940–2952. [Google Scholar] [CrossRef]
- Sun, C.-M.; Hall, J.A.; Blank, R.B.; Bouladoux, N.; Oukka, M.; Mora, J.R.; Belkaid, Y. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J. Exp. Med. 2007, 204, 1775–1785. [Google Scholar] [CrossRef]
- Wawrzyniak, M.; O’Mahony, L.; Akdis, M. Role of Regulatory Cells in Oral Tolerance. Allergy Asthma Immunol. Res. 2017, 9, 107–115. [Google Scholar] [CrossRef]
- Yu, W.; Freeland, D.M.H.; Nadeau, K.C. Food allergy: Immune mechanisms, diagnosis and immunotherapy. Nat. Rev. Immunol. 2016, 16, 751–765. [Google Scholar] [CrossRef]
- Moorman, C.D.; Sohn, S.J.; Phee, H. Emerging Therapeutics for Immune Tolerance: Tolerogenic Vaccines, T cell Therapy, and IL-2 Therapy. Front. Immunol. 2021, 12, 657768. [Google Scholar] [CrossRef] [PubMed]
- Weiner, H.L.; Mackin, G.A.; Matsui, M.; Orav, E.J.; Khoury, S.J.; Dawson, D.M.; Hafler, D.A. Double-Blind Pilot Trial of Oral Tolerization with Myelin Antigens in Multiple Sclerosis. Science 1993, 259, 1321–1324. [Google Scholar] [CrossRef]
- Benson, J.M.; Stuckman, S.S.; Cox, K.L.; Wardrop, R.M.; Gienapp, I.E.; Cross, A.; Trotter, J.L.; Whitacre, C.C. Oral administration of myelin basic protein is superior to myelin in suppressing established relapsing experimental autoimmune encephalomyelitis. J. Immunol. 1999, 162, 6247–6254. [Google Scholar]
- Bielekova, B.; Goodwin, B.; Richert, N.; Cortese, I.; Kondo, T.; Afshar, G.; Gran, B.; Eaton, J.; Antel, J.; Frank, J.A.; et al. Encephalitogenic potential of the myelin basic protein peptide (amino acids 83–99) in multiple sclerosis: Results of a phase II clinical trial with an altered peptide ligand. Nat. Med. 2000, 6, 1167–1175. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.-U.; Lee, W.-K.; Ryoo, J.-W.; Kim, S.-H.; Kim, J.; Youn, J.; Min, S.-Y.; Bae, E.-Y.; Hwang, S.-Y.; Park, S.-H.; et al. Suppression of collagen-induced arthritis by single administration of poly(lactic-co-glycolic acid) nanoparticles entrapping type II collagen: A novel treatment strategy for induction of oral tolerance. Arthritis Care Res. 2002, 46, 1109–1120. [Google Scholar] [CrossRef] [PubMed]
- Dhadwar, S.S.; Kiernan, J.; Wen, J.; Hortelano, G. Repeated oral administration of chitosan/DNA nanoparticles delivers functional FVIII with the absence of antibodies in hemophilia A mice. J. Thromb. Haemost. 2010, 8, 2743–2750. [Google Scholar] [CrossRef]
- Getts, D.R.; McCarthy, D.P.; Miller, S.D. Exploiting Apoptosis for Therapeutic Tolerance Induction. J. Immunol. 2013, 191, 5341–5346. [Google Scholar] [CrossRef]
- Kontos, S.; Kourtis, I.C.; Dane, K.Y.; Hubbell, J.A. Engineering antigens for in situ erythrocyte binding induces T-cell deletion. Proc. Natl. Acad. Sci. USA 2012, 110, E60–E68. [Google Scholar] [CrossRef]
- Pishesha, N.; Bilate, A.M.; Wibowo, M.C.; Huang, N.-J.; Li, Z.; Deshycka, R.; Bousbaine, D.; Li, H.; Patterson, H.C.; Dougan, S.K.; et al. Engineered erythrocytes covalently linked to antigenic peptides can protect against autoimmune disease. Proc. Natl. Acad. Sci. USA 2017, 114, 3157–3162. [Google Scholar] [CrossRef]
- Yau, F.C.; Balu-Iyer, S.V. Effect of an Active Phosphatidylserine Species on Antigen-Specific Tolerance Induction towards Factor VIII in Hemophilia A Mice. FASEB J. 2017, 31, 674.10. [Google Scholar] [CrossRef]
- Pujol-Autonell, I.; Serracant-Prat, A.; Cano-Sarabia, M.; Ampudia, R.M.; Rodríguez-Fernández, S.; Sanchez, A.; Izquierdo, C.; Stratmann, T.; Puig-Domingo, M.; Maspoch, D.; et al. Use of Autoantigen-Loaded Phosphatidylserine-Liposomes to Arrest Autoimmunity in Type 1 Diabetes. PLoS ONE 2015, 10, e0127057. [Google Scholar] [CrossRef] [Green Version]
- Roberts, R.A.; Eitas, T.K.; Byrne, J.D.; Johnson, B.M.; Short, P.J.; McKinnon, K.P.; Reisdorf, S.; Luft, J.; DeSimone, J.M.; Ting, J.P. Towards programming immune tolerance through geometric manipulation of phosphatidylserine. Biomaterials 2015, 72, 1–10. [Google Scholar] [CrossRef]
- Sun, J.; Shi, J.; Li, J.; Wu, M.; Li, Y.; Jia, S.; Ma, C.; Wang, X.; Li, Z.; Hu, N.; et al. The Effect of Immunosuppressive Adjuvant Kynurenine on Type 1 Diabetes Vaccine. Front. Immunol. 2021, 12, 681328. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; He, Y.; Cheng, K.; Zhang, D.; Miao, S.; Zhang, A.; Meng, F.; Miao, F.; Zhang, J. Killer artificial antigen-presenting cells deplete alloantigen-specific T cells in a murine model of alloskin transplantation. Immunol. Lett. 2011, 138, 144–155. [Google Scholar] [CrossRef] [PubMed]
- Macauley, M.; Pfrengle, F.; Rademacher, C.; Nycholat, C.M.; Gale, A.J.; Von Drygalski, A.; Paulson, J.C. Antigenic liposomes displaying CD22 ligands induce antigen-specific B cell apoptosis. J. Clin. Investig. 2013, 123, 3074–3083. [Google Scholar] [CrossRef]
- Benito-Villalvilla, C.; Pérez-Diego, M.; Angelina, A.; Kisand, K.; Rebane, A.; Subiza, J.L.; Palomares, O. Allergoid–mannan conjugates reprogram monocytes into tolerogenic dendritic cells via epigenetic and metabolic rewiring. J. Allergy Clin. Immunol. 2022, 149, 212–222.e9. [Google Scholar] [CrossRef]
- Nagy, N.A.; de Haas, A.M.; Geijtenbeek, T.B.H.; van Ree, R.; Tas, S.W.; van Kooyk, Y.; de Jong, E.C. Therapeutic Liposomal Vaccines for Dendritic Cell Activation or Tolerance. Front. Immunol. 2021, 12, 674048. [Google Scholar] [CrossRef]
- Dalod, M.; Chelbi, R.; Malissen, B.; Lawrence, T. Dendritic cell maturation: Functional specialization through signaling specificity and transcriptional programming. EMBO J. 2014, 33, 1104–1116. [Google Scholar] [CrossRef]
- Castenmiller, C.; Keumatio-Doungtsop, B.C.; van Ree, R.; de Jong, E.C.; van Kooyk, Y. Tolerogenic Immunotherapy: Targeting DC Surface Receptors to Induce Antigen-Specific Tolerance. Front. Immunol. 2021, 12, 643240. [Google Scholar] [CrossRef]
- Collin, M.; Bigley, V. Human dendritic cell subsets: An update. Immunology 2018, 154, 3–20. [Google Scholar] [CrossRef]
- Menges, M.; Rößner, S.; Voigtländer, C.; Schindler, H.; Kukutsch, N.A.; Bogdan, C.; Erb, K.J.; Schuler, G.; Lutz, M.B. Repetitive Injections of Dendritic Cells Matured with Tumor Necrosis Factor α Induce Antigen-specific Protection of Mice from Autoimmunity. J. Exp. Med. 2001, 195, 15–22. [Google Scholar] [CrossRef]
- Lutz, M.B.; Schuler, G. Immature, semi-mature and fully mature dendritic cells: Which signals induce tolerance or immunity? Trends Immunol. 2002, 23, 445–449. [Google Scholar] [CrossRef]
- Fucikova, J.; Palova-Jelinkova, L.; Bartunkova, J.; Spisek, R. Induction of Tolerance and Immunity by Dendritic Cells: Mechanisms and Clinical Applications. Front. Immunol. 2019, 10, 2393. [Google Scholar] [CrossRef] [PubMed]
- Mosanya, C.H.; Isaacs, J.D. Tolerising cellular therapies: What is their promise for autoimmune disease? Ann. Rheum. Dis. 2019, 78, 297–310. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Qian, S.; Liang, X.; Wang, L.; Woodward, J.E.; Giannoukakis, N.; Robbins, P.D.; Bertera, S.; Trucco, M.; Fung, J.J.; et al. Prevention of Diabetes in NOD Mice by Administration of Dendritic Cells Deficient in Nuclear Transcription Factor-κB Activity. Diabetes 2003, 52, 1976–1985. [Google Scholar] [CrossRef] [PubMed]
- Verginis, P.; Li, H.S.; Carayanniotis, G. Tolerogenic Semimature Dendritic Cells Suppress Experimental Autoimmune Thyroiditis by Activation of Thyroglobulin-Specific CD4+CD25+T Cells. J. Immunol. 2005, 174, 7433–7439. [Google Scholar] [CrossRef] [PubMed]
- Benham, H.; Nel, H.J.; Law, S.C.; Mehdi, A.M.; Street, S.; Ramnoruth, N.; Pahau, H.; Lee, B.T.; Ng, J.; Brunck, M.E.G.; et al. Citrullinated peptide dendritic cell immunotherapy in HLA risk genotype–positive rheumatoid arthritis patients. Sci. Transl. Med. 2015, 7, 290ra87. [Google Scholar] [CrossRef]
- Pistoia, V.; Raffaghello, L. Mesenchymal stromal cells and autoimmunity. Int. Immunol. 2017, 29, 49–58. [Google Scholar] [CrossRef]
- Giannoukakis, N.; Phillips, B.; Finegold, D.; Harnaha, J.; Trucco, M. Phase I (Safety) Study of Autologous Tolerogenic Dendritic Cells in Type 1 Diabetic Patients. Diabetes Care 2011, 34, 2026–2032. [Google Scholar] [CrossRef]
- Zubizarreta, I.; Flórez-Grau, G.; Vila, G.; Cabezón, R.; España, C.; Andorra, M.; Saiz, A.; Llufriu, S.; Sepulveda, M.; Sola-Valls, N.; et al. Immune tolerance in multiple sclerosis and neuromyelitis optica with peptide-loaded tolerogenic dendritic cells in a phase 1b trial. Proc. Natl. Acad. Sci. USA 2019, 116, 8463–8470. [Google Scholar] [CrossRef]
- Jauregui-Amezaga, A.; Cabezón, R.; Ramírez-Morros, A.; España, C.; Rimola, J.; Bru, C.; Pinó-Donnay, S.; Gallego, M.; Masamunt, M.C.; Ordas, I.; et al. Intraperitoneal Administration of Autologous Tolerogenic Dendritic Cells for Refractory Crohn’s Disease: A Phase I Study. J. Crohn’s Colitis 2015, 9, 1071–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, G.M.; Anderson, A.E.; Diboll, J.; Reece, R.; Eltherington, O.; Harry, R.A.; Fouweather, T.; MacDonald, C.; Chadwick, T.; McColl, E.; et al. Autologous tolerogenic dendritic cells for rheumatoid and inflammatory arthritis. Ann. Rheum. Dis. 2017, 76, 234. [Google Scholar] [CrossRef] [PubMed]
- Harry, R.A.; Anderson, A.E.; Isaacs, J.D.; Hilkens, C.M.U. Generation and characterisation of therapeutic tolerogenic dendritic cells for rheumatoid arthritis. Ann. Rheum. Dis. 2010, 69, 2042–2050. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Jung, H.H.; Lee, C.K. Generation, Characteristics and Clinical Trials of Ex Vivo Generated Tolerogenic Dendritic Cells. Yonsei Med. J. 2018, 59, 807–815. [Google Scholar] [CrossRef]
- Price, J.D.; Tarbell, K.V. The role of dendritic cell subsets and innate immunity in the pathogenesis of type 1 diabetes and other autoimmune diseases. Front. Immunol. 2015, 6, 288. [Google Scholar] [CrossRef]
- Zhou, X.; Bailey-Bucktrout, S.L.; Jeker, L.T.; Penaranda, C.; Martínez-Llordella, M.; Ashby, M.; Nakayama, M.; Rosenthal, W.; Bluestone, J.A. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat. Immunol. 2009, 10, 1000–1007. [Google Scholar] [CrossRef]
- Hoffmann, P.; Boeld, T.J.; Eder, R.; Huehn, J.; Floess, S.; Wieczorek, G.; Olek, S.; Dietmaier, W.; Andreesen, R.; Edinger, M. Loss of FOXP3 expression in natural human CD4+CD25+ regulatory T cells upon repetitive in vitro stimulation. Eur. J. Immunol. 2009, 39, 1088–1097. [Google Scholar] [CrossRef]
- Mahnke, K.; Qian, Y.; Knop, J.; Enk, A.H. Induction of CD4+/CD25+ regulatory T cells by targeting of antigens to immature dendritic cells. Blood 2003, 101, 4862–4869. [Google Scholar] [CrossRef]
- Idoyaga, J.; Cheong, C.; Suda, K.; Suda, N.; Kim, Y.; Lee, H.; Park, C.G.; Steinman, R.M. Cutting Edge: Langerin/CD207 Receptor on Dendritic Cells Mediates Efficient Antigen Presentation on MHC I and II Products In Vivo. J. Immunol. 2008, 180, 3647–3650. [Google Scholar] [CrossRef]
- Petzold, C.; Schallenberg, S.; Stern, J.N.; Kretschmer, K. Targeted Antigen Delivery to DEC-205+Dendritic Cells for Tolerogenic Vaccination. Rev. Diabet. Stud. 2012, 9, 305–318. [Google Scholar] [CrossRef]
- Ring, S.; Maas, M.; Nettelbeck, D.M.; Enk, A.H.; Mahnke, K. Targeting of Autoantigens to DEC205+ Dendritic Cells In Vivo Suppresses Experimental Allergic Encephalomyelitis in Mice. J. Immunol. 2013, 191, 2938–2947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Idoyaga, J.; Fiorese, C.; Zbytnuik, L.; Lubkin, A.; Miller, J.; Malissen, B.; Mucida, D.; Merad, M.; Steinman, R.M. Specialized role of migratory dendritic cells in peripheral tolerance induction. J. Clin. Investig. 2013, 123, 844–854. [Google Scholar] [CrossRef] [PubMed]
- Wadwa, M.; Klopfleisch, R.; Buer, J.; Westendorf, A.M. Targeting antigens to DEC-205 on dendritic cells induces immune protection in experimental colitis in mice. Eur. J. Microbiol. Immunol. 2016, 6, 1–8. [Google Scholar] [CrossRef]
- Mukhopadhaya, A.; Hanafusa, T.; Jarchum, I.; Chen, Y.-G.; Iwai, Y.; Serreze, D.V.; Steinman, R.M.; Tarbell, K.V.; DiLorenzo, T.P. Selective delivery of β cell antigen to dendritic cells in vivo leads to deletion and tolerance of autoreactive CD8+ T cells in NOD mice. Proc. Natl. Acad. Sci. USA 2008, 105, 6374–6379. [Google Scholar] [CrossRef] [PubMed]
- Spiering, R.; Margry, B.; Keijzer, C.; Petzold, C.; Hoek, A.; Wagenaar-Hilbers, J.; van der Zee, R.; van Eden, W.; Kretschmer, K.; Broere, F. DEC205+ Dendritic Cell–Targeted Tolerogenic Vaccination Promotes Immune Tolerance in Experimental Autoimmune Arthritis. J. Immunol. 2015, 194, 4804–4813. [Google Scholar] [CrossRef]
- Inaba, K.; Swiggard, W.J.; Inaba, M.; Meltzer, J.; Miryza, A.; Sasagawa, T.; Nussenzweig, M.C.; Steinman, R.U. Tissue Distribution of the DEC-205 Protein That Is Detected by the Monoclonal Antibody NLDC-145. I. Expression on Dendritic Cells and Other Subsets of Mouse Leukocytes. Cell. Immunol. 1995, 163, 148–156. [Google Scholar] [CrossRef]
- Kato, M.; McDonald, K.J.; Khan, S.; Ross, I.L.; Vuckovic, S.; Chen, K.; Munster, D.; MacDonald, K.; Hart, D.N.J. Expression of human DEC-205 (CD205) multilectin receptor on leukocytes. Int. Immunol. 2006, 18, 857–869. [Google Scholar] [CrossRef]
- Sancho, D.; Reis e Sousa, C. Signaling by myeloid C-type lectin receptors in immunity and homeostasis. Annu. Rev. Immunol. 2012, 30, 491–529. [Google Scholar] [CrossRef]
- Cvetkovic, J.; Ilic, N.; Gruden-Movsesijan, A.; Tomic, S.; Mitic, N.; Pinelli, E.; Sofronic-Milosavljevic, L. DC-SIGN signalling induced by Trichinella spiralis products contributes to the tolerogenic signatures of human dendritic cells. Sci. Rep. 2020, 10, 20283. [Google Scholar] [CrossRef]
- Marciani, D.J. Effects of immunomodulators on the response induced by vaccines against autoimmune diseases. Autoimmunity 2017, 50, 393–402. [Google Scholar] [CrossRef]
- Ilarregui, J.M.; Rabinovich, G.A. Tolerogenic Dendritic Cells in the Control of Autoimmune Neuroinflammation: An Emerging Role of Protein-Glycan Interactions. Neuroimmunomodulation 2010, 17, 157–160. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Fan, X.; Huang, H.; Dang, Q.; Lei, H.; Li, Y. A Single Microorganism Epitope Attenuates the Development of Murine Autoimmune Arthritis: Regulation of Dendritic Cells via the Mannose Receptor. Front. Immunol. 2018, 9, 1528. [Google Scholar] [CrossRef] [PubMed]
- Sirvent, S.; Soria, I.; Cirauqui, C.; Cases, B.; Manzano, A.I.; Diez-Rivero, C.M.; Reche, P.A.; López-Relaño, J.; Martínez-Naves, E.; Cañada, F.J.; et al. Novel vaccines targeting dendritic cells by coupling allergoids to nonoxidized mannan enhance allergen uptake and induce functional regulatory T cells through programmed death ligand 1. J. Allergy Clin. Immunol. 2016, 138, 558–567.e11. [Google Scholar] [CrossRef] [PubMed]
- Loschko, J.; Heink, S.; Hackl, D.; Dudziak, D.; Reindl, W.; Korn, T.; Krug, A. Antigen Targeting to Plasmacytoid Dendritic Cells via Siglec-H Inhibits Th Cell-Dependent Autoimmunity. J. Immunol. 2011, 187, 6346–6356. [Google Scholar] [CrossRef]
- Perdicchio, M.; Ilarregui, J.M.; Verstege, M.I.; Cornelissen, L.A.M.; Schetters, S.T.T.; Engels, S.; Ambrosini, M.; Kalay, H.; Veninga, H.; Haan, J.M.M.D.; et al. Sialic acid-modified antigens impose tolerance via inhibition of T-cell proliferation and de novo induction of regulatory T cells. Proc. Natl. Acad. Sci. USA 2016, 113, 3329–3334. [Google Scholar] [CrossRef]
- Hesse, L.; Feenstra, R.; Ambrosini, M.; de Jager, W.A.; Petersen, A.; Vietor, H.; Unger, W.W.J.; van Kooyk, Y.; Nawijn, M.C. Subcutaneous immunotherapy using modified Phl p5a-derived peptides efficiently alleviates allergic asthma in mice. Allergy 2019, 74, 2495–2498. [Google Scholar] [CrossRef]
- Jackson, N.A.C.; Kester, K.E.; Casimiro, D.; Gurunathan, S.; DeRosa, F. The promise of mRNA vaccines: A biotech and industrial perspective. NPJ Vaccines 2020, 5, 11. [Google Scholar] [CrossRef]
- Karikó, K.; Muramatsu, H.; Ludwig, J.; Weissman, D. Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Res. 2011, 39, e142. [Google Scholar] [CrossRef]
- Karikó, K.; Buckstein, M.; Ni, H.; Weissman, D. Suppression of RNA Recognition by Toll-like Receptors: The Impact of Nucleoside Modification and the Evolutionary Origin of RNA. Immunity 2005, 23, 165–175. [Google Scholar] [CrossRef]
- Krienke, C.; Kolb, L.; Diken, E.; Streuber, M.; Kirchhoff, S.; Bukur, T.; Akilli-Öztürk, Ö.; Kranz, L.M.; Berger, H.; Petschenka, J.; et al. A noninflammatory mRNA vaccine for treatment of experimental autoimmune encephalomyelitis. Science 2021, 371, 145–153. [Google Scholar] [CrossRef]
- Bar-Or, A.; Vollmer, T.; Antel, J.; Arnold, D.L.; Bodner, C.A.; Campagnolo, D.; Gianettoni, J.; Jalili, F.; Kachuck, N.; Lapierre, Y.; et al. Induction of Antigen-Specific Tolerance in Multiple Sclerosis After Immunization with DNA Encoding Myelin Basic Protein in a Randomized, Placebo-Controlled Phase 1/2 Trial. Arch. Neurol. 2007, 64, 1407–1415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fissolo, N.M.; Costa, C.; Nurtdinov, R.N.; Bustamante, M.F.; Llombart, V.; Mansilla, M.J.; Espejo, C.; Montalban, X.; Comabella, M. Treatment with MOG-DNA vaccines induces CD4+CD25+FoxP3+ regulatory T cells and up-regulates genes with neuroprotective functions in experimental autoimmune encephalomyelitis. J. Neuroinflamm. 2012, 9, 139. [Google Scholar] [CrossRef] [PubMed]
- Schif-Zuck, S.; Wildbaum, G.; Karin, N. Coadministration of plasmid DNA constructs encoding an encephalitogenic determinant and IL-10 elicits regulatory T cell-mediated protective immunity in the central nervous system. J. Immunol. 2006, 177, 8241–8247. [Google Scholar] [CrossRef]
- Garren, H.; Robinson, W.H.; Krasulová, E.; Havrdová, E.; Nadj, C.; Selmaj, K.; Losy, J.; Nadj, I.; Radue, E.-W.; Kidd, B.A.; et al. Phase 2 trial of a DNA vaccine encoding myelin basic protein for multiple sclerosis. Ann. Neurol. 2008, 63, 611–620. [Google Scholar] [CrossRef]
- Smith, C.E.; Miller, S.D. Multi-peptide coupled-cell tolerance ameliorates ongoing relapsing EAE associated with multiple pathogenic autoreactivities. J. Autoimmun. 2006, 27, 218–231. [Google Scholar] [CrossRef] [PubMed]
- Kendal, A.R.; Waldmann, H. Infectious tolerance: Therapeutic potential. Curr. Opin. Immunol. 2010, 22, 560–565. [Google Scholar] [CrossRef] [PubMed]
- Walsh, P.T.; Taylor, D.K.; Turka, L.A. Tregs and transplantation tolerance. J. Clin. Investig. 2004, 114, 1398–1403. [Google Scholar] [CrossRef]
- Guerrini, G.; Magrì, D.; Gioria, S.; Medaglini, D.; Calzolai, L. Characterization of nanoparticles-based vaccines for COVID-19. Nat. Nanotechnol. 2022, 17, 570–576. [Google Scholar] [CrossRef]
- Chen, J.; Chen, J.; Xu, Q. Current Developments and Challenges of mRNA Vaccines. Annu. Rev. Biomed. Eng. 2022, 24, 85–109. [Google Scholar] [CrossRef]
- Bauman, J.; Burris, H.; Clarke, J.; Patel, M.; Cho, D.; Gutierrez, M.; Julian, R.; Scott, A.; Cohen, P.; Frederick, J.; et al. 798 Safety, tolerability, and immunogenicity of mRNA-4157 in combination with pembrolizumab in subjects with unresectable solid tumors (KEYNOTE-603): An update. J. Immunother. Cancer 2020, 8 (Suppl. S3), A846. [Google Scholar] [CrossRef]
- Jiang, W.; Gupta, R.K.; Deshpande, M.C.; Schwendeman, S.P. Biodegradable poly(lactic-co-glycolic acid) microparticles for injectable delivery of vaccine antigens. Adv. Drug Deliv. Rev. 2005, 57, 391–410. [Google Scholar] [CrossRef] [PubMed]
- Cappellano, G.; Woldetsadik, A.D.; Orilieri, E.; Shivakumar, Y.; Rizzi, M.; Carniato, F.; Gigliotti, C.L.; Boggio, E.; Clemente, N.; Comi, C.; et al. Subcutaneous inverse vaccination with PLGA particles loaded with a MOG peptide and IL-10 decreases the severity of experimental autoimmune encephalomyelitis. Vaccine 2014, 32, 5681–5689. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.F.A.; Goth, S.R.; Dong, B.; Pessah, I.N.; Matsumura, F. Aryl hydrocarbon receptor signaling mediates expression of indoleamine 2,3-dioxygenase. Biochem. Biophys. Res. Commun. 2008, 375, 331–335. [Google Scholar] [CrossRef]
- Kenison, J.E.; Jhaveri, A.; Li, Z.; Khadse, N.; Tjon, E.; Tezza, S.; Nowakowska, D.; Plasencia, A.; Stanton, V.P., Jr.; Sherr, D.H.; et al. Tolerogenic nanoparticles suppress central nervous system inflammation. Proc. Natl. Acad. Sci. USA 2020, 117, 32017–32028. [Google Scholar] [CrossRef]
- Rojas, C.; Campos-Mora, M.; Cárcamo, I.; Villalón, N.; Elhusseiny, A.; Contreras-Kallens, P.; Refisch, A.; Gálvez-Jirón, F.; Emparán, I.; Montoya-Riveros, A.; et al. T regulatory cells-derived extracellular vesicles and their contribution to the generation of immune tolerance. J. Leukoc. Biol. 2020, 108, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Di Bonito, P.; Chiozzini, C.; Arenaccio, C.; Anticoli, S.; Manfredi, F.; Olivetta, E.; Ferrantelli, F.; Falcone, E.; Ruggieri, A.; Federico, M. Antitumor HPV E7-specific CTL activity elicited by in vivo engineered exosomes produced through DNA inoculation. Int. J. Nanomed. 2017, 12, 4579. [Google Scholar] [CrossRef]
- Barberis, E.; Vanella, V.V.; Falasca, M.; Caneparo, V.; Cappellano, G.; Raineri, D.; Ghirimoldi, M.; de Giorgis, V.; Puricelli, C.; Vaschetto, R.; et al. Circulating Exosomes Are Strongly Involved in SARS-CoV-2 Infection. Front. Mol. Biosci. 2021, 8, 29. [Google Scholar] [CrossRef]
- Cappellano, G.; Raineri, D.; Rolla, R.; Giordano, M.; Puricelli, C.; Vilardo, B.; Manfredi, M.; Cantaluppi, V.; Sainaghi, P.P.; Castello, L.; et al. Circulating Platelet-Derived Extracellular Vesicles Are a Hallmark of Sars-Cov-2 Infection. Cells 2021, 10, 85. [Google Scholar] [CrossRef]
- Martins, P.; Machado, D.; Theizen, T.H.; Guarnieri, J.P.O.; Bernardes, B.G.; Gomide, G.P.; Corat, M.A.F.; Abbehausen, C.; Proenca-Modena, J.L.; Melo, C.F.O.R.; et al. Outer Membrane Vesicles from Neisseria Meningitidis (Proteossome) Used for Nanostructured Zika Virus Vaccine Production. Sci. Rep. 2018, 8, 8290. [Google Scholar] [CrossRef]
- Manolova, V.; Flace, A.; Bauer, M.; Schwarz, K.; Saudan, P.; Bachmann, M.F. Nanoparticles target distinct dendritic cell populations according to their size. Eur. J. Immunol. 2008, 38, 1404–1413. [Google Scholar] [CrossRef]
- Benne, N.; van Duijn, J.; Kuiper, J.; Jiskoot, W.; Slütter, B. Orchestrating immune responses: How size, shape and rigidity affect the immunogenicity of particulate vaccines. J. Control. Release 2016, 234, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Shima, F.; Uto, T.; Akagi, T.; Baba, M.; Akashi, M. Size effect of amphiphilic poly(γ-glutamic acid) nanoparticles on cellular uptake and maturation of dendritic cells in vivo. Acta Biomater. 2013, 9, 8894–8901. [Google Scholar] [CrossRef] [PubMed]
- Mant, A.; Chinnery, F.; Elliott, T.; Williams, A.P. The pathway of cross-presentation is influenced by the particle size of phagocytosed antigen. Immunology 2012, 136, 163–175. [Google Scholar] [CrossRef]
- Brewer, J.M.; Pollock, K.G.J.; Tetley, L.; Russell, D.G. Vesicle Size Influences the Trafficking, Processing, and Presentation of Antigens in Lipid Vesicles. J. Immunol. 2004, 173, 6143–6150. [Google Scholar] [CrossRef]
- Tran, K.K.; Shen, H. The role of phagosomal pH on the size-dependent efficiency of cross-presentation by dendritic cells. Biomaterials 2009, 30, 1356–1362. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Kwon, J.E.; Cho, M.-L. Immunological pathogenesis of inflammatory bowel disease. Intest. Res. 2018, 16, 26–42. [Google Scholar] [CrossRef]
- Zhuang, Q.; Cai, H.; Cao, Q.; Li, Z.; Liu, S.; Ming, Y. Tolerogenic Dendritic Cells: The Pearl of Immunotherapy in Organ Transplantation. Front. Immunol. 2020, 11, 552988. [Google Scholar] [CrossRef]
- Slepicka, P.F.; Yazdanifar, M.; Bertaina, A. Harnessing Mechanisms of Immune Tolerance to Improve Outcomes in Solid Organ Transplantation: A Review. Front. Immunol. 2021, 12, 688460. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Tolerance Type | Mechanism |
|---|---|
| Deletional tolerance: induction of apoptosis (or anergy) of the antigen-specific lymphocytes | |
| Lack of danger signals | Low expression of MHC I and/or MHC II molecules by APCs → weak signal one. Low expression of co-stimulatory molecules (CD80, CD86, CD28) → weak signal two. Low levels of pro-inflammatory and T cell-polarizing cytokines (IFN-γ, IL-1, IL-2, IL-4, IL-17) → weak signal three. |
| Excessive triggering of antigen receptors | Expression of high levels of autoantigens |
| Expression of death receptors | Tissues expressing high levels of FasL delete activated lymphocytes expressing Fas. |
| Dominant tolerance: activity of several types of regulatory (suppressive) cells | |
| Increased production of anti-inflammatory cytokines | Increased synthesis and release of IL-10, TGF-β, IL-4, and NO, result in immune cell anergy and differentiation into several types of regulatory cells. |
| Increased expression of co-inhibitory molecules | Increased expression of CTLA-4, PD1, and PD-L1, by both APCs and T cells, mediate a downregulation of T-cell activity despite adequate antigen presentation. |
| Direct killing of immune cells (fratricide) | Perforin- and granzyme-mediated killing of immune cells by several types of cytotoxic lymphocytes. |
| Metabolic induction of regulatory T cells | IDO-mediated conversion of tryptophan into kynurenines, AhR-mediated differentiation into Tregs, and functional suppression of activated T cells. Creation of a “purinergic halo” around immune cells through a CD39- and CD73- mediated increase in immunosuppressive extracellular adenosine. Promotion of Treg differentiation through DC RALD2, turning vitamin A into the immunoregulator, retinoic acid. |
| Study Group | Year | Study Type * | Animals/Subjects/ Cell Types under Study | Vaccine Type | Clinical Application | Results |
|---|---|---|---|---|---|---|
| Clemente-Casares et al. [23] | 2016 | Preclinical in vivo | NOD mice | Systemic delivery of uncoated nanoparticles or nanoparticles coated with a pMHC that is recognized by the diabetogenic BDC2.5-specific T-cell receptor (TCR). | Animal model of autoimmune diabetes | Expansion of memory-like (CD44hiCD62Llow) FOXP3− TR1-like T cells, leading to the suppression of autoantigen-loaded APCs and the differentiation of B cells into autoimmune disease-suppressing B cells. |
| Mice expressing a transgenic hybrid MHCII molecule composed of the peptide-binding domain of human HLA-DR4 and the membrane-proximal domain of mouse IE. | Systemic delivery of nanoparticles diplaying mouse collagen (mCII)259–273/DR4-IE. | CIA | Expansion of TR1-like T cells, reduction of joint inflammation in arthritic mice. | |||
| Mice expressing a transgenic hybrid MHCII molecule composed of the peptide-binding domain of human HLA-DR4 and the membrane-proximal domain of mouse IE. | Systemic delivery of nanoparticles coated with human MOG97–108/DR4-IE. | EAE | Systemic expansion of cognate TR1-like T cells, EAE blunting. | |||
| Tsai et al. [24] | 2010 | Preclinical in vivo | NOD mice | IV injectrion of iron oxide nanoparticles coated with pMHC displaying IGRP. | Animal model of autoimmune diabetes | Expansion of autoregulatory T cells, suppression of local autoantigen presentation by APCs, disease prevention in prediabetic mice, and restoration of glycemic control in diabetic animals. |
| Capini et al. [26] | 2009 | Preclinical in vivo | Mice primed with Ag to induce inflammatory arthritis | IV or SC injection of egg phosphatidylcholine liposomes loaded with OVA or methylated BSA and a lipophilic NF-κB inhibitor (curcumin, quercetin, or Bay11-7082). | AIA | Suppression of preexisting immune responses in an Ag-specific manner, in situ suppression of APC responsiveness to NF-κB, and induction of Ag-specific FoxP3+ regulatory T cells, reduction of joint severity scores. |
| Tostanoski et al. [28] | 2016 | Preclinical in vivo | Ten to eleven week-old female mice primed with MOG35–55 and pertussis toxin to induce EAE | Intranodal injection of polymer particles encapsulating MOG35–55 and rapamycin. | EAE | Local LN reorganization, reduced inflammation, systemic expansion of Tregs, reduced T cell infiltration to the CNS, reversal of paralysis after a single treatment. |
| Weiner et al. [33] | 1993 | Double-blind clinical trial | Thirty subjects with RR–MS | Daily oral administration of capsules of bovime myelin or control protein. | RR–MS | Reduction of the number of T cells that were reactive to MBP in the myelin-treated group, disease exacerbation in 6 out of 15 treated subjects versus 12 out of 15 controls, no toxicity or side effects in etheir group. |
| Benson et al. [34] | 1999 | Preclinical in vivo | Mice immunized with myelin antigens to induce REAE | Oral administration of high doses of myelin or MBP either before disease induction or during the course of the disease. | REAE | No reduction of in vitro T cell responses, nor protection from disease after oral administration of heterogeneous myelin, versus a reduction in IL-2-, IFN-γ-, and IL-5-secreting MBP-specific T cells and suppression of REAE after repeated oral administration of homogeneous MBP. |
| Bielekova et al. [35] | 2000 | Phase II clinical trial | Twenty-four patients with RR–MS | Weekly subcutaneous administration of 50 mg of CGP77116, an altered peptide ligand mimicking MBP83–99. | RR–MS | Exacerbation of MS in three patients, trial interruption due to poor tolerance of the administered peptide. |
| Weiner [34] ** | / | Multicenter clinical trial controlled for patients’ gender and steroid treatments | Five hundred patients with RR–MS | Daily oral administration of 300 mg of bovine myelin or casein. | RR–MS | No clinical improvement after bovine myelin administration. Results not published. |
| Kim et al. [36] | 2002 | Preclinical in vivo | Mice immunized with CII to induce CIA 14 days after vaccine administration | Prophylactic oral administration of PLGA nanoparticles, entrapping CII 14 days before immunization. | CIA | Higher level of TGF-β mRNA expression in Peyer’s patches, lower level of TNF-α mRNA expression in draining lymph nodes in treated mice, presence of serum anti-CII IgG antibodies and CII-specific T cell proliferation, reduced incidence and severity of CIA. |
| Dhadwar et al. [37] | 2010 | Preclinical in vivo | FVIII KO mice | Oral administration of chitosan nanoparticles containing canine FVIII DNA on two biconsecutive days. | Animal model of hemophilia | Decrease in aPTT, stable clot formation, restoration of FVIII activity, disappearance of FVIII inhibitors and non-neutralizing anti-FVIII antibodies. |
| Kontos et al. [38] | 2012 | Preclinical in vivo | NOD mice receiving transgenic diabetogenic CD4+ T cells to induce rapid diabetes onset | IV administration of a peptide antigen fused to an erythrocyte-binding antibody. | Animal model of autoimmune diabetes | Prevention of diabetes onset. |
| Yau et al. [41] | 2017 | Preclinical in vivo | FVIII KO mice | Six weekly SC injections of FVIII in the absence and presence of liposomes bearing s-PS, followed by a re-challenge with four weekly SC injections of solely FVIII in half of the animals, and OVA in the other half 24 h after liposome treatment. | Animal model of hemophilia | Reduction in both the total anti-FVIII titers and neutralizing titers, robust immune response to the irrelevant antigen OVA, irrespective of treatment, with s-PS liposomes or free FVIII. |
| Pujol-Autonell et al. [42] | 2015 | Preclinical in vivo | Normoglycemic NOD mice at 8 weeks of age (at least 12 per group) | Single IP dose of 3.5 mg of PS-liposomes (empty or insulin peptide-filled) or a saline solution, followed by monitoring until 30 weeks of age. | Animal model of autoimmune diabetes | Induction of tolerogenic dendritic cells, impairment of antigen-specific autoreactive T cell proliferation, reduction of insulitis severity and prevention of T1D, significant expansion of antigen-specific CD4+ T cells in the spleen, pancreatic, and mediastinal lymph nodes in mice treated with insulin-filled PS-liposomes compared with those receiving empty PS-liposomes or saline. |
| Roberts et al. [43] | 2015 | Preclinical in vivo | Mice challenged with a SC MOG35–55 peptide using a complete Freund’s adjuvant and heat-killed mycobacterium tuberculosis to induce EAE | Four-day-long IV administration of 50 μg of liposomal PS, PS-loaded PLGA nanorods, or blank PLGA nanoparticles seven days after the MOG35–55 challenge. | EAE | Significant suppression of IL-6 and IL-12 by DCs and of IFN-γ, IL-2, IL-6, and TNF-α by T cells, decreased disease burden, more pronounced in the mouse group receiving PS-loaded PLGA nanorods versus liposomal PS. |
| Sun et al. [44] | 2021 | Preclinical in vivo | Female NOD mice at 4–6 weeks of age (12 animals per group) | Double SC immunization with GAD65 phage vaccine alone or mixed with 200 mg of Kyn. | Animal model of autoimmune diabetes | In the group receiving a GAD65 phage vaccine mixed with Kyn, there was an enhancement of Th2-mediated immune responses, regulation of the Th1/Th2 imbalance, increased secretion of Th2 cytokines and of CD4+ CD25+ Foxp3+ T cells, suppression of DC maturation and GAD65-specific T lymphocyte proliferation, and prevention of hyperglycemia in 60% of mice for at least one month. |
| Shen et al. [45] | 2011 | Preclinical in vivo | H-2K(d) mice grafted with skin squares from another mouse strain (H-2K(b)) | IV administration of latex beads covalently coupled to H-2K(b)/peptide monomers and anti-Fas mAb after skin engraftment. | Allograft rejection | Prolongation of alloskin graft survival for 6 days, 60% decrease of H-2K(b) antigen-alloreactive T cells. |
| Macauley et al. [46] | 2013 | Preclinical in vivo | FVIII-deficient mice challenged with human FVIII | Liposomal nanoparticles displaying CD22 ligands (STALs) and FVIII. | Hemophilia mouse model | Prevention of anti-FVIII inhibitory antibody formation, prevention of bleeding in the tail-cut assay after human FVIII administration. |
| Benito-Villalvilla et al. [47] | 2022 | Preclinical in vitro | Monocytes from nonatopic and allergic human subjects | Culture of monocytes with allergoid–mannan conjugates. | Allergy | Differentiation of monocytes into stable tolerogenic DCs, production of fewer cytokines, a lower TNF-α/IL-10 ratio, and higher expression of the tolerogenic molecules PDL1, IDO, SOCS1, SOCS3, and IL10 after LPS stimulation, induction of higher numbers of functional FOXP3+ Tregs, shift of glucose metabolism due to the Warburg effect, lactate production due to mitochondrial oxidative phosphorylation, epigenetic reprogramming within tolerogenic loci, increased expression of the anti-inflammatory miRNA-146a/b, and decreased expression of proinflammatory miRNA-155. |
| Ma et al. [56] | 2003 | Preclinical in vivo | Female six to seven week-old NOD mice | Single IV injection of MBDCs cultured with ODNs containing a consensus of NF-κB binding sites that inhibit NF-κB activity, stimulated with LPS, and primed with lysate of pancreatic islets for the last 48 h of culture. | Animal model of autoimmune diabetes | Suppression of costimulatory molecule expression, IL-12 production, and immunostimulatory capacity in presenting allo- and islet-associated antigens using NF-κB ODN DC, prevention of diabetes onset, pancreatic T-cell hyporesponsiveness in islet antigens with low production of IFN-γ and IL-2. |
| Verginis et al. [57] | 2005 | Preclinical in vivo | Female six to eight-week old female mice challenged with Tg | IV administration of TNF-α-treated, semimature BMDCs pulsed with Tg or OVA. | EAT | Inhibition of the subsequent development of Tg-induced EAT after administration of DCs pulsed with Tg, but not with OVA. |
| Benham et al. [58] | 2015 | Single-center, open-labeled, human phase I trial | Eighteen HLA risk genotype-positive RA patients with citrullinated peptide-specific autoimmunity and 16 RA patients as controls | Single ID administration of Rheumavax: autologous DCs modified with a NF-kB inhibitor that is exposed to four citrullinated peptide antigens. | RA | Reduction of effector T cells and an increased ratio of Treg to effector T cells, reduction in serum IL-15, IL-29, CX3CL1, and CXCL11, and reduced T cell IL-6 responses to vimentin447–455–Cit450 relative to controls, reduction in disease activity scores one month after treatment, good tolerance with minimal side effects. |
| Bell et al. [63] | 2017 | Unblinded, randomised, controlled, dose escalation phase I trial | Patients with knee RA, three patients per cohort | Arthroscopic injection of 1 × 106, 3 × 106 or 10 × 106 autologous DCs differentiated from PBMCs obtained by leukapheresis and tolerized using dexamethasone and vitamin D. | RA | Stabilization of knee symptoms in two patients receiving 10 × 106 tolDC but no systemic clinical or immunomodulatory effects were detectable. |
| Harry et al. [64] | 2010 | Preclinical in vitro | Monocyte-derived DCs from RA patients and controls | Culture of monocyte-derived DCs with the immunosuppressive drugs dexamethasone, vitamin D₃, and the immunomodulator monophosphoryl lipid A. | RA | Induction of a tolerogenic phenotype in DCs from RA patients (reduced costimulatory molecules, low production of proinflammatory cytokines, and impaired stimulation of autologous antigen-specific T cells), comparable to healthy control tolDCs, refractoriness to further challenge with proinflammatory mediators. |
| Giannoukakis et al. [60] | 2011 | Randomized, double-blind, phase I study | Ten insulin-requiring type 1 diabetic patients between 18 and 60 years of age | ID abdominal administration of 10 million autologous BMDCs, unmanipulated or engineered ex vivo toward an immunosuppressive state (with a mixture of antisense oligonucleotides targeting the primary transcripts of CD40, CD80, and CD86) once every two weeks for a total of four administrations. | T1D | Increased frequency of peripheral B220+ CD11c − B cells, but no statistically relevant differences in immune populations or biochemical, hematological, and immune biomarkers compared with baseline results. |
| Jauregui-Amezaga et al. [62] | 2015 | Phase I, single-centre, sequential-cohorts, dose-range study | Nine refractory-CD patients | Single versus three biweekly IP injection of tolDCs (treated with dexamethasone and vitamin A) at escalating doses (2 × 106/5 × 106/10 × 106), one-year follow-up. | CD | Clinical remission in 11% of participants, clinical response in 22% of participants, and lesion improvement in 33% of patients. An increase of circulating Tregs and decrease in IFN-γ levels. Withdrawal suffered by three patients due to CD symptoms worsening. |
| Zubizarreta et al. [61] | 2019 | Human phase 1b clinical trial | Twelve patients, 8 with MS and 4 with NMOSD | IV administration of three doses of autologous DCs differentiated from PBMCs obtained by leukapheresis and tolerized with dexamethasone. | MS and NMOSDs | Good tolerance, significant increase in the production of IL-10 and IFN-γ levels, increased frequency of Tr1 cells and switch towards Th2 responses by week 12 of follow-up. |
| NCT02354911 | 2015–2019 | Randomized, quadruple-blind, placebo-controlled, cross-over, phase II clinical trial | 24 subjects with T1D | Abdominal ID injection of autologous DCs harvested by leukapheresis and engineered ex vivo via incubation with antisense DNA oligonucleotides that target the primary transcripts of CD40, CD80, and CD86 to convert to active immunoregulators (four separate injections at 2 week intervals). | T1D | Primary endpoint: change from baseline with a mean of 2 h, AUC for plasma C-peptide at 12 and 24 months. Secondary endpoints: adverse events, changes in HbA1c profile, insulin needed, immunological changes. |
| NCT02622763 | 2015–2019 | Randomized, single-blinded, phase I clinical trial | Three patients with refractory CD | Intralesional administration of tolDCs. | CD | Primary endpoints: adverse events, clinical response. Secondary endpoints: clinical remission, quality of life, lesion severity. |
| NCT03337165 | 2016–2019 | Open-label, phase I clinical trial | Ten patients with RA | Single intra-articular injection (into the knee joint) of autologous moDCs generated in the presence of IFN-α/GM-CSF and tolerized with dexamethasone. | RA | Primary endpoint: adverse events. Secondary endpoint: change in clinical severity. |
| NCT02903537 (TOLERVIT-MS) | 2017–2021 | Non-randomized, open-label, phase I clinical trial | Sixteen participants with RR–MS | Intranodal administration of autologous moDCs tolerized with vitamin-D3 and pulsed with myelin peptides. | MS | Primary endpoints: adverse events, neurologic, and imaging changes. Secondary endpoints: relapse rate, clinical efficacy, immunological changes |
| NCT02618902 (MS-tolDC) | 2017–2021 | Open-label, dose-escalating, phase I clinical trial | Nine subjects with MS | ID injection of tolDCs in the subclavicular region. | MS | Primary endpoints: safety and feasibility. Secondary endpoints: clinical impact, lesion severity, immunological changes. |
| ONE Study ATDC Trial (NCT02252055) | 2015–2018 | Phase I/II monocentric trial | Eleven patients with renal insufficiency receiving a first kidney transplant from a living donor | IV administration of autologous tolDCs the day before transplantation. | Organ transplantation | Primary endopint: incidence of biopsy-confirmed acute rejection Secondary endpoints: incidence of short- and long-term complications (including malignancy), immunological conditions, total immunosuppressive burden |
| NCT02283671 | 2015–2019 | Phase I, open-label clinical trial | Twenty patients with MS or NMO | IV administration of tolDCs loaded with myelin peptides every two weeks for a total of three administrations per patient. | MS, NMO | Primary endpoint: tolerability and safety Secondary endopoints: disease severity, changes in immunological profile |
| Mahnke et al. [69] | 2003 | Preclinical in vivo | Mice challenged with OVA | SC injection of OVA coupled with anti-DEC-205 mAb. | Antigen-induced hypersensitivity reactions | In vivo induction of a tolerogenic phenotype in DCs, suppression of CD4+ T-cell-mediated hypersensitivity reactions (reduced IL-2 production), reduction of CD8+ T-cell–mediated allergic reactions. |
| Ring et al. [72] | 2013 | Preclinical in vivo | Mice challenged with MOG to induce EAE | IV injection of scFv specific for DEC-205 fused MOG 7, administered three days before the induction of EAE, or one and four days after the induction of EAE. | EAE | Elevated numbers of highly activated, IL-10–producing CD4 + CD25 + Foxp3+ Tregs, increased levels of TGF-β, protection from EAE or EAE abrogation in 90% of the vaccinated mice, compared with isotype controls and uninjected mice. |
| Wadwa et al. [74] | 2016 | Preclinical in vivo | VILLIN-HA transgenic mice receiving HA- specific CD4+ Foxp3− T cells IV to induce intestinal inflammation | IP injection of antibody–antigen complex consisting of the immunogenic HA110–120 peptide coupled to an anti-DEC-205 mAb 2 and one day before adoptive transfer of HA-specific CD4+Foxp3− T cells. | IBD | Reduction of intestinal inflammation, conversion of naive HA-specific CD4 + Foxp3 − T cells into HA-specific CD4 + Foxp3 + Tregs. |
| Mukhopadhaya et al. [75] | 2008 | Preclinical in vivo | NOD mice | Mimotope of a β cell peptide coupled with an anti-DEC-205 mAb. | Animal model of autoimmune diabetes | Deletion of autoreactive CD8+ T cells, absence of immune response after a rechallenge with the mimotope peptide in the adjuvant. |
| Spiering et al. [76] | 2015 | Preclinical in vivo | Mice receiving IP injection of human PG70–84 to induce PGIA | IP injection of human PG70–84 coupled with anti-DEC-205 mAb, 10 or 20 days prior to the induction of PGIA. | PGIA | No alterations in the proportion of Foxp3+ Treg cells, but reduced numbers of IL-17+ and IFN-γ+ cells, impaired germinal center formation, and reduced serum levels of PG-specific IgG2a antibodies. |
| Yang et al. [83] | 2018 | Preclinical in vivo | Mice challenged with collagen to induce CIA | Vaccination or treatment with a LACK156–173 epitope expression plasmid or polypeptide. | CIA | Amelioration of arthritis severity, improvement in the balance of effector T cells in synovial tissue towards a Th2 polarization compared with untreated arthritis controls, decreased expression of TLR4 expression, decreased macrophage activation. |
| Sirvent et al. [84] | 2016 | In vitro and in vivo | hmoDCs from patients with grass pollen allergy | Treatment of hmoDCs with glutaraldehyde-polymerized grass pollen allergoids coupled to nonoxidized mannan (PM), glutaraldehyde-polymerized allergoids (P), native grass pollen extracts (N), or oxidized PM. | Grass pollen allergy | Induction of a tolerogenic phenotype in hmoDCs treated with PM compared with P or N, higher levels of IL-6 and IL-10 and lower IL-4 levels, induction of hmoDC-induced generation of Th1 cells and a Th2/Treg cell shift in favor of Tregs, abolition of these effects after oxidation of PM (due to alteration of the carbohydrate structure). |
| Mouse models | Subcutaneous immunization of mice with PM, P, N, or oxidized PM. | Comparing mice immunized with PM to those immunized with P or N. Higher percentage of Tregs in splenocytes, higher ratio of serum IgG2a/IgE levels that are specific to native grass pollen extracts, higher IL-10/IL-4 ratios after in vitro stimulation of mouse splenocytes with the native grass pollen extract, abolition of these effects after oxidation of PM (due to alteration of the carbohydrate structure). | ||||
| Loschko et al. [85] | 2011 | Preclinical in vivo | Mice challenged with MOG on the same day and two days after immunization | IP injection of OVA coupled with anti-Siglec mAb. | EAE | Reduction of Th1/Th17 cell polarization, but no generation or expansion of MOG-specific Tregs nor deviation to Th2 or Tr1 cells, EAE onset delay and reduction of disease severity. |
| Hesse et al. [87] | 2019 | Preclinical in vivo | Grass pollen-sensitized mice | SC injection of unmodified or sialylated PhI-p5a peptides before grass pollen challenge. | Airway inflammation due to grass pollen allergy | Increased T-cell activation, enhanced numbers of FoxP3+ T cells both in vitro and in vivo, and increased suppression of Th2 cells and eosinophilic inflammation in lung tissue after peptide sialylation compared with unmodified peptides. |
| Fissolo et al. [93] | 2012 | Preclinical in vivo | Mice immunized with MOG35–55 peptide to induce EAE | IM administration of a DNA vaccine encoding MOG prophylactically or therapeutically. | EAE | Reduction of clinical and histopathological signs of EAE in both prophylactic and therapeutic settings, dampening of antigen-specific proinflammatory Th1 and Th17 immune responses, expansion of Tregs in the periphery, upregulation in the CNS of genes encoding neurotrophic factors and proteins involved in remyelination. |
| Bar-Or et al. [92] | 2007 | Randomized, double-blind, multi-centered, placebo-controlled clinical trial | Thirty patients with RR–MS or SP–MSMS patients | IM injection of a DNA vaccine encoding full-length MBP (BHT-3009) at weeks 1, 3, 5, and 9 at three testing doses (0.5 mg, 1.5 mg, and 3 mg). | MS | Good tolerability and safety, marked decrease in proliferation of IFN-γ-producing CD4+ T cells, reduction in titers of myelin-specific autoantibodies in CSF, reduction of inflammatory lesions on brain MRIs. |
| Schif-Zuck et al. [94] | 2022 | Preclinical in vivo | Seven week-old female mice immunized with SC MBP68–86 (six mice per group) | Treatment with plasmid DNA encoding MBP68–86, or plasmid DNA encoding IL-10, or both plasmids coadministered. | EAE | Fast EAE remission only in rats given coadministration of the IL-10-encoding DNA plasmid together with the MBP-encoding plasmids, improvement of EAE-induced CNS lesions at histology (elevation in Ag-specific T cells producing IL-10, increase in apoptosis of cells around high endothelial venules, induction of Tr1-induced active tolerance). |
| Garren et al. [95] | 2008 | Randomized, placebo-controlled, multi-centered phase II trial | Two hundred and eighty-nine RR–MS patients randomized into three groups (1:1:1) | IM injection of placebo, 0.5 mg BHT-3009 (a DNA vaccine encoding full-length MBP), or 1.5 mg BHT-3009 at weeks 0, 2, 4, and every four weeks thereafter until week 44. | MS | Reduction of the rate of new enhancing MRI lesions after treatment with the lower dose (0.5 mg), lack of efficacy at higher doses. |
| Smith et al. [96] | 2006 | Preclinical in vivo | Eight to ten-week-old female mice immunized with SC encephalitogenic peptides to induce EAE | IV administration of splenocytes coupled with a peptide cocktail of four distinct encephalitogenic epitopes immediately after immunization or at the peak of acute disease. | EAE | Inhibition of active EAE initiation, prevention of activation of autoreactive Th1 cells and subsequent infiltration of inflammatory cells into the CNS, prevention of clinical relapses due to epitope spreading and increased production of the anti-inflammatory cytokines TGF-β and/or IL-10 in both the periphery and the CNS when administered at the peak of the acute disease. |
| Cappellano et al. [103] | 2014 | Preclinical in vivo | Four to eight-week old female mice immunized to induce EAE | SC injection of PLGA nanoparticles loaded with MOG 35–55 and recombinant IL-10 either 30 and 15 days before EAE induction or 8 and 22 days after EAE induction. | EAE | Inhibition of EAE development after prophylactic vaccination and significant amelioration of EAE after therapeutic vaccination, decrease in histopathological lesions, reduced secretion of IL-17 and IFN-γ. |
| Kenison et al. [105] | 2020 | Preclinical in vivo | Eight to ten-week old female mice immunized with SC MOG35–55 to induce EAE | Administration of NLPs loaded with an AhR agonist and a T cell epitope from MOG35–55; either SC once a week beginning on day one after disease induction, IV once on day seven for disease prevention, or once on day fifteen for disease reversal. | EAE | EAE suppression, expansion of MOG35–55-specific FoxP3+ Tregs and Tr1 cells, reduction in CNS-infiltrating effector T cells, amelioration of chronic progressive EAE. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puricelli, C.; Boggio, E.; Gigliotti, C.L.; Stoppa, I.; Sutti, S.; Rolla, R.; Dianzani, U. Cutting-Edge Delivery Systems and Adjuvants in Tolerogenic Vaccines: A Review. Pharmaceutics 2022, 14, 1782. https://doi.org/10.3390/pharmaceutics14091782
Puricelli C, Boggio E, Gigliotti CL, Stoppa I, Sutti S, Rolla R, Dianzani U. Cutting-Edge Delivery Systems and Adjuvants in Tolerogenic Vaccines: A Review. Pharmaceutics. 2022; 14(9):1782. https://doi.org/10.3390/pharmaceutics14091782
Chicago/Turabian StylePuricelli, Chiara, Elena Boggio, Casimiro Luca Gigliotti, Ian Stoppa, Salvatore Sutti, Roberta Rolla, and Umberto Dianzani. 2022. "Cutting-Edge Delivery Systems and Adjuvants in Tolerogenic Vaccines: A Review" Pharmaceutics 14, no. 9: 1782. https://doi.org/10.3390/pharmaceutics14091782
APA StylePuricelli, C., Boggio, E., Gigliotti, C. L., Stoppa, I., Sutti, S., Rolla, R., & Dianzani, U. (2022). Cutting-Edge Delivery Systems and Adjuvants in Tolerogenic Vaccines: A Review. Pharmaceutics, 14(9), 1782. https://doi.org/10.3390/pharmaceutics14091782