

Topoisomeric Membrane-Active Peptides: A Review of the Last Two Decades



Abstract
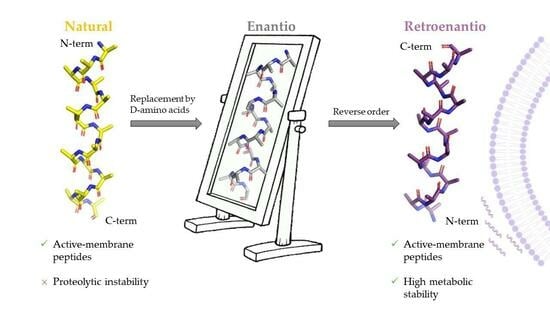
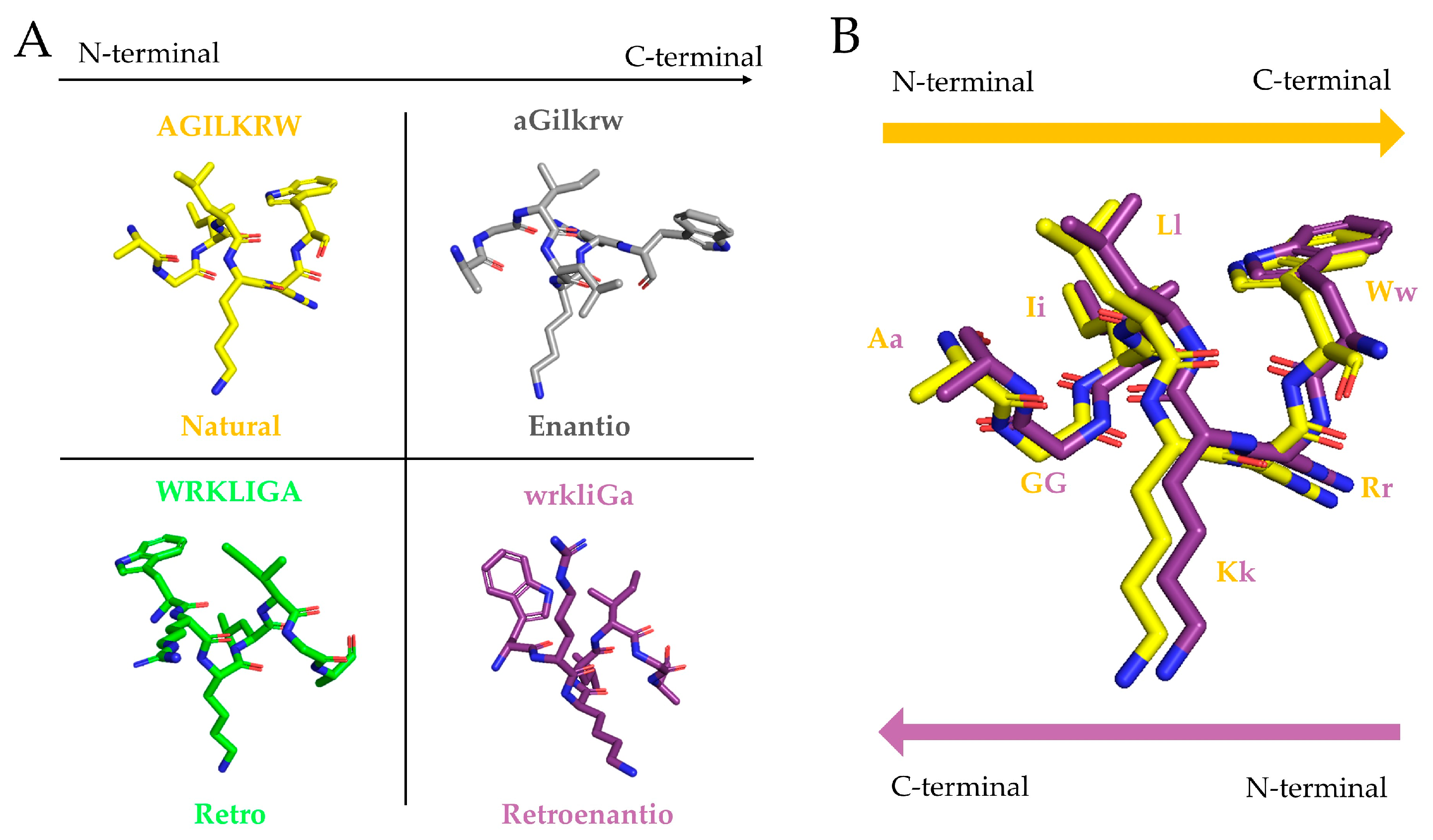
:
1. Introduction
2. Origins of Topoisomer Peptides
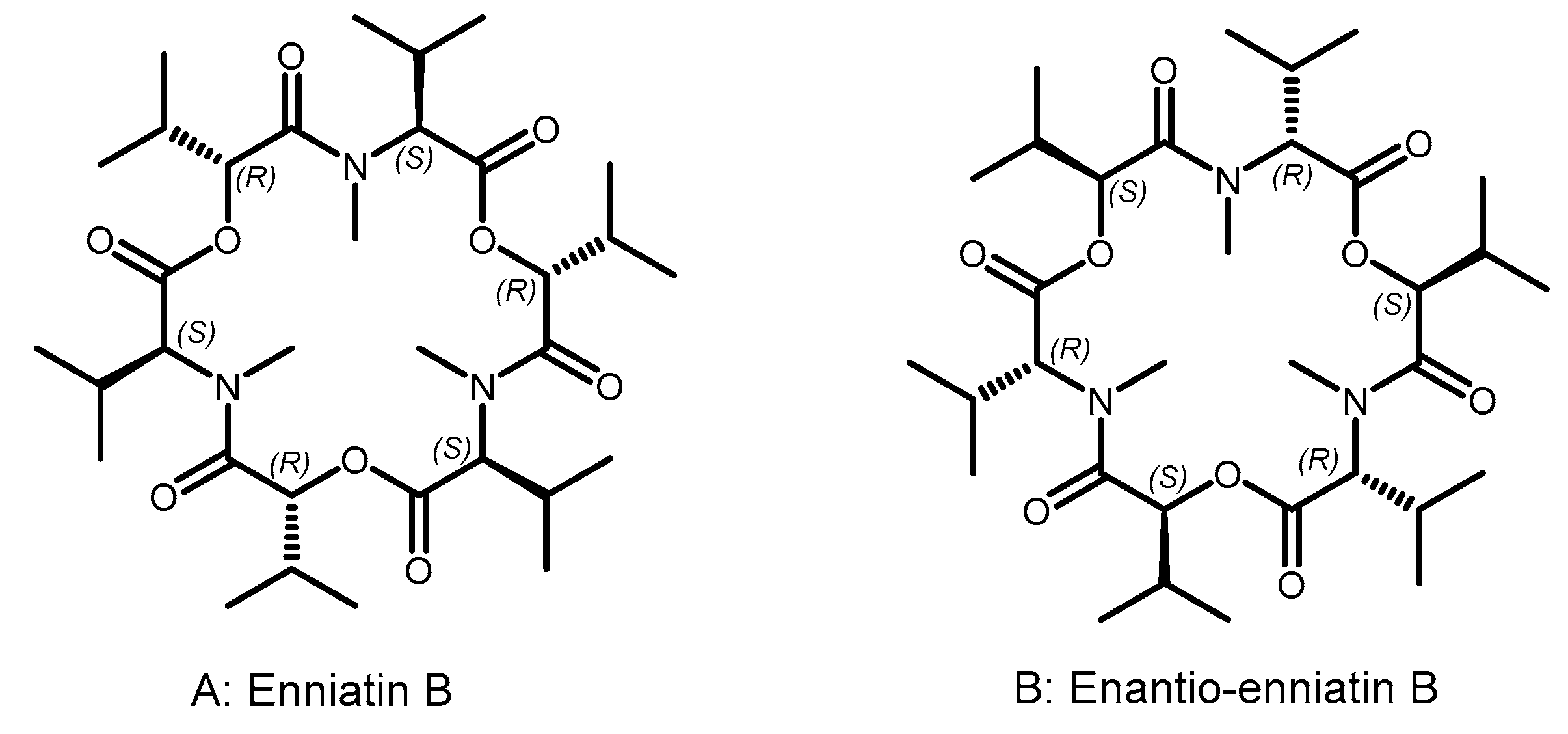
“Indeed, if one turns the formulas […] 60° in the plane of the figure, all the like asymmetric centers coincide, while each ester group will take the place of the N-methyl amide group and vice versa. […] There should therefore be a close matching of both these topochemically similar antipodes to the same stereoselective receptor…”.
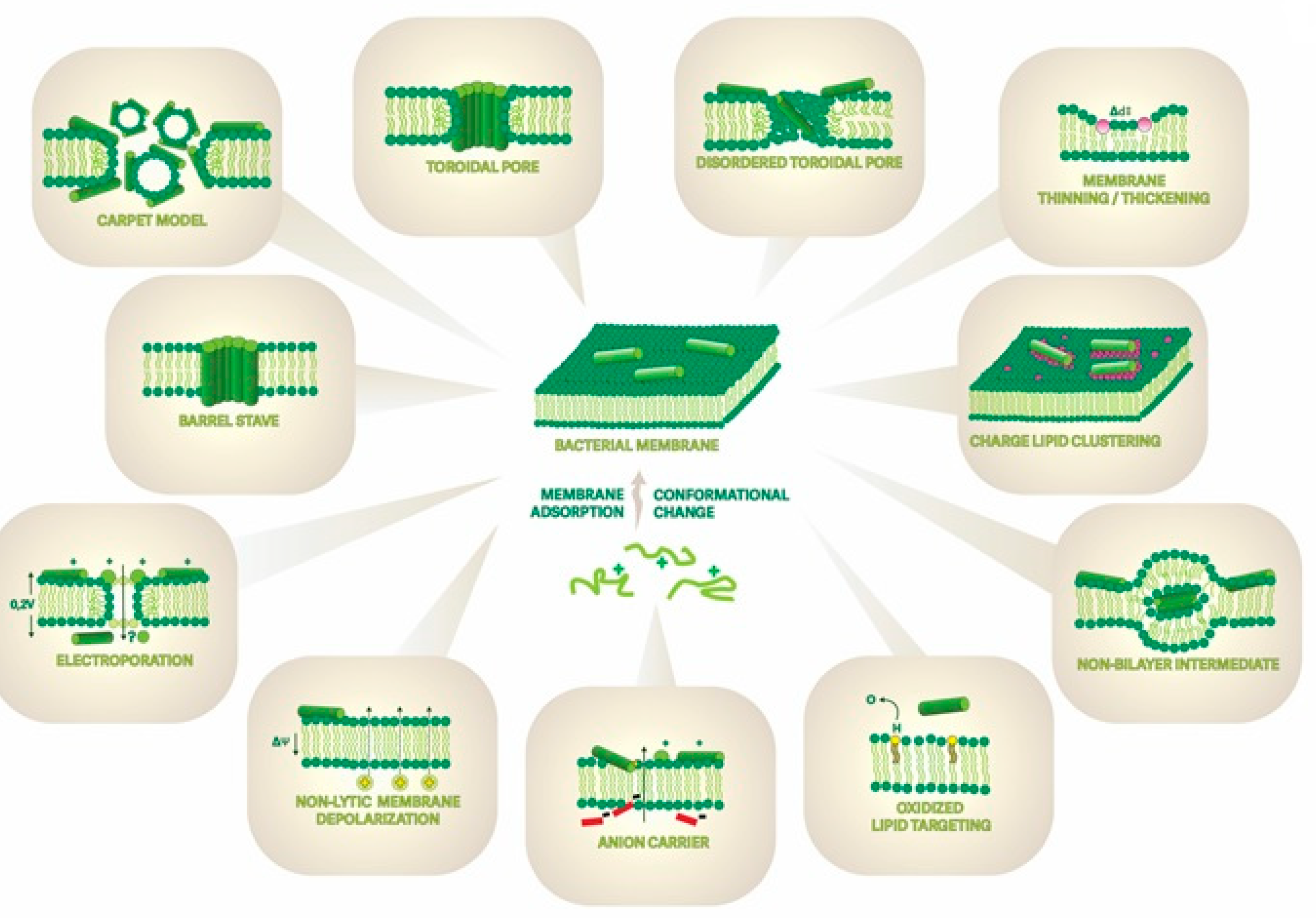
3. Membrane-Active Peptides and Their Mechanisms of Interaction
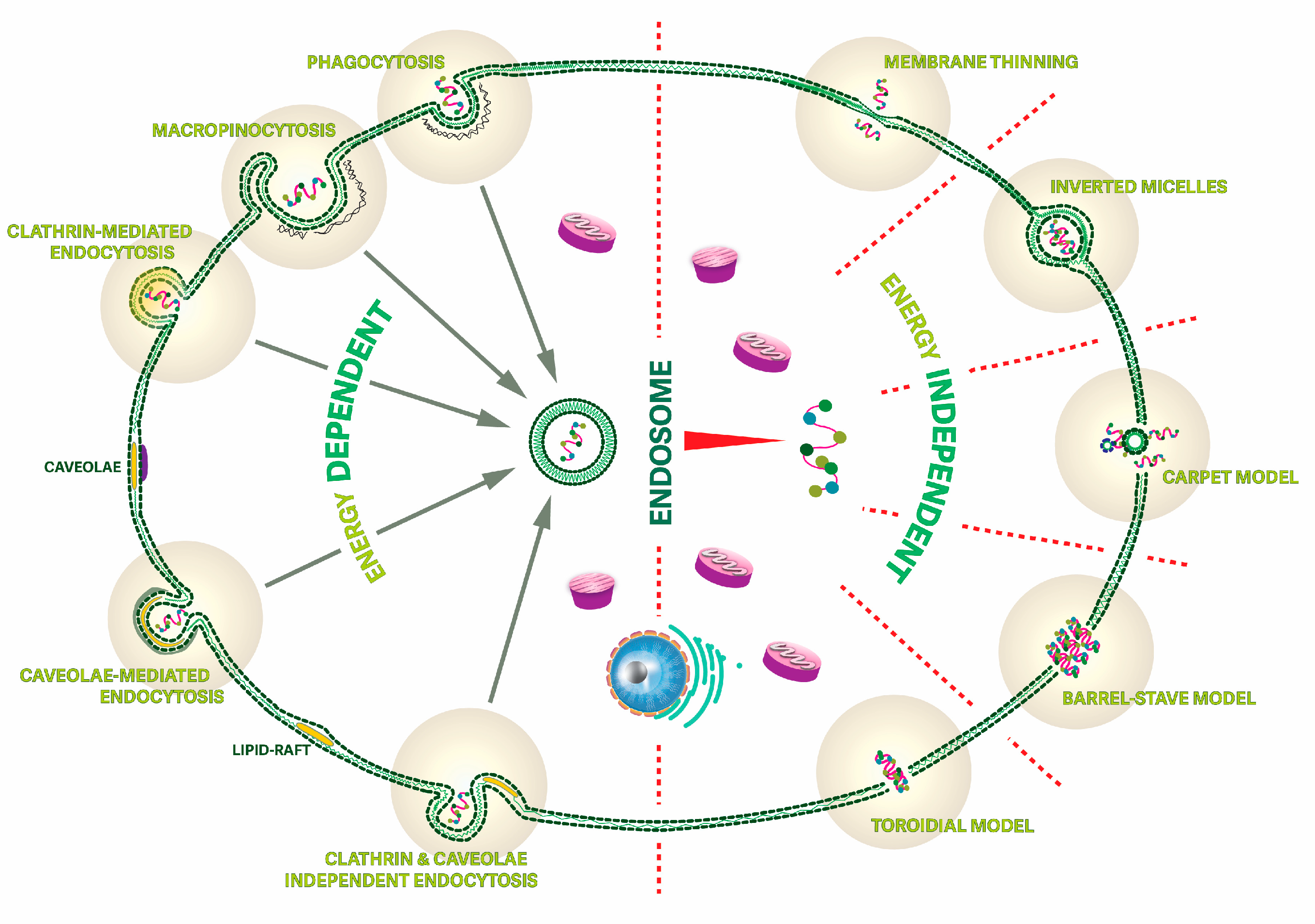
4. Membrane-Active Topoisomeric Peptides
4.1. AMP Topoisomers for Facing the AMR Challenge

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name a | Sequence b | Topoisomer Class | Active Against | Observations c | References |
|---|---|---|---|---|---|
| Retro-indolicin | RRWPWWPWKWPLI | r | G+ (S. aureus), G− (E. coli) | Same MIC values. | [44] |
| Inverso-indolicin | ilpwkwpwwpwr | e | G+ (S. aureus), G− (E. coli) | Same MIC values and increased stability. | [44] |
| Retroinverso-indolicin | rrwpwwpwkwpli | re | G+ (S. aureus), G− (E. coli) | Same MIC values and increased stability. | [44] |
| Retro-[Trp4,6,8,9,11Phe]-indolicidin | RRFPFFPFKFPLI | r | G+ (S. aureus), G− (E. coli) | Same MIC values. | [44] |
| Inverso-[Trp4,6,8,9,11Phe]-indolicidin | ilpfkfpffpfrr | e | G+ (S. aureus), G− (E. coli) | Same MIC values and increased stability. | [44] |
| Retroinverso-[Trp4,6,8,9,11Phe]-indolicidin | rrfpffpfkfpli | re | G+ (S. aureus), G− (E. coli) | Same MIC values and increased stability. | [44] |
| D-V681 | kwksflktfksavktvlhtalkaiss | e | G+/G− | Same MIC values and increased stability. | [158] |
| D-V13KD | kwksflktfksakktvlhtalkaiss | e | G+/G− | Enhanced AMP activity and increased stability. | [158] |
| D-BMAP-28 * | GGlrslGrkilrawkkyGpiivpiiriG | e | Leishmania major (protozoa) | Enhanced AMP activity. | [151] |
| RI-BMAP-28 * | GiriipviipGykkwarlikrGlsrlGG | re | Leishmania major (protozoa) | Enhanced AMP activity. | [151] |
| D-Ano-NH2 | Gllkriktll | e | G+/G− | Same MIC values. | [159] |
| D-GL13K | Gkiiklkaslkll | e | G+ | Enhanced AMP activity. | [160,161] |
| retro-HHC10 | WRIWKWWRK | r | G+/G− | Same MIC values. | [162] |
| inverso-HHC10 | krwwkwirw | e | G+/G− | Same MIC values and increased stability. | [162] |
| retro-inverso-HHC10 | wriwkwwrk | re | G+/G− | Same MIC values. | [162] |
| inverso-CysHHC10 | ckrwwkwirw | e | G+/G− | Same MIC values. | [162,163,164] |
| IK8-all D | irikirik | e | G+/G− | Enhanced AMP activity. | [165] |
| IK12-all D | irvkirvkirvk | e | G+/G− | Enhanced AMP activity. | [165] |
| D-MPI | idwkklldaakqil | e | G+/G− | Same MIC values. | [166] |
| r-CAMEL | LVKLVAGIKKFLKWK | r | G+/G− | Curtailed AMP activity | [157] |
| r-citropin 1.1 | LGGIVSAVKKIVDFLG | r | G+/G− | Curtailed AMP activity. | [157] |
| r-omiganan | KRRWPWWPWRLI | r | G+/G− | Enhanced AMP activity. | [157,167] |
| r-pexiganan | KKLIKVFAKGFKKAKKLFKGIG | r | G− | Same MIC values. | [157] |
| r-temporin A | LIGSLVRGILPLF | r | G+ | Curtailed AMP activity | [157] |
| D-RR4 | wlrrikawlrrika | e | G− | Same MIC values. | [168] |
| RI-73 | lwGvwrrvidwlr | re | G+ (S. aureus) | Same MIC values. | [150] |
| D2D | kk(1nal)fk(1nal)knle | e | G+/G− | Enhanced AMP activity. | [169] |
| (ri)-r(P)ApoBSPro * | GsllkvprkpspiifklkGpklavhp | re | G− | Same MIC values and increased stability. | [170] |
| Ctn retro * | FPITVGIVMPKKFIKKLRKKVSKKVKKFFKKFRK | r | G− | Curtailed AMP activity | [31] |
| Ctn enantio * | krfkkffkkvkksvkkrlkkifkkpmviGvtipf | e | G− | Curtailed AMP activity and increased stability. | [31] |
| Ctn retroenantio * | fpitvGivmpkkfikklrkkvskkvkkffkkfrk | re | G− | Curtailed AMP activity and increased stability. | [31] |
| Ctn[15-34] retro * | FPITVGIVMPKKFIKKLRKK | r | G− | Same MIC values. | [31] |
| Ctn[15-34] enantio * | kkrlkkifkkpmviGvtipf | e | G− | Same MIC values and increased stability. | [31] |
| Ctn[15-34] retroenantio * | fpitvGivmpkkfikklrkk | re | G− | Same MIC values and increased stability. | [31] |
| D-Caerin | GllsvlGsvakhvlphvvpviaehl | e | G+ | Enhanced AMP activity. | [171] |
4.2. CPP Topoisomers and Drug Delivery Challenges
| Name a | Sequence b | Cargo | Topoisomer Class | In Vitro Cell Lines Tested | In Vivo Models | References |
| Tat-D | Grkkrrqrrrppq | Peptides, small molecules | e | Caco-2, ATCC, HTB-37, Calu-3, ATCC, HTB55 | - | [192,209] |
| Tatri | qpprrrqrrkkrG | Peptides, small molecules | re | Caco-2, ATCC, HTB-37, Calu-3, ATCC, HTB55 | - | [192] |
| D-Tat 49-57 | rkkrrqrrr | Small molecules, nanoparticles, proteins. | e | Jurkat T | - | [191,210] |
| D-Tat 57-49 | rrrqrrkkr | Small molecules, nanoparticles, proteins. | re | Jurkat T, MCF-7 | Peritoneal-TA3/St tumor-bearing mice | [191,210,211] |
| D-dfTAT | (ckrkkrrqrrrG)2 (disulfide bridge) | - | e | HeLa, MCH58 and HDF | - | [212] |
| D-penetratin | rqikiwfqnrrmkwkk | Insulin | e | Caco-2, HepG2 and IEC-6 | o.a. mice | [193,194,213] |
| D-penetraMax | kwfkiqmqirrwknkr | Insulin | e | - | o.a. mice | [194] |
| D-polyarginine | (rrrr)x | Insulin, peptide, nucleic acid | e, re | Caco-2, HT-29, Fetal hepatocytes, HeLa, Mc57 fibrosarcoma and Jurkat T | i.v. male Sprague-Dawley rats, i.v. and i.p. ducklings | [195,196,202,214,215,216] |
| DAngiopep | yeetkfnnrkGrsGGyfft | Nanoparticles | re | bEnd.3 and U87 | Glioma model in mice | [198,217] |
| THRre | pwvpswmpprht | Small molecules, nanoparticles | re | bEnd.3 cells | - | [28,199,200,218] |
| JNKD | tdqsrpvqpflnlttprkprpprrrqrrkkrG | - | re | Primary cortical neuronal cultures | i.p. Sprague-Dawley P7 rat pups | [216,219] |
| D-R9F2C | rrrrrrrrrffc | Oligonucleotides | e | HeLa | - | [220] |
| D-SAP | (vrlppp)3 | - | e | HeLa | - | [221] |
| β-syn 36D | GvlyvGsktr | - | e | SH-SY5Y5 | Drosophila model, mixed with the food | [222] |
| retro-inverso β-syn 36 | rtksGvylvG | - | re | SH-SY5Y5 | Drosophila model, mixed with the food | [222] |
| RI-HER-2 | vcsaGftyrGepnpmseftdtnytvlapchl (cyclic disulfide form) | - | re | BT-474, SK-BR-3, MDA-468, and TS/A | Combination with RI-VEGF-P4, s.c. mice | [223] |
| RI-VEGF-P4 | fsmecimrikphqGqhiGcqmti (cyclic disulfide form) | - | re | BT-474, SK-BR-3, MDA-468, and TS/A | Combination with RI-HER-2, s.c. mice | [223] |
| iMP | inlkalaalakkil | Peptides | e | U373MG | - | [201] |
| rMP | LIKKALAALAKLNI | Peptides | r | U373MG | - | [201] |
| riMP | likkalaalaklni | Peptides | re | U373MG | - | [201] |
| iMitP | inlkklakl(Aib)kkil | Peptides | e | U373MG | - | [201] |
| rMitP | LIKK(Aib)ALAALAKLNI | Peptides | r | U373MG | - | [201] |
| riMitP | likk(Aib)lkalkklni | Peptides | re | U373MG | - | [201] |
| riDOM | qqrkrkiwsilaplGttlvklvaGic | - | re | Lipid vesicles | - | [224] |
| R.I.-p1932 | qpkGppppGqpknGGqpppG | - | re | PE/CA PJ15 and hGF | - | [225] |
| NrTP5 | ykqchkkGGkkGsG | - | e | HeLa and BHK21 | - | [226] |
| RI-C2 | arkGrsntfidc | siRNA | re | M17, PC12, L929, or S2103 | i.v. C57BL/6 mice model | [29] |
| D-K4 | kkkk | Peptide nucleic acid | e, re | Fetal hepatocytes | i.v. and i.p. ducklings | [215] |
| RICK | kwllrwlsrllrwlarwlG | Nanoparticles | re | U87 | - | [227] |
| D-CADY-K | Glwralwrllrslwrllwk | Nanoparticles | e | U87 | - | [227] |
| retro-D-HAI | hrpyiah | - | re | B cells | i.p. mice, s.c. rabbit | [228] |
| DVS | svafpsyrhrsfwsv | Small molecules | re | HUVEC and U87 | i.v. intracranial tumor model mice | [229] |
| CHA-061 | hsfriitsitlrGrrrrrrrrr | - | re | HK-2 | Streptozotocin (STZ)-induced diabetes mouse model (via i.p. injection) | [230] |
| DCDX | GreirtGraerwsekf | Liposomes | re | bEnd.3 and U87 | i.p. male mice | [207] |
| DPepH3 | aGilkrw | Proteins, antibodies | e | HBEC-5i | - | [59] |
| DRT-017 | wliymyayvaGilkrw | - | re | bEnd.3 | i.v. mouse | [208] |
| DA7R | rpplwta | Peptides | re | HUVECs and U87 | s.c. mice | [231] |
| OPBP-1 | rvysf | Peptides | re | HUVECs and U87 | s.c. mice | [231] |
4.3. ACP Topoisomers for Mitigating Side Effects in Cancer Treatments
| Name a | Sequence b | Topoisomer Class | In Vitro Cell Lines Tested | References |
|---|---|---|---|---|
| DPMI-α | tnwyanlekllr | e | U87, U251, HCT116 p53+/+ and HCT116 p53−/− | [259,275] |
| DPMI-β | tawyanfekllr | e | U87, U251, HCT116 p53+/+ and HCT116 p53−/− | [259,275] |
| DPMI-γ | dwwplafeallr | e | U87, U251, HCT116 p53+/+ and HCT116 p53−/− | [275] |
| D(LPR) | lpr | re | HUVEC | [262,263,276] |
| D-SP5 | prpspkmGvsvs | re | SGC7901 | [277] |
| retro-tuftsin | RPKT | r | A549 and HL-60 | [278] |
| D(RGD) | dGr | re | U87MG, C6and Hela | [264,265,279] |
| RI-BK | rfpsfGppr | re | HUVEC and C6 | [280] |
| RI-VAP | pavrtns | re | U87MG, HUVEC and HL7702 | [267] |
| D-VAP | sntrvap | e | U87MG, HUVEC and HL7702 | [267] |
| DWSW | wswGpys | re | U87 and HUVEC | [281] |
| RI-3 | yr(Aib)r | re | RBL-2H3 and RBL-2H3/ETFR, osteosarcoma Saos-2 | [282] |
| D(CendR) | rppreGr c | e | HUVEC, C6, U87 and A549 | [270] |
| D(CendR) | rGerppr c | re | HUVEC, C6, U87 and A549 | [270] |
| DT7 | hrpyiah | re | HepG2 | [283] |
| D-FP21 | ftctGqiGprapdGyvldrty | re | HO8910 and HEK 293 T | [274,284] |
| retro-inverso FSH β 33–53 peptide | ftctkqikprapdkyvldrty | re | A2780 | [285] |
| [D]-NRC-03 | GrrkrkwlrriGkGvkiiGGaaldhl | e | HMEC, HDF and HUVEC | [286] |
| RIF7 | rqwllfi | re | A549 | [287] |
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Escribá, P.V.; González-Ros, J.M.; Goñi, F.M.; Kinnunen, P.K.J.; Vigh, L.; Sánchez-Magraner, L.; Fernández, A.M.; Busquets, X.; Horváth, I.; Barceló-Coblijn, G. Membranes: A Meeting Point for Lipids, Proteins and Therapies. J. Cell. Mol. Med. 2008, 12, 829–875. [Google Scholar] [CrossRef] [PubMed]
- Singer, S.J. Some Early History of Membrane Molecular Biology. Annu. Rev. Physiol. 2004, 66, 1–27. [Google Scholar] [CrossRef]
- Lucio, M.; Lima, J.L.F.C.; Reis, S. Drug-Membrane Interactions: Significance for Medicinal Chemistry. Curr. Med. Chem. 2010, 17, 1795–1809. [Google Scholar] [CrossRef]
- Seddon, A.M.; Casey, D.; Law, R.V.; Gee, A.; Templer, R.H.; Ces, O. Drug Interactions with Lipid Membranes. Chem. Soc. Rev. 2009, 38, 2509–2519. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.C.L.; Harris, J.L.; Khanna, K.K.; Hong, J.H. A Comprehensive Review on Current Advances in Peptide Drug Development and Design. Int. J. Mol. Sci. 2019, 20, 2383. [Google Scholar] [CrossRef]
- Erak, M.; Bellmann-Sickert, K.; Els-Heindl, S.; Beck-Sickinger, A.G. Peptide Chemistry Toolbox—Transforming Natural Peptides into Peptide Therapeutics. Bioorg. Med. Chem. 2018, 26, 2759–2765. [Google Scholar] [CrossRef]
- Agamennone, M.; Fantacuzzi, M.; Vivenzio, G.; Scala, M.C.; Campiglia, P.; Superti, F.; Sala, M. Antiviral Peptides as Anti-Influenza Agents. Int. J. Mol. Sci. 2022, 23, 11433. [Google Scholar] [CrossRef]
- Mazurkiewicz-Pisarek, A.; Baran, J.; Ciach, T. Antimicrobial Peptides: Challenging Journey to the Pharmaceutical, Biomedical, and Cosmeceutical Use. Int. J. Mol. Sci. 2023, 24, 9031. [Google Scholar] [CrossRef]
- Kalmouni, M.; Al-Hosani, S.; Magzoub, M. Cancer Targeting Peptides. Cell. Mol. Life Sci. 2019, 76, 2171–2183. [Google Scholar] [CrossRef]
- Kurrikoff, K.; Aphkhazava, D.; Langel, Ü. The Future of Peptides in Cancer Treatment. Curr. Opin. Pharmacol. 2019, 47, 27–32. [Google Scholar] [CrossRef]
- Lv, Y.; Chen, X.; Chen, Z.; Shang, Z.; Li, Y.; Xu, W.; Mo, Y.; Wang, X.; Xu, D.; Li, S.; et al. Melittin Tryptophan Substitution with a Fluorescent Amino Acid Reveals the Structural Basis of Selective Antitumor Effect and Subcellular Localization in Tumor Cells. Toxins 2022, 14, 428. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, K. The Natriuretic Peptide System in Heart Failure: Diagnostic and Therapeutic Implications. Pharmacol. Ther. 2021, 227, 107863. [Google Scholar] [CrossRef]
- Liao, W.; Fan, H.; Davidge, S.T.; Wu, J. Egg White–Derived Antihypertensive Peptide IRW (Ile-Arg-Trp) Reduces Blood Pressure in Spontaneously Hypertensive Rats via the ACE2/Ang (1-7)/Mas Receptor Axis. Mol. Nutr. Food Res. 2019, 63, 1900063. [Google Scholar] [CrossRef] [PubMed]
- Ribarič, S. Peptides as Potential Therapeutics for Alzheimer’s Disease. Molecules 2018, 23, 283. [Google Scholar] [CrossRef] [PubMed]
- Jeremic, D.; Jiménez-Díaz, L.; Navarro-López, J.D. Past, Present and Future of Therapeutic Strategies against Amyloid-β Peptides in Alzheimer’s Disease: A Systematic Review. Ageing Res. Rev. 2021, 72, 101496. [Google Scholar] [CrossRef] [PubMed]
- Sasabe, J.; Suzuki, M. Distinctive Roles of D-Amino Acids in the Homochiral World: Chirality of Amino Acids Modulates Mammalian Physiology and Pathology. Keio J. Med. 2018, 68, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Kreil, G. D-Amino Acids in Animal Peptides. Annu. Rev. Biochem. 1997, 66, 337–345. [Google Scholar] [CrossRef]
- Amiche, M.; Sagan, S.; Mor, A.; Delfour, A.; Nicolas, P. Dermenkephalin (Tyr-D-Met-Phe-His-Leu-Met-Asp-NH2): A Potent and Fully Specific Agonist for the Delta Opioid Receptor. Mol. Pharmacol. 1989, 35, 774–779. [Google Scholar] [PubMed]
- Broccardo, M.; Erspamer, V.; Falconieri, G.; Improta, G.; Linari, G.; Melchiorri, P.; Montecucchi, P. Pharmacological Data on Dermorphins, a New Class of Potent Opioid Peptides from Amphibian Skin. Br. J. Pharmacol. 1981, 73, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Montecucchi, P.C.; De Castiglione, R.; Piani, S.; Gozzini, L.; Erspamer, V. Amino Acid Composition and Sequence of Dermorphin, a Novel Opiate-like Peptide from the Skin of Phyllomedusa Sauvagei. Int. J. Pept. Protein Res. 1981, 17, 275–283. [Google Scholar] [CrossRef]
- Mor, A.; Delfour, A.; Sagan, S.; Amiche, M.; Pradelles, P.; Rossier, J.; Nicolas, P. Isolation of Dermenkephalin from Amphibian Skin, a High-Affinity (δ-Selective Opioid Heptapeptide Containing a D-Amino Acid Residue. FEBS Lett. 1989, 255, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Teramoto, T.; Kuwada, M.; Niidome, T.; Sawada, K.; Nishizawa, Y.; Katayama, K. A Novel Peptide from Funnel Web Spider Venom, ω-Aga-TK, Selectively Blocks P-Type Calcium Channels. Biochem. Biophys. Res. Commun. 1993, 196, 134–140. [Google Scholar] [CrossRef]
- Kamatani, Y.; Minakata, H.; Kenny, P.T.M.; Iwashita, T.; Watanabe, K.; Funase, K.; Sun, X.P.; Yongsiri, A.; Kim, K.H.; Novales-Li, P.; et al. Achatin-I, an Endogenous Neuroexcitatory Tetrapeptide from Achatina Fulica Férussac Containing A d-Amino Acid Residue. Biochem. Biophys. Res. Commun. 1989, 160, 1015–1020. [Google Scholar] [CrossRef]
- Fujimoto, K.; Kubota, I.; Yasuda-Kamatani, Y.; Minakata, H.; Nomoto, K.; Yoshida, M.; Harada, A.; Muneoka, Y.; Kobayashi, M. Purification of Achatin-I from the Atria of the African Giant Snail, Achatina Fulica, and Its Possible Function. Biochem. Biophys. Res. Commun. 1991, 177, 847–853. [Google Scholar] [CrossRef] [PubMed]
- Doti, N.; Mardirossian, M.; Sandomenico, A.; Ruvo, M.; Caporale, A. Recent Applications of Retro-Inverso Peptides. Int. J. Mol. Sci. 2021, 22, 8677. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.Y.; Oh, J.E.; Lee, K.H. Effect of D-Amino Acid Substitution on the Stability, the Secondary Structure, and the Activity of Membrane-Active Peptide. Biochem. Pharmacol. 1999, 58, 1775–1780. [Google Scholar] [CrossRef] [PubMed]
- Lucana, M.C.; Arruga, Y.; Petrachi, E.; Roig, A.; Lucchi, R.; Oller-Salvia, B. Protease-Resistant Peptides for Targeting and Intracellular Delivery of Therapeutics. Pharmaceutics 2021, 13, 2065. [Google Scholar] [CrossRef] [PubMed]
- Prades, R.; Oller-Salvia, B.; Schwarzmaier, S.M.; Selva, J.; Moros, M.; Balbi, M.; Grazú, V.; De La Fuente, J.M.; Egea, G.; Plesnila, N.; et al. Applying the Retro-Enantio Approach To Obtain a Peptide Capable of Overcoming the Blood–Brain Barrier. Angew. Chem. Int. Ed. 2015, 54, 3967–3972. [Google Scholar] [CrossRef]
- Javed, H.; Menon, S.A.; Al-Mansoori, K.M.; Al-Wandi, A.; Majbour, N.K.; Ardah, M.T.; Varghese, S.; Vaikath, N.N.; Haque, M.E.; Azzouz, M.; et al. Development of Nonviral Vectors Targeting the Brain as a Therapeutic Approach For Parkinson’s Disease and Other Brain Disorders. Mol. Ther. 2016, 24, 746–758. [Google Scholar] [CrossRef]
- Zheng, Y.; Mao, K.; Chen, S.; Zhu, H. Chirality Effects in Peptide Assembly Structures. Front. Bioeng. Biotechnol. 2021, 9, 703004. [Google Scholar] [CrossRef] [PubMed]
- Carrera-Aubesart, A.; Defaus, S.; Pérez-Peinado, C.; Sandín, D.; Torrent, M.; Jiménez, M.Á.; Andreu, D. Examining Topoisomers of a Snake-Venom-Derived Peptide for Improved Antimicrobial and Antitumoral Properties. Biomedicines 2022, 10, 2110. [Google Scholar] [CrossRef]
- Xi, W.; Hansmann, U.H.E. The Effect of Retro-Inverse D-Amino Acid Aβ-Peptides on Aβ-Fibril Formation. J. Chem. Phys. 2019, 150, 095101. [Google Scholar] [CrossRef]
- Neves, V.; Aires-Da-Silva, F.; Morais, M.; Gano, L.; Ribeiro, E.; Pinto, A.; Aguiar, S.; Gaspar, D.; Fernandes, C.; Correia, J.D.G.; et al. Novel Peptides Derived from Dengue Virus Capsid Protein Translocate Reversibly the Blood-Brain Barrier through a Receptor-Free Mechanism. ACS Chem. Biol. 2017, 12, 1257–1268. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger, L. The PyMOL Molecular Graphics System, Version 1.2r3pre; Schrodinger LLC: New York, NY, USA, 2008; 3p. [Google Scholar]
- Stewart, J.M.; Woolley, D.W. All-D-Bradykinin and the Problem of Peptide Antimetabolites. Nature 1965, 206, 619–620. [Google Scholar] [CrossRef]
- Vogler, K.; Studer, R.O.; Lergier, W.; Lanz, P. Synthese von All-D-Val5-Angiotensin II-Asp1-β-Amid. Helv. Chim. Acta 1965, 48, 1407–1414. [Google Scholar] [CrossRef] [PubMed]
- Shemyakin, M.M.; Ovchinnikov, Y.A.; Ivanov, V.T.; Evstratov, A.V. Topochemical Approach in Studies of the Structure-Activity Relation: Enantio-Enniatin B. Nature 1967, 213, 412–413. [Google Scholar] [CrossRef]
- Merrifield, R.B.; Juvvadi, P.; Andreu, D.; Ubach, J.; Boman, A.; Boman, H.G. Retro and Retroenantio Analogs of Cecropin-Melittin Hybrids. Proc. Natl. Acad. Sci. USA 1995, 92, 3449–3453. [Google Scholar] [CrossRef]
- Shemyakin, M.M.; Ovchinnikov, Y.A.; Ivanov, V.T. Topochemical Investigations on Peptide Systems. Angew. Chem. Int. Ed. Engl. 1969, 8, 492–499. [Google Scholar] [CrossRef]
- Merrifield, E.L.; Mitchell, S.A.; Ubach, J.; Boman, H.G.; Andreu, D.; Merrifield, R.B. D-Enantiomers of 15-Residue Cecropin A-Melittin Hybrids. Int. J. Pept. Protein Res. 1995, 46, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Wade, D.; Andreu, D.; Mitchell, S.A.; Silveira, A.M.V.; Boman, A.; Boman, H.G.; Merrifield, R.B. Antibacterial Peptides Designed as Analogs or Hybrids of Cecropins and Melittin. Int. J. Pept. Protein Res. 1992, 40, 429–436. [Google Scholar] [CrossRef]
- Wade, D.; Boman, A.; Wåhlint, B.; Drain, C.M.; Andreu, D.; Boman, H.G.; Merrifield, R.B. All-D Amino Acid-Containing Channel-Forming Antibiotic Peptides. Proc. Natl. Acad. Sci. USA 1990, 87, 4761–4765. [Google Scholar] [CrossRef]
- Vunnam, S.; Juvvadi, P.; Rotondi, K.S.; Merrifield, R.B. Synthesis and Study of Normal, Enantio, Retro, and Retroenantio Isomers of Cecropin A-Melittin Hybrids, Their End Group Effects and Selective Enzyme Inactivation. J. Pept. Res. 1998, 51, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Staubitz, P.; Peschel, A.; Nieuwenhuizen, W.F.; Otto, M.; Götz, F.; Jung, G.; Jack, R.W. Structure–Function Relationships in the Tryptophan-Rich, Antimicrobial Peptide Indolicidin. J. Pept. Sci. 2001, 7, 552–564. [Google Scholar] [CrossRef]
- Ruvo, M.; Fassina, G. End-Group Modified Retro-Inverso Isomers of Tripeptide Oxytocin Analogues: Binding to Neurophysin II and Enhancement of Its Self-Association Properties. Int. J. Pept. Protein Res. 1995, 45, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.; Pothoulakis, C.; Wai Koon, H. Antimicrobial Peptides and Colitis. Curr. Pharm. Des. 2013, 19, 40–47. [Google Scholar]
- Savini, F.; Loffredo, M.R.; Troiano, C.; Bobone, S.; Malanovic, N.; Eichmann, T.O.; Caprio, L.; Canale, V.C.; Park, Y.; Mangoni, M.L.; et al. Binding of an Antimicrobial Peptide to Bacterial Cells: Interaction with Different Species, Strains and Cellular Components. Biochim. Biophys. Acta-Biomembr. 2020, 1862, 183291. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Song, Y. Mechanism of Antimicrobial Peptides: Antimicrobial, Anti-Inflammatory and Antibiofilm Activities. Int. J. Mol. Sci. 2021, 22, 11401. [Google Scholar] [CrossRef] [PubMed]
- Epand, R.M.; Walker, C.; Epand, R.F.; Magarvey, N.A. Molecular Mechanisms of Membrane Targeting Antibiotics. Biochim. Biophys. Acta-Biomembr. 2016, 1858, 980–987. [Google Scholar] [CrossRef]
- Kang, S.J.; Park, S.J.; Mishig-Ochir, T.; Lee, B.J. Antimicrobial Peptides: Therapeutic Potentials. Expert Rev. Anti. Infect. Ther. 2014, 12, 1477–1486. [Google Scholar] [CrossRef]
- Thapa, R.K.; Diep, D.B.; Tønnesen, H.H. Topical Antimicrobial Peptide Formulations for Wound Healing: Current Developments and Future Prospects. Acta Biomater. 2020, 103, 52–67. [Google Scholar] [CrossRef]
- Lai, Z.; Yuan, X.; Chen, H.; Zhu, Y.; Dong, N.; Shan, A. Strategies Employed in the Design of Antimicrobial Peptides with Enhanced Proteolytic Stability. Biotechnol. Adv. 2022, 59, 107962. [Google Scholar] [CrossRef]
- Derossi, D.; Joliot, A.H.; Chassaing, G.; Prochiantz, A. The Third Helix of the Antennapedia Homeodomain Translocates through Biological Membranes. J. Biol. Chem. 1994, 269, 10444–10450. [Google Scholar] [CrossRef]
- Derossi, D.; Calvet, S.; Trembleau, A.; Brunissen, A.; Chassaing, G.; Prochiantz, A. Cell Internalization of the Third Helix of the Antennapedia Homeodomain Is Receptor-Independent. J. Biol. Chem. 1996, 271, 18188–18193. [Google Scholar] [CrossRef]
- De Figueiredo, I.R.; Freire, J.M.; Flores, L.; Veiga, A.S.; Castanho, M.A.R.B. Cell-Penetrating Peptides: A Tool for Effective Delivery in Gene-Targeted Therapies. IUBMB Life 2014, 66, 182–194. [Google Scholar] [CrossRef]
- Kardani, K.; Milani, A.; Shabani, S.H.; Bolhassani, A. Cell Penetrating Peptides: The Potent Multi-Cargo Intracellular Carriers. Expert Opin. Drug Deliv. 2019, 16, 1227–1258. [Google Scholar] [CrossRef]
- Porosk, L.; Gaidutšik, I.; Langel, Ü. Approaches for the Discovery of New Cell-Penetrating Peptides. Expert Opin. Drug Discov. 2020, 16, 553–565. [Google Scholar] [CrossRef]
- Sajid, M.I.; Moazzam, M.; Stueber, R.; Park, S.E.; Cho, Y.; Malik, N.u.A.; Tiwari, R.K. Applications of Amphipathic and Cationic Cyclic Cell-Penetrating Peptides: Significant Therapeutic Delivery Tool. Peptides 2021, 141, 170542. [Google Scholar] [CrossRef]
- Cavaco, M.; Valle, J.; da Silva, R.; Correia, J.D.G.; Castanho, M.A.R.B.; Andreu, D.; Neves, V. D PepH3, an Improved Peptide Shuttle for Receptor-Independent Transport Across the Blood-Brain Barrier. Curr. Pharm. Des. 2020, 26, 1495–1506. [Google Scholar] [CrossRef]
- Guichard, G.; Benkirane, N.; Zeder-Lutz, G.; Van Regenmortel, M.H.; Briand, J.P.; Muller, S. Antigenic Mimicry of Natural L-Peptides with Retro-Inverso-Peptidomimetics. Proc. Natl. Acad. Sci. USA 1994, 91, 9765–9769. [Google Scholar] [CrossRef]
- Röckendorf, N.; Nehls, C.; Gutsmann, T. Design of Membrane Active Peptides Considering Multi-Objective Optimization for Biomedical Application. Membranes 2022, 12, 180. [Google Scholar] [CrossRef]
- Drexelius, M.; Reinhardt, A.; Grabeck, J.; Cronenberg, T.; Nitsche, F.; Huesgen, P.F.; Maier, B.; Neundorf, I. Multistep Optimization of a Cell-Penetrating Peptide towards Its Antimicrobial Activity. Biochem. J. 2021, 478, 63–78. [Google Scholar] [CrossRef]
- Hollmann, A.; Martinez, M.; Maturana, P.; Semorile, L.C.; Maffia, P.C. Antimicrobial Peptides: Interaction with Model and Biological Membranes and Synergism with Chemical Antibiotics. Front. Chem. 2018, 6, 204. [Google Scholar] [CrossRef]
- Nyström, L.; Malmsten, M. Membrane Interactions and Cell Selectivity of Amphiphilic Anticancer Peptides. Curr. Opin. Colloid Interface Sci. 2018, 38, 1–17. [Google Scholar] [CrossRef]
- Gawde, U.; Chakraborty, S.; Waghu, F.H.; Barai, R.S.; Khanderkar, A.; Indraguru, R.; Shirsat, T.; Idicula-Thomas, S. CAMPR4: A Database of Natural and Synthetic Antimicrobial Peptides. Nucleic Acids Res. 2023, 51, D377–D383. [Google Scholar] [CrossRef]
- Tripathi, A.K.; Vishwanatha, J.K. Role of Anti-Cancer Peptides as Immunomodulatory Agents: Potential and Design Strategy. Pharmaceutics 2022, 14, 2686. [Google Scholar] [CrossRef]
- Matsuzaki, K. Why and How Are Peptide–Lipid Interactions Utilized for Self-Defense? Magainins and Tachyplesins as Archetypes. Biochim. Biophys. Acta-Biomembr. 1999, 1462, 1–10. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Haney, E.F.; Vogel, H.J. The Expanding Scope of Antimicrobial Peptide Structures and Their Modes of Action. Trends Biotechnol. 2011, 29, 464–472. [Google Scholar] [CrossRef]
- Gallo, M.; Defaus, S.; Andreu, D. 1988–2018: Thirty Years of Drug Smuggling at the Nano Scale. Challenges and Opportunities of Cell-Penetrating Peptides in Biomedical Research. Arch. Biochem. Biophys. 2019, 661, 74–86. [Google Scholar] [CrossRef]
- Sánchez-Navarro, M.; Giralt, E. Peptide Shuttles for Blood-Brain Barrier Drug Delivery. Pharmaceutics 2022, 14, 1874. [Google Scholar] [CrossRef]
- Todorovski, T.; Kalafatovic, D.; Andreu, D. Antiviral Peptide-Based Conjugates: State of the Art and Future Perspectives. Pharmaceutics 2023, 15, 357. [Google Scholar] [CrossRef]
- Ramaker, K.; Henkel, M.; Krause, T.; Röckendorf, N.; Frey, A. Cell Penetrating Peptides: A Comparative Transport Analysis for 474 Sequence Motifs. Drug Deliv. 2018, 25, 928–937. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Sayers, E.J.; Watson, P.; Jones, A.T. Contrasting Roles for Actin in the Cellular Uptake of Cell Penetrating Peptide Conjugates. Sci. Rep. 2018, 8, 7318. [Google Scholar] [CrossRef] [PubMed]
- Gestin, M.; Dowaidar, M.; Langel, Ü. Uptake Mechanism of Cell-Penetrating Peptides. In Peptides and Peptide-Based Biomaterials and Their Biomedical Applications; Sunna, A., Care, A., Bergquist, P., Eds.; Springer: New York, NY, USA, 2017; Volume 1030, pp. 255–264. [Google Scholar]
- Mueller, J.; Kretzschmar, I.; Volkmer, R.; Boisguerin, P. Comparison of Cellular Uptake Using 22 CPPs in 4 Different Cell Lines. Bioconjug. Chem. 2008, 19, 2363–2374. [Google Scholar] [CrossRef]
- Walrant, A.; Cardon, S.; Burlina, F.; Sagan, S. Membrane Crossing and Membranotropic Activity of Cell-Penetrating Peptides: Dangerous Liaisons? Acc. Chem. Res. 2017, 50, 2968–2975. [Google Scholar] [CrossRef] [PubMed]
- Kulbacka, J.; Choromańska, A.; Rossowska, J.; Weżgowiec, J.; Saczko, J.; Rols, M.P. Cell Membrane Transport Mechanisms: Ion Channels and Electrical Properties of Cell Membranes. In Transport across Natural and Modified Biological Membranes and Its Implications in Physiology and Therapy; Kulbacka, J., Satkauskas, S., Eds.; Springer: New York, NY, USA, 2017; Volume 227, pp. 39–58. [Google Scholar]
- Sugano, K.; Kansy, M.; Artursson, P.; Avdeef, A.; Bendels, S.; Di, L.; Ecker, G.F.; Faller, B.; Fischer, H.; Gerebtzoff, G.; et al. Coexistence of Passive and Carrier-Mediated Processes in Drug Transport. Nat. Rev. Drug Discov. 2010, 9, 597–614. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Yee, S.W.; Kim, R.B.; Giacomini, K.M. SLC Transporters as Therapeutic Targets: Emerging Opportunities. Nat. Rev. Drug Discov. 2015, 14, 543–560. [Google Scholar] [CrossRef] [PubMed]
- Perland, E.; Fredriksson, R. Classification Systems of Secondary Active Transporters. Trends Pharmacol. Sci. 2017, 38, 305–315. [Google Scholar] [CrossRef]
- Alleva, K.; Chara, O.; Amodeo, G. Aquaporins: Another Piece in the Osmotic Puzzle. FEBS Lett. 2012, 586, 2991–2999. [Google Scholar] [CrossRef]
- Yukutake, Y.; Hirano, Y.; Suematsu, M.; Yasui, M. Rapid and Reversible Inhibition of Aquaporin-4 by Zinc. Biochemistry 2009, 48, 12059–12061. [Google Scholar] [CrossRef]
- Gerbeau, P.; Amodeo, G.; Henzler, T.; Santoni, V.; Ripoche, P.; Maurel, C. The Water Permeability of Arabidopsis Plasma Membrane Is Regulated by Divalent Cations and PH. Plant J. 2002, 30, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Cleal, K.; He, L.; Watson, P.D.; Jones, A.T. Endocytosis, Intracellular Traffic and Fate of Cell Penetrating Peptide Based Conjugates and Nanoparticles. Curr. Pharm. Des. 2013, 19, 2878–2894. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.J.; Hinner, M.J. Getting across the Cell Membrane: An Overview for Small Molecules, Peptides, and Proteins. In Site-Specific Protein Labeling; Gautier, A., Hinner, M., Eds.; Humana Press: New York, NY, USA, 2015; Volume 1266, pp. 29–53. [Google Scholar]
- Mayor, S.; Pagano, R.E. Pathways of Clathrin-Independent Endocytosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Desale, K.; Kuche, K.; Jain, S. Cell-Penetrating Peptides (CPPs): An Overview of Applications for Improving the Potential of Nanotherapeutics. Biomater. Sci. 2021, 9, 1153–1188. [Google Scholar] [CrossRef] [PubMed]
- Madani, F.; Lindberg, S.; Langel, U.; Futaki, S.; Gräslund, A.; Langel, Ü.; Futaki, S.; Gräslund, A. Mechanisms of Cellular Uptake of Cell-Penetrating Peptides. J. Biophys. 2011, 2011, 414729. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.P.; Langer, R.; Jensen, K.F. Intracellular Delivery by Membrane Disruption: Mechanisms, Strategies, and Concepts. Chem. Rev. 2018, 118, 7409–7531. [Google Scholar] [CrossRef] [PubMed]
- Last, N.B.; Schlamadinger, D.E.; Miranker, A.D. A Common Landscape for Membrane-Active Peptides. Protein Sci. 2013, 22, 870–882. [Google Scholar] [CrossRef]
- Sun, D.; Forsman, J.; Woodward, C.E. Current Understanding of the Mechanisms by Which Membrane-Active Peptides Permeate and Disrupt Model Lipid Membranes. Curr. Top. Med. Chem. 2016, 16, 170–186. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.Y.; Yan, Z.B.; Meng, Y.M.; Hong, X.Y.; Shao, G.; Ma, J.J.; Cheng, X.R.; Liu, J.; Kang, J.; Fu, C.Y. Antimicrobial Peptides: Mechanism of Action, Activity and Clinical Potential. Mil. Med. Res. 2021, 8, 48. [Google Scholar] [CrossRef]
- Zasloff, M. Antimicrobial Peptides of Multicellular Organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Yeaman, M.R.; Yount, N.Y. Mechanisms of Antimicrobial Peptide Action and Resistance. Pharmacol. Rev. 2003, 55, 27–55. [Google Scholar] [CrossRef]
- Brogden, K.A. Antimicrobial Peptides: Pore Formers or Metabolic Inhibitors in Bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar] [CrossRef]
- Lee, T.-H.; Hall, K.N.; Aguilar, M.-I. Antimicrobial Peptide Structure and Mechanism of Action: A Focus on the Role of Membrane Structure. Curr. Top. Med. Chem. 2016, 16, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Kizhakkedathu, J.N.; Straus, S.K. Antimicrobial Peptides: Diversity, Mechanism of Action and Strategies to Improve the Activity and Biocompatibility In Vivo. Biomolecules 2018, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Hale, J.D.F.; Hancock, R.E.W. Alternative Mechanisms of Action of Cationic Antimicrobial Peptides on Bacteria. Expert Rev. Anti. Infect. Ther. 2007, 5, 951–959. [Google Scholar] [CrossRef]
- Lu, J.; Xu, H.; Xia, J.; Ma, J.; Xu, J.; Li, Y.; Feng, J. D- and Unnatural Amino Acid Substituted Antimicrobial Peptides with Improved Proteolytic Resistance and Their Proteolytic Degradation Characteristics. Front. Microbiol. 2020, 11, 2869. [Google Scholar] [CrossRef] [PubMed]
- Cavaco, M.; Andreu, D.; Castanho, M.A.R.B. The Challenge of Peptide Proteolytic Stability Studies: Scarce Data, Difficult Readability, and the Need for Harmonization. Angew. Chem. 2021, 133, 1710–1712. [Google Scholar] [CrossRef]
- Klompas, M. Overuse of Broad-Spectrum Antibiotics for Pneumonia. JAMA Intern. Med. 2020, 180, 485–486. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.A.; Centor, R.M.; Humphrey, L.L.; Jokela, J.A.; Andrews, R.; Qaseem, A. Appropriate Use of Short-Course Antibiotics in Common Infections: Best Practice Advice from the American College of Physicians. Ann. Intern. Med. 2021, 174, 822–827. [Google Scholar] [CrossRef]
- Interagency Coordination Group on Antimicrobial Resistance. No Time to Wait: Securing the Future from Drug-Resistant Infections. Report to the Secretary-General of the United Nations; WHO: Geneva, Switzerland, 2019. [Google Scholar]
- Yu, T.; Jiang, G.; Gao, R.; Chen, G.; Ren, Y.; Liu, J.; van der Mei, H.C.; Busscher, H.J. Circumventing Antimicrobial-Resistance and Preventing Its Development in Novel, Bacterial Infection-Control Strategies. Expert Opin. Drug Deliv. 2020, 17, 1151–1164. [Google Scholar] [CrossRef]
- Ciotti, M.; Ciccozzi, M.; Pieri, M.; Bernardini, S. The COVID-19 Pandemic: Viral Variants and Vaccine Efficacy. Crit. Rev. Clin. Lab. Sci. 2021, 59, 66–75. [Google Scholar] [CrossRef]
- Hadj Hassine, I. COVID-19 Vaccines and Variants of Concern: A Review. Rev. Med. Virol. 2022, 32, e2313. [Google Scholar] [CrossRef] [PubMed]
- Chams, N.; Chams, S.; Badran, R.; Shams, A.; Araji, A.; Raad, M.; Mukhopadhyay, S.; Stroberg, E.; Duval, E.J.; Barton, L.M.; et al. COVID-19: A Multidisciplinary Review. Front. Public Health 2020, 8, 383. [Google Scholar] [CrossRef] [PubMed]
- Parasher, A. COVID-19: Current Understanding of Its Pathophysiology, Clinical Presentation and Treatment. Postgrad. Med. J. 2021, 97, 312–320. [Google Scholar] [CrossRef]
- Department of Health and Human Services. COVID-19 U.S. Impact on Antimicrobial Resistance, Special Report 2022; Department of Health and Human Services, CDC: Atlanta, GA, USA, 2022. [Google Scholar]
- Chedid, M.; Waked, R.; Haddad, E.; Chetata, N.; Saliba, G.; Choucair, J. Antibiotics in Treatment of COVID-19 Complications: A Review of Frequency, Indications, and Efficacy. J. Infect. Public Health 2021, 14, 570–576. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.G.; Ahammad, S.Z. COVID-19 and Antimicrobial Resistance: A Cross-Study. Sci. Total Environ. 2022, 807, 150873. [Google Scholar] [CrossRef]
- De Oliveira, D.M.P.; Forde, B.M.; Kidd, T.J.; Harris, P.N.A.; Schembri, M.A.; Beatson, S.A.; Paterson, D.L.; Walker, M.J. Antimicrobial Resistance in ESKAPE Pathogens. Clin. Microbiol. Rev. 2020, 33, e00181-19. [Google Scholar] [CrossRef] [PubMed]
- Aloke, C.; Achilonu, I. Coping with the ESKAPE Pathogens: Evolving Strategies, Challenges and Future Prospects. Microb. Pathog. 2023, 175, 105963. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, G.; Midiri, A.; Gerace, E.; Biondo, C. Bacterial Antibiotic Resistance: The Most Critical Pathogens. Pathogens 2021, 10, 1310. [Google Scholar] [CrossRef] [PubMed]
- Manniello, M.D.; Moretta, A.; Salvia, R.; Scieuzo, C.; Lucchetti, D.; Vogel, H.; Sgambato, A.; Falabella, P. Insect Antimicrobial Peptides: Potential Weapons to Counteract the Antibiotic Resistance. Cell. Mol. Life Sci. 2021, 78, 4259–4282. [Google Scholar] [CrossRef]
- Moravej, H.; Moravej, Z.; Yazdanparast, M.; Heiat, M.; Mirhosseini, A.; Moosazadeh Moghaddam, M.; Mirnejad, R. Antimicrobial Peptides: Features, Action, and Their Resistance Mechanisms in Bacteria. Microb. Drug Resist. 2018, 24, 747–767. [Google Scholar] [CrossRef] [PubMed]
- Mwangi, J.; Hao, X.; Lai, R.; Zhang, Z.-Y. Antimicrobial Peptides: New Hope in the War against Multidrug Resistance. Zool. Res. 2019, 40, 488–505. [Google Scholar] [CrossRef] [PubMed]
- Lima, P.G.; Oliveira, J.T.A.; Amaral, J.L.; Freitas, C.D.T.; Souza, P.F.N. Synthetic Antimicrobial Peptides: Characteristics, Design, and Potential as Alternative Molecules to Overcome Microbial Resistance. Life Sci. 2021, 278, 119647. [Google Scholar] [CrossRef]
- Valenti, G.E.; Alfei, S.; Caviglia, D.; Domenicotti, C.; Marengo, B. Antimicrobial Peptides and Cationic Nanoparticles: A Broad-Spectrum Weapon to Fight Multi-Drug Resistance Not Only in Bacteria. Int. J. Mol. Sci. 2022, 23, 6108. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, R.D.; Dubos, R.J. Fractionation of the Bactericidal Agent from Cultures of a Soil Bacillus. J. Biol. Chem. 1940, 132, 791–792. [Google Scholar] [CrossRef]
- Dubos, R.J. Studies on a Bactericidal Agent Extracted from a Soil Bacillus: I. Preparation of the Agent. Its Activity In Vitro. J. Exp. Med. 1939, 70, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Dubos, R.J. Studies on a Bactericidal Agent Extracted from a Soil Bacillus: II. Protective Effect of the Bactericidal Agent against Experimental Pneumococcus Infections in Mice. J. Exp. Med. 1939, 70, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Phoenix, D.A.; Dennison, S.R.; Harris, F. (Eds.) Antimicrobial Peptides: Their History, Evolution, and Functional Promiscuity. In Antimicrobial Peptides; John Wiley & Sons, Ltd.: London, UK, 2013; pp. 1–37. [Google Scholar]
- Fernandez De Caleya, R.; Gonzalez-Pascual, B.; García-Olmedo, F.; Carbonero, P. Susceptibility of Phytopathogenic Bacteria to Wheat Purothionins In Vitro. Appl. Microbiol. 1972, 23, 998–1000. [Google Scholar] [CrossRef]
- Kiss, G.; Michl, H. Uber Das Giftsekret Der Gelbbauchunke, Bombina variegata L. Toxicon 1962, 1, 33–34. [Google Scholar] [CrossRef]
- Simmaco, M.; Kreil, G.; Barra, D. Bombinins, Antimicrobial Peptides from Bombina Species. Biochim. Biophys. Acta-Biomembr. 2009, 1788, 1551–1555. [Google Scholar] [CrossRef]
- Hultmark, D.; Steiner, H.; Rasmuson, T.; Boman, H.G. Insect Immunity. Purification and Properties of Three Inducible Bactericidal Proteins from Hemolymph of Immunized Pupae of Hyalophora Cecropia. Eur. J. Biochem. 1980, 106, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Steiner, H.; Hultmark, D.; Engström, Å.; Bennich, H.; Boman, H.G. Sequence and Specificity of Two Antibacterial Proteins Involved in Insect Immunity. Nature 1981, 292, 246–248. [Google Scholar] [CrossRef] [PubMed]
- Selsted, M.E.; Brown, D.M.; DeLange, R.J.; Lehrer, R.I. Primary Structures of MCP-1 and MCP-2, Natural Peptide Antibiotics of Rabbit Lung Macrophages. J. Biol. Chem. 1983, 258, 14485–14489. [Google Scholar] [CrossRef] [PubMed]
- Selsted, M.E.; Szklarek, D.; Lehrer, R.I. Purification and Antibacterial Activity of Antimicrobial Peptides of Rabbit Granulocytes. Infect. Immun. 1984, 45, 150–154. [Google Scholar] [CrossRef]
- Selsted, M.E.; Brown, D.M.; DeLange, R.J.; Harwig, S.S.; Lehrer, R.I. Primary Structures of Six Antimicrobial Peptides of Rabbit Peritoneal Neutrophils. J. Biol. Chem. 1985, 260, 4579–4584. [Google Scholar] [CrossRef]
- Ganz, T.; Selsted, M.E.; Szklarek, D.; Harwig, S.S.; Daher, K.; Bainton, D.F.; Lehrer, R.I. Defensins. Natural Peptide Antibiotics of Human Neutrophils. J. Clin. Investig. 1985, 76, 1427–1435. [Google Scholar] [CrossRef] [PubMed]
- Zasloff, M. Magainins, a Class of Antimicrobial Peptides from Xenopus Skin: Isolation, Characterization of Two Active Forms, and Partial CDNA Sequence of a Precursor. Proc. Natl. Acad. Sci. USA 1987, 84, 5449–5453. [Google Scholar] [CrossRef] [PubMed]
- Selsted, M.E.; Tang, Y.Q.; Morris, W.L.; McGuire, P.A.; Novotny, M.J.; Smith, W.; Henschen, A.H.; Cullor, J.S. Purification, Primary Structures, and Antibacterial Activities of Beta-Defensins, a New Family of Antimicrobial Peptides from Bovine Neutrophils. J. Biol. Chem. 1993, 268, 6641–6648. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.Q.; Yuan, J.; Ösapay, G.; Ösapay, K.; Tran, D.; Miller, C.J.; Ouellette, A.J.; Selsted, M.E. A Cyclic Antimicrobial Peptide Produced in Primate Leukocytes by the Ligation of Two Truncated α-Defensins. Science 1999, 286, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Brogden, K.A.; Ackermann, M.; Huttner, K.M. Small, Anionic, and Charge-Neutralizing Propeptide Fragments of Zymogens Are Antimicrobial. Antimicrob. Agents Chemother. 1997, 41, 1615–1617. [Google Scholar] [CrossRef]
- Tam, J.P.; Wang, S.; Wong, K.H.; Tan, W.L. Antimicrobial Peptides from Plants. Pharmaceuticals 2015, 8, 711–757. [Google Scholar] [CrossRef]
- Masso-Silva, J.A.; Diamond, G. Antimicrobial Peptides from Fish. Pharmaceuticals 2014, 7, 265–310. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Peinado, C.; Defaus, S.; Andreu, D. Hitchhiking with Nature: Snake Venom Peptides to Fight Cancer and Superbugs. Toxins 2020, 12, 255. [Google Scholar] [CrossRef]
- Santana, F.L.; Estrada, K.; Ortiz, E.; Corzo, G. Reptilian β-Defensins: Expanding the Repertoire of Known Crocodylian Peptides. Peptides 2021, 136, 170473. [Google Scholar] [CrossRef] [PubMed]
- Andreu, D.; Rivas, L. Animal Antimicrobial Peptides: An Overview. Pept. Sci. 1998, 47, 415–433. [Google Scholar] [CrossRef]
- Mangoni, M.L.; Shai, Y. Temporins and Their Synergism against Gram-Negative Bacteria and in Lipopolysaccharide Detoxification. Biochim. Biophys. Acta-Biomembr. 2009, 1788, 1610–1619. [Google Scholar] [CrossRef]
- Simons, A.; Alhanout, K.; Duval, R.E. Bacteriocins, Antimicrobial Peptides from Bacterial Origin: Overview of Their Biology and Their Impact against Multidrug-Resistant Bacteria. Microorganisms 2020, 8, 639. [Google Scholar] [CrossRef]
- Mookherjee, N.; Anderson, M.A.; Haagsman, H.P.; Davidson, D.J. Antimicrobial Host Defence Peptides: Functions and Clinical Potential. Nat. Rev. Drug Discov. 2020, 19, 311–332. [Google Scholar] [CrossRef] [PubMed]
- Harris, F.; Dennison, S.; Phoenix, D. Anionic Antimicrobial Peptides from Eukaryotic Organisms. Curr. Protein Pept. Sci. 2009, 10, 585–606. [Google Scholar] [CrossRef]
- Boparai, J.K.; Sharma, P.K. Mini Review on Antimicrobial Peptides, Sources, Mechanism and Recent Applications. Protein Pept. Lett. 2020, 27, 4–16. [Google Scholar] [CrossRef]
- Savini, F.; Bobone, S.; Roversi, D.; Mangoni, M.L.; Stella, L. From Liposomes to Cells: Filling the Gap between Physicochemical and Microbiological Studies of the Activity and Selectivity of Host-Defense Peptides. Pept. Sci. 2018, 110, e24041. [Google Scholar] [CrossRef]
- Pfalzgraff, A.; Brandenburg, K.; Weindl, G. Antimicrobial Peptides and Their Therapeutic Potential for Bacterial Skin Infections and Wounds. Front. Pharmacol. 2018, 9, 281. [Google Scholar] [CrossRef]
- Kumar, P.; Takayesu, A.; Abbasi, U.; Kalathottukaren, M.T.; Abbina, S.; Kizhakkedathu, J.N.; Straus, S.K. Antimicrobial Peptide-Polymer Conjugates with High Activity: Influence of Polymer Molecular Weight and Peptide Sequence on Antimicrobial Activity, Proteolysis, and Biocompatibility. ACS Appl. Mater. Interfaces 2017, 9, 37575–37586. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Pletzer, D.; Haney, E.F.; Rahanjam, N.; Cheng, J.T.J.; Yue, M.; Aljehani, W.; Hancock, R.E.W.; Kizhakkedathu, J.N.; Straus, S.K. Aurein-Derived Antimicrobial Peptides Formulated with Pegylated Phospholipid Micelles to Target Methicillin-Resistant Staphylococcus Aureus Skin Infections. ACS Infect. Dis. 2019, 5, 443–453. [Google Scholar] [CrossRef]
- Lynn, M.A.; Kindrachuk, J.; Marr, A.K.; Jenssen, H.; Panté, N.; Elliott, M.R.; Napper, S.; Hancock, R.E.; McMaster, W.R. Effect of BMAP-28 Antimicrobial Peptides on Leishmania Major Promastigote and Amastigote Growth: Role of Leishmanolysin in Parasite Survival. PLoS Negl. Trop. Dis. 2011, 5, e1141. [Google Scholar] [CrossRef]
- Kelly, B.L.; Nelson, T.N.; McMaster, W.R. Stage-Specific Expression in Leishmania Conferred by 3′ Untranslated Regions of L. Major Leishmanolysin Genes (GP63). Mol. Biochem. Parasitol. 2001, 116, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Selsted, M.E.; Novotny, M.J.; Morris, W.L.; Tang, Y.Q.; Smith, W.; Cullor, J.S. Indolicidin, a Novel Bactericidal Tridecapeptide Amide from Neutrophils. J. Biol. Chem. 1992, 267, 4292–4295. [Google Scholar] [CrossRef]
- Falcao, C.B.; Pérez-Peinado, C.; De La Torre, B.G.; Mayol, X.; Zamora-Carreras, H.; Jiménez, M.Á.; Rádis-Baptista, G.; Andreu, D. Structural Dissection of Crotalicidin, a Rattlesnake Venom Cathelicidin, Retrieves a Fragment with Antimicrobial and Antitumor Activity. J. Med. Chem. 2015, 58, 8553–8563. [Google Scholar] [CrossRef]
- Pérez-Peinado, C.; Dias, S.A.; Mendonça, D.A.; Castanho, M.A.R.B.; Veiga, A.S.; Andreu, D. Structural Determinants Conferring Unusual Long Life in Human Serum to Rattlesnake-Derived Antimicrobial Peptide Ctn[15-34]. J. Pept. Sci. 2019, 25, e3195. [Google Scholar] [CrossRef]
- Pérez-Peinado, C.; Defaus, S.; Sans-Comerma, L.; Valle, J.; Andreu, D. Decoding the Human Serum Interactome of Snake-Derived Antimicrobial Peptide Ctn[15–34]: Toward an Explanation for Unusually Long Half-Life. J. Proteom. 2019, 204, 103372. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, D.; Jaśkiewicz, M.; Migoń, D.; Bauer, M.; Sikora, K.; Sikorska, E.; Kamysz, E.; Kamysz, W. Retro Analog Concept: Comparative Study on Physico-Chemical and Biological Properties of Selected Antimicrobial Peptides. Amino Acids 2017, 49, 1755–1771. [Google Scholar] [CrossRef]
- Chen, Y.; Vasil, A.I.; Rehaume, L.; Mant, C.T.; Burns, J.L.; Vasil, M.L.; Hancock, R.E.W.; Hodges, R.S. Comparison of Biophysical and Biologic Properties of α-Helical Enantiomeric Antimicrobial Peptides. Chem. Biol. Drug Des. 2006, 67, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Won, A.; Khan, M.; Gustin, S.; Akpawu, A.; Seebun, D.; Avis, T.J.; Leung, B.O.; Hitchcock, A.P.; Ianoul, A. Investigating the Effects of L- to D-Amino Acid Substitution and Deamidation on the Activity and Membrane Interactions of Antimicrobial Peptide Anoplin. Biochim. Biophys. Acta-Biomembr. 2011, 1808, 1592–1600. [Google Scholar] [CrossRef]
- Hirt, H.; Hall, J.W.; Larson, E.; Gorr, S.U. A D-Enantiomer of the Antimicrobial Peptide GL13K Evades Antimicrobial Resistance in the Gram Positive Bacteria Enterococcus Faecalis and Streptococcus Gordonii. PLoS ONE 2018, 13, e0194900. [Google Scholar] [CrossRef] [PubMed]
- Hirt, H.; Gorr, S.U. Antimicrobial Peptide GL13K Is Effective in Reducing Biofilms of Pseudomonas Aeruginosa. Antimicrob. Agents Chemother. 2013, 57, 4903–4910. [Google Scholar] [CrossRef] [PubMed]
- Cleophas, R.T.C.; Riool, M.; Quarles Van Ufford, H.C.; Zaat, S.A.J.; Kruijtzer, J.A.W.; Liskamp, R.M.J. Convenient Preparation of Bactericidal Hydrogels by Covalent Attachment of Stabilized Antimicrobial Peptides Using Thiol-Ene Click Chemistry. ACS Macro Lett. 2014, 3, 477–480. [Google Scholar] [CrossRef]
- Buckholtz, G.A.; Reger, N.A.; Anderton, W.D.; Schimoler, P.J.; Roudebush, S.L.; Meng, W.S.; Miller, M.C.; Gawalt, E.S. Reducing Escherichia Coli Growth on a Composite Biomaterial by a Surface Immobilized Antimicrobial Peptide. Mater. Sci. Eng. C 2016, 65, 126–134. [Google Scholar] [CrossRef]
- Cleophas, R.T.C.; Sjollema, J.; Busscher, H.J.; Kruijtzer, J.A.W.; Liskamp, R.M.J. Characterization and Activity of an Immobilized Antimicrobial Peptide Containing Bactericidal PEG-Hydrogel. Biomacromolecules 2014, 15, 3390–3395. [Google Scholar] [CrossRef]
- Ong, Z.Y.; Cheng, J.; Huang, Y.; Xu, K.; Ji, Z.; Fan, W.; Yang, Y.Y. Effect of Stereochemistry, Chain Length and Sequence Pattern on Antimicrobial Properties of Short Synthetic β-Sheet Forming Peptide Amphiphiles. Biomaterials 2014, 35, 1315–1325. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, M.; Qiu, S.; Wang, J.; Peng, J.; Zhao, P.; Zhu, R.; Wang, H.; Li, Y.; Wang, K.; et al. Antimicrobial Activity and Stability of the D-Amino Acid Substituted Derivatives of Antimicrobial Peptide Polybia-MPI. AMB Express 2016, 6, 122. [Google Scholar]
- Czechowicz, P.; Jaśkiewicz, M.; Neubauer, D.; Gościniak, G.; Kamysz, W. Anticandidal Activity of Omiganan and Its Retro Analog Alone and in Combination with Fluconazole. Probiotics Antimicrob. Proteins 2021, 13, 1173–1182. [Google Scholar] [CrossRef]
- Mohamed, M.F.; Brezden, A.; Mohammad, H.; Chmielewski, J.; Seleem, M.N. A Short D-Enantiomeric Antimicrobial Peptide with Potent Immunomodulatory and Antibiofilm Activity against Multidrug-Resistant Pseudomonas Aeruginosa and Acinetobacter Baumannii. Sci. Rep. 2017, 7, 6953. [Google Scholar] [CrossRef]
- Greco, I.; Hansen, J.E.; Jana, B.; Molchanova, N.; Oddo, A.; Thulstrup, P.W.; Damborg, P.; Guardabassi, L.; Hansen, P.R. Structure–Activity Study, Characterization, and Mechanism of Action of an Antimicrobial Peptoid D2 and Its D- and L-Peptide Analogues. Molecules 2019, 24, 1121. [Google Scholar] [CrossRef]
- Cesaro, A.; Torres, M.D.T.; Gaglione, R.; Dell’Olmo, E.; Di Girolamo, R.; Bosso, A.; Pizzo, E.; Haagsman, H.P.; Veldhuizen, E.J.A.; de la Fuente-Nunez, C.; et al. Synthetic Antibiotic Derived from Sequences Encrypted in a Protein from Human Plasma. ACS Nano 2022, 16, 1880–1895. [Google Scholar] [CrossRef] [PubMed]
- López-Sanmartín, M.; Rengel, R.; López-López, M.; Lebrón, J.A.; Molina-Márquez, A.; de la Rosa, I.; López-Cornejo, P.; Cuesta, A.; Vigara, J.; León, R. D-Amino Acid Peptides as Antimicrobial Agents against Vibrio-Associated Diseases in Aquaculture. Aquaculture 2023, 569, 739362. [Google Scholar]
- Schenk, M.; Mueller, C. The Mucosal Immune System at the Gastrointestinal Barrier. Best Pract. Res. Clin. Gastroenterol. 2008, 22, 391–409. [Google Scholar] [CrossRef]
- Tetro, N.; Moushaev, S.; Rubinchik-Stern, M.; Eyal, S. The Placental Barrier: The Gate and the Fate in Drug Distribution. Pharm. Res. 2018, 35, 71. [Google Scholar]
- Pardridge, W.M. The Blood-Brain Barrier: Bottleneck in Brain Drug Development. NeuroRx 2005, 2, 3–14. [Google Scholar] [CrossRef]
- Futaki, S.; Suzuki, T.; Ohashi, W.; Yagami, T.; Tanaka, S.; Ueda, K.; Sugiura, Y. Arginine-Rich Peptides: An Abundant Source of Membrane-Permeable Peptides Having Potential as Carriers for Intracellular Protein Delivery. J. Biol. Chem. 2001, 276, 5836–5840. [Google Scholar] [CrossRef]
- Liu, Y.; Mei, L.; Xu, C.; Yu, Q.; Shi, K.; Zhang, L.; Wang, Y.; Zhang, Q.; Gao, H.; Zhang, Z.; et al. Dual Receptor Recognizing Cell Penetrating Peptide for Selective Targeting, Efficient Intratumoral Diffusion and Synthesized Anti-Glioma Therapy. Theranostics 2016, 6, 177–191. [Google Scholar] [CrossRef] [PubMed]
- Futaki, S.; Ohashi, W.; Suzuki, T.; Niwa, M.; Tanaka, S.; Ueda, K.; Harashima, H.; Sugiura, Y. Stearylated Arginine-Rich Peptides: A New Class of Transfection Systems. Bioconjug. Chem. 2001, 12, 1005–1011. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, T. Cationic Peptide. In Cell-Penetrating Peptides: Design, Development and Applications; Oba, M., Demizu, Y., Eds.; John Wiley & Sons, Ltd.: London, UK, 2023; pp. 45–55. [Google Scholar]
- Pujals, S.; Sabidó, E.; Tarragó, T.; Giralt, E. All-D Proline-Rich Cell-Penetrating Peptides: A Preliminary In Vivo Internalization Study. Biochem. Soc. Trans. 2007, 35, 794–796. [Google Scholar] [PubMed]
- Fernández-Carneado, J.; Kogan, M.J.; Pujals, S.; Giralt, E. Amphipathic Peptides and Drug Delivery. Pept. Sci. 2004, 76, 196–203. [Google Scholar]
- Oba, M. Amphipathic Peptide. In Cell-Penetrating Peptides: Design, Development and Applications; Oba, M., Demizu, Y., Eds.; John Wiley & Sons, Ltd.: London, UK, 2023; pp. 57–67. [Google Scholar]
- Demizu, Y. Hydrophobic CPPs. In Cell-Penetrating Peptides: Design, Development and Applications; Oba, M., Demizu, Y., Eds.; John Wiley & Sons, Ltd.: London, UK, 2023; pp. 69–77. [Google Scholar]
- Schmidt, S.; Adjobo-Hermans, M.J.W.; Kohze, R.; Enderle, T.; Brock, R.; Milletti, F. Identification of Short Hydrophobic Cell-Penetrating Peptides for Cytosolic Peptide Delivery by Rational Design. Bioconjug. Chem. 2017, 28, 382–389. [Google Scholar] [PubMed]
- Jirka, S.M.G.; Heemskerk, H.; Tanganyika-De Winter, C.L.; Muilwijk, D.; Pang, K.H.; De Visser, P.C.; Janson, A.; Karnaoukh, T.G.; Vermue, R.; ’T Hoen, P.A.C.; et al. Peptide Conjugation of 2′-O-Methyl Phosphorothioate Antisense Oligonucleotides Enhances Cardiac Uptake and Exon Skipping in Mdx Mice. Nucleic Acid Ther. 2014, 24, 25–36. [Google Scholar]
- Martín, I.; Teixidó, M.; Giralt, E. Design, Synthesis and Characterization of a New Anionic Cell-Penetrating Peptide: SAP(E). ChemBioChem 2011, 12, 896–903. [Google Scholar]
- Kam, A.; Loo, S.; Fan, J.S.; Sze, S.K.; Yang, D.; Tam, J.P. Roseltide RT7 Is a Disulfide-Rich, Anionic, and Cell-Penetrating Peptide That Inhibits Proteasomal Degradation. J. Biol. Chem. 2019, 294, 19604–19615. [Google Scholar]
- Szabó, I.; Yousef, M.; Soltész, D.; Bató, C.; Mező, G.; Bánóczi, Z. Redesigning of Cell-Penetrating Peptides to Improve Their Efficacy as a Drug Delivery System. Pharmaceutics 2022, 14, 907. [Google Scholar]
- Palm, C.; Jayamanne, M.; Kjellander, M.; Hällbrink, M. Peptide Degradation Is a Critical Determinant for Cell-Penetrating Peptide Uptake. Biochim. Biophys. Acta-Biomembr. 2007, 1768, 1769–1776. [Google Scholar]
- Kalafatovic, D.; Giralt, E. Cell-Penetrating Peptides: Design Strategies beyond Primary Structure and Amphipathicity. Molecules 2017, 22, 1929. [Google Scholar]
- Vivès, E.; Brodin, P.; Lebleu, B. A Truncated HIV-1 Tat Protein Basic Domain Rapidly Translocates through the Plasma Membrane and Accumulates in the Cell Nucleus. J. Biol. Chem. 1997, 272, 16010–16017. [Google Scholar]
- Wender, P.A.; Mitchell, D.J.; Pattabiraman, K.; Pelkey, E.T.; Steinman, L.; Rothbard, J.B. The Design, Synthesis, and Evaluation of Molecules That Enable or Enhance Cellular Uptake: Peptoid Molecular Transporters. Proc. Natl. Acad. Sci. USA 2000, 97, 13003–13008. [Google Scholar] [PubMed]
- Seisel, Q.; Lakumpa, I.; Josse, E.; Vivès, E.; Varilh, J.; Taulan-Cadars, M.; Boisguérin, P. Highway to Cell: Selection of the Best Cell-Penetrating Peptide to Internalize the CFTR-Stabilizing ICAL36 Peptide. Pharmaceutics 2022, 14, 808. [Google Scholar] [PubMed]
- Nielsen, E.J.B.; Yoshida, S.; Kamei, N.; Iwamae, R.; Khafagy, E.S.; Olsen, J.; Rahbek, U.L.; Pedersen, B.L.; Takayama, K.; Takeda-Morishita, M. In Vivo Proof of Concept of Oral Insulin Delivery Based on a Co-Administration Strategy with the Cell-Penetrating Peptide Penetratin. J. Control. Release 2014, 189, 19–24. [Google Scholar] [PubMed]
- Kamei, N.; Shigei, C.; Hasegawa, R.; Takeda-Morishita, M. Exploration of the Key Factors for Optimizing the In Vivo Oral Delivery of Insulin by Using a Noncovalent Strategy with Cell-Penetrating Peptides. Biol. Pharm. Bull. 2018, 41, 239–246. [Google Scholar]
- Garcia, J.; Fernández-Blanco, Á.; Teixidó, M.; Sánchez-Navarro, M.; Giralt, E. D-Polyarginine Lipopeptides as Intestinal Permeation Enhancers. ChemMedChem 2018, 13, 2045–2052. [Google Scholar] [PubMed]
- Kamei, N.; Yamaoka, A.; Fukuyama, Y.; Itokazu, R.; Takeda-Morishita, M. Noncovalent Strategy with Cell-Penetrating Peptides to Facilitate the Brain Delivery of Insulin through the Blood–Brain Barrier. Biol. Pharm. Bull. 2018, 41, 546–554. [Google Scholar] [PubMed]
- Wang, C.; Lin, G.; Shen, Z.; Wang, R. Angiopep-2 as an Exogenous Chemical Exchange Saturation Transfer Contrast Agent in Diagnosis of Alzheimer’s Disease. J. Healthc. Eng. 2022, 2022, 7480519. [Google Scholar] [PubMed]
- Xie, R.; Wu, Z.; Zeng, F.; Cai, H.; Wang, D.; Gu, L.; Zhu, H.; Lui, S.; Guo, G.; Song, B.; et al. Retro-Enantio Isomer of Angiopep-2 Assists Nanoprobes across the Blood-Brain Barrier for Targeted Magnetic Resonance/Fluorescence Imaging of Glioblastoma. Signal Transduct. Target. Ther. 2021, 6, 309. [Google Scholar]
- Díaz-Perlas, C.; Oller-Salvia, B.; Sánchez-Navarro, M.; Teixidó, M.; Giralt, E. Branched BBB-Shuttle Peptides: Chemoselective Modification of Proteins to Enhance Blood–Brain Barrier Transport. Chem. Sci. 2018, 9, 8409–8415. [Google Scholar]
- Bukchin, A.; Sanchez-Navarro, M.; Carrera, A.; Teixidó, M.; Carcaboso, A.M.; Giralt, E.; Sosnik, A. Amphiphilic Polymeric Nanoparticles Modified with a Retro-Enantio Peptide Shuttle Target the Brain of Mice. Chem. Mater. 2020, 32, 7679–7693. [Google Scholar]
- Jones, S.; Howl, J. Enantiomer-Specific Bioactivities of Peptidomimetic Analogues of Mastoparan and Mitoparan: Characterization of Inverso Mastoparan as a Highly Efficient Cell Penetrating Peptide. Bioconjug. Chem. 2012, 23, 47–56. [Google Scholar] [PubMed]
- Verdurmen, W.P.R.; Bovee-Geurts, P.H.; Wadhwani, P.; Ulrich, A.S.; Hällbrink, M.; Van Kuppevelt, T.H.; Brock, R. Preferential Uptake of L- versus D-Amino Acid Cell-Penetrating Peptides in a Cell Type-Dependent Manner. Chem. Biol. 2011, 18, 1000–1010. [Google Scholar]
- Li, J.P.; Kusche-Gullberg, M. Heparan Sulfate: Biosynthesis, Structure, and Function. Int. Rev. Cell Mol. Biol. 2016, 325, 215–273. [Google Scholar] [PubMed]
- Nakase, I.; Niwa, M.; Takeuchi, T.; Sonomura, K.; Kawabata, N.; Koike, Y.; Takehashi, M.; Tanaka, S.; Ueda, K.; Simpson, J.C.; et al. Cellular Uptake of Arginine-Rich Peptides: Roles for Macropinocytosis and Actin Rearrangement. Mol. Ther. 2004, 10, 1011–1022. [Google Scholar]
- Zhan, C.; Li, B.; Hu, L.; Wei, X.; Feng, L.; Fu, W.; Lu, W. Micelle-Based Brain-Targeted Drug Delivery Enabled by a Nicotine Acetylcholine Receptor Ligand. Angew. Chem. Int. Ed. 2011, 50, 5482–5485. [Google Scholar]
- Wei, X.; Zhan, C.; Shen, Q.; Fu, W.; Xie, C.; Gao, J.; Peng, C.; Zheng, P.; Lu, W. A D-Peptide Ligand of Nicotine Acetylcholine Receptors for Brain-Targeted Drug Delivery. Angew. Chem. Int. Ed. 2015, 54, 3023–3027. [Google Scholar]
- Han, B.; Xie, W.; Zhang, Y.; Zhou, S.; Yang, J.; Wang, R.; Sun, Y.; Wang, X.; Xu, J.; Chen, D.; et al. The Influx/Efflux Mechanisms of d-Peptide Ligand of NAChRs across the Blood–Brain Barrier and Its Therapeutic Value in Treating Glioma. J. Control. Release 2020, 327, 384–396. [Google Scholar] [PubMed]
- Gallo, M.; Moreno, E.; Defaus, S.; Ortega-Alvaro, A.; Gonzalez, A.; Robledo, P.; Cavaco, M.; Neves, V.; Castanho, M.A.R.B.; Casadó, V.; et al. Orally Active Peptide Vector Allows Using Cannabis to Fight Pain While Avoiding Side Effects. J. Med. Chem. 2021, 64, 6937–6948. [Google Scholar] [PubMed]
- Meloni, B.P.; Craig, A.J.; Milech, N.; Hopkins, R.M.; Watt, P.M.; Knuckey, N.W. The Neuroprotective Efficacy of Cell-Penetrating Peptides TAT, Penetratin, Arg-9, and Pep-1 in Glutamic Acid, Kainic Acid, and In Vitro Ischemia Injury Models Using Primary Cortical Neuronal Cultures. Cell. Mol. Neurobiol. 2014, 34, 173–181. [Google Scholar]
- Snyder, E.L.; Meade, B.R.; Saenz, C.C.; Dowdy, S.F. Treatment of Terminal Peritoneal Carcinomatosis by a Transducible P53-Activating Peptide. PLoS Biol. 2004, 2, 0186–0193. [Google Scholar]
- Liu, L.; Xie, H.J.; Mu, L.M.; Liu, R.; Su, Z.B.; Cui, Y.N.; Xie, Y.; Lu, W.L. Functional Chlorin Gold Nanorods Enable to Treat Breast Cancer by Photothermal/Photodynamic Therapy. Int. J. Nanomed. 2018, 13, 8119–8135. [Google Scholar]
- Najjar, K.; Erazo-Oliveras, A.; Brock, D.J.; Wang, T.Y.; Pellois, J.P. An L- to D-Amino Acid Conversion in an Endosomolytic Analog of the Cell-Penetrating Peptide TAT Influences Proteolytic Stability, Endocytic Uptake, and Endosomal Escape. J. Biol. Chem. 2017, 292, 847–861. [Google Scholar] [PubMed]
- Birch, D.; Christensen, M.V.; Staerk, D.; Franzyk, H.; Nielsen, H.M. Stereochemistry as a Determining Factor for the Effect of a Cell-Penetrating Peptide on Cellular Viability and Epithelial Integrity. Biochem. J. 2018, 475, 1773–1788. [Google Scholar]
- Liu, X.; Liu, C.; Zhang, W.; Xie, C.; Wei, G.; Lu, W. Oligoarginine-Modified Biodegradable Nanoparticles Improve the Intestinal Absorption of Insulin. Int. J. Pharm. 2013, 448, 159–167. [Google Scholar] [PubMed]
- Ndeboko, B.; Ramamurthy, N.; Lemamy, G.J.; Jamard, C.; Nielsen, P.E.; Cova, L. Role of Cell-Penetrating Peptides in Intracellular Delivery of Peptide Nucleic Acids Targeting Hepadnaviral Replication. Mol. Ther.-Nucleic Acids 2017, 9, 162–169. [Google Scholar]
- Edwards, A.B.; Cross, J.L.; Anderton, R.S.; Knuckey, N.W.; Meloni, B.P. Poly-Arginine R18 and R18D (D-Enantiomer) Peptides Reduce Infarct Volume and Improves Behavioural Outcomes Following Perinatal Hypoxic-Ischaemic Encephalopathy in the P7 Rat. Mol. Brain 2018, 11, 8. [Google Scholar]
- Wei, X.; Zhan, C.; Chen, X.; Hou, J.; Xie, C.; Lu, W. Retro-Inverso Isomer of Angiopep-2: A Stable d -Peptide Ligand Inspires Brain-Targeted Drug Delivery. Mol. Pharm. 2014, 11, 3261–3268. [Google Scholar]
- Bukchin, A.; Sanchez-Navarro, M.; Carrera, A.; Resa-Pares, C.; Castillo-Ecija, H.; Balaguer-Lluna, L.; Teixidó, M.; Olaciregui, N.G.; Giralt, E.; Carcaboso, A.M.; et al. Amphiphilic Polymeric Nanoparticles Modified with a Protease-Resistant Peptide Shuttle for the Delivery of SN-38 in Diffuse Intrinsic Pontine Glioma. ACS Appl. Nano Mater. 2021, 4, 1314–1329. [Google Scholar]
- Bonny, C.; Oberson, A.; Negri, S.; Sauser, C.; Schorderet, D.F. Cell-Permeable Peptide Inhibitors of JNKNovel Blockers of β-Cell Death. Diabetes 2001, 50, 77–82. [Google Scholar] [PubMed]
- Youngblood, D.S.; Hatlevig, S.A.; Hassinger, J.N.; Iversen, P.L.; Moulton, H.M. Stability of Cell-Penetrating Peptide-Morpholino Oligomer Conjugates in Human Serum and in Cells. Bioconjug. Chem. 2007, 18, 50–60. [Google Scholar]
- Pujals, S.; Fernández-Carneado, J.; Ludevid, M.D.; Giralt, E. D-SAP: A New, Noncytotoxic, and Fully Protease Resistant Cell-Penetrating Peptide. ChemMedChem 2008, 3, 296–301. [Google Scholar]
- Shaltiel-Karyo, R.; Frenkel-Pinter, M.; Egoz-Matia, N.; Frydman-Marom, A.; Shalev, D.E.; Segal, D.; Gazit, E. Inhibiting α-Synuclein Oligomerization by Stable Cell-Penetrating β-Synuclein Fragments Recovers Phenotype of Parkinson’s Disease Model Flies. PLoS ONE 2010, 5, e13863. [Google Scholar]
- Foy, K.C.; Liu, Z.; Phillips, G.; Miller, M.; Kaumaya, P.T.P. Combination Treatment with HER-2 and VEGF Peptide Mimics Induces Potent Anti-Tumor and Anti-Angiogenic Responses In Vitro and In Vivo. J. Biol. Chem. 2011, 286, 13626–13637. [Google Scholar]
- Québatte, G.; Kitas, E.; Seelig, J. RiDOM, a Cell Penetrating Peptide. Interaction with Phospholipid Bilayers. Biochim. Biophys. Acta-Biomembr. 2014, 1838, 968–977. [Google Scholar]
- Radicioni, G.; Stringaro, A.; Molinari, A.; Nocca, G.; Longhi, R.; Pirolli, D.; Scarano, E.; Iavarone, F.; Manconi, B.; Cabras, T.; et al. Characterization of the Cell Penetrating Properties of a Human Salivary Proline-Rich Peptide. Biochim. Biophys. Acta-Biomembr. 2015, 1848, 2868–2877. [Google Scholar]
- Rodrigues, M.; Andreu, D.; Santos, N.C. Uptake and Cellular Distribution of Nucleolar Targeting Peptides (NrTPs) in Different Cell Types. Pept. Sci. 2015, 104, 101–109. [Google Scholar]
- Vaissière, A.; Aldrian, G.; Konate, K.; Lindberg, M.F.; Jourdan, C.; Telmar, A.; Seisel, Q.; Fernandez, F.; Viguier, V.; Genevois, C.; et al. A Retro-Inverso Cell-Penetrating Peptide for SiRNA Delivery. J. Nanobiotechnol. 2017, 15, 34. [Google Scholar]
- Arranz-Gibert, P.; Ciudad, S.; Seco, J.; García, J.; Giralt, E.; Teixidó, M. Immunosilencing Peptides by Stereochemical Inversion and Sequence Reversal: Retro-D-Peptides. Sci. Rep. 2018, 8, 6446. [Google Scholar]
- Ren, Y.; Zhan, C.; Gao, J.; Zhang, M.; Wei, X.; Ying, M.; Liu, Z.; Lu, W. A D-Peptide Ligand of Integrins for Simultaneously Targeting Angiogenic Blood Vasculature and Glioma Cells. Mol. Pharm. 2018, 15, 592–601. [Google Scholar] [PubMed]
- Chai, Z.; Wu, T.; Dai, A.; Huynh, P.; Koentgen, F.; Krippner, G.; Ren, S.; Cooper, M.E. Targeting the CDA1/CDA1BP1 Axis Retards Renal Fibrosis in Experimental Diabetic Nephropathy. Diabetes 2019, 68, 395–408. [Google Scholar] [PubMed]
- Jiao, L.; Dong, Q.; Zhai, W.; Zhao, W.; Shi, P.; Wu, Y.; Zhou, X.; Gao, Y. A PD-L1 and VEGFR2 Dual Targeted Peptide and Its Combination with Irradiation for Cancer Immunotherapy. Pharmacol. Res. 2022, 182, 106343. [Google Scholar] [CrossRef]
- Knezevic, C.E.; Clarke, W. Cancer Chemotherapy: The Case for Therapeutic Drug Monitoring. Ther. Drug Monit. 2020, 42, 6–19. [Google Scholar]
- Springfeld, C.; Jäger, D.; Büchler, M.W.; Strobel, O.; Hackert, T.; Palmer, D.H.; Neoptolemos, J.P. Chemotherapy for Pancreatic Cancer. Presse Med. 2019, 48, e159–e174. [Google Scholar]
- Bukowski, K.; Kciuk, M.; Kontek, R. Mechanisms of Multidrug Resistance in Cancer Chemotherapy. Int. J. Mol. Sci. 2020, 21, 3233. [Google Scholar] [CrossRef]
- Arina, A.; Gutiontov, S.I.; Weichselbaum, R.R. Radiotherapy and Immunotherapy for Cancer: From “Systemic” to “Multisite". Clin. Cancer Res. 2020, 26, 2777–2782. [Google Scholar] [CrossRef]
- Vinod, S.K.; Hau, E. Radiotherapy Treatment for Lung Cancer: Current Status and Future Directions. Respirology 2020, 25, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Podder, T.K.; Fredman, E.T.; Ellis, R.J. Advances in Radiotherapy for Prostate Cancer Treatment. In Mechanisms of Multidrug Resistance in Cancer Chemotherapy; Schatten, H., Ed.; Springer: New York, NY, USA, 2018; Volume 1126, pp. 31–47. [Google Scholar]
- Dijksteel, G.S.; Ulrich, M.M.W.; Middelkoop, E.; Boekema, B.K.H.L. Review: Lessons Learned from Clinical Trials Using Antimicrobial Peptides (AMPs). Front. Microbiol. 2021, 12, 616979. [Google Scholar]
- Riley, R.S.; June, C.H.; Langer, R.; Mitchell, M.J. Delivery Technologies for Cancer Immunotherapy. Nat. Rev. Drug Discov. 2019, 18, 175–196. [Google Scholar]
- Hoda, M. Potential Alternatives to Conventional Cancer Therapeutic Approaches: The Way Forward. Curr. Pharm. Biotechnol. 2021, 22, 1141–1148. [Google Scholar] [PubMed]
- Chiangjong, W.; Chutipongtanate, S.; Hongeng, S. Anticancer Peptide: Physicochemical Property, Functional Aspect and Trend in Clinical Application. Int. J. Oncol. 2020, 57, 678–696. [Google Scholar] [CrossRef] [PubMed]
- Johansson, A.; Dar, H.; van ’t Veer, L.J.; Tobin, N.P.; Perez-Tenorio, G.; Nordenskjöld, A.; Johansson, U.; Hartman, J.; Skoog, L.; Yau, C.; et al. Twenty-Year Benefit from Adjuvant Goserelin and Tamoxifen in Premenopausal Patients with Breast Cancer in a Controlled Randomized Clinical Trial. J. Clin. Oncol. 2022, 40, 4071–4082. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.C.; Meethal, S.V.; Bowen, R.L.; Atwood, C.S. Leuprolide Acetate: A Drug of Diverse Clinical Applications. Expert Opin. Investig. Drugs 2007, 16, 1851–1863. [Google Scholar] [CrossRef]
- Lamberts, S.W.J.; Hofland, L.J. Octreotide, 40 Years Later. Eur. J. Endocrinol. 2019, 181, R173–R183. [Google Scholar] [CrossRef]
- de la Torre, B.G.; Albericio, F. Peptide Therapeutics 2.0. Molecules 2020, 25, 2293. [Google Scholar] [CrossRef]
- D’Hondt, M.; Bracke, N.; Taevernier, L.; Gevaert, B.; Verbeke, F.; Wynendaele, E.; De Spiegeleer, B. Related Impurities in Peptide Medicines. J. Pharm. Biomed. Anal. 2014, 101, 2–30. [Google Scholar] [CrossRef]
- Hilchie, A.L.; Hoskin, D.W.; Power Coombs, M.R. Anticancer Activities of Natural and Synthetic Peptides. Adv. Exp. Med. Biol. 2019, 1117, 131–147. [Google Scholar]
- Hwang, J.S.; Kim, S.G.; Shin, T.H.; Jang, Y.E.; Kwon, D.H.; Lee, G. Development of Anticancer Peptides Using Artificial Intelligence and Combinational Therapy for Cancer Therapeutics. Pharmaceutics 2022, 14, 997. [Google Scholar] [CrossRef]
- Matijass, M.; Neundorf, I. Cell-Penetrating Peptides as Part of Therapeutics Used in Cancer Research. Med. Drug Discov. 2021, 10, 100092. [Google Scholar] [CrossRef]
- Norouzi, P.; Mirmohammadi, M.; Houshdar Tehrani, M.H. Anticancer Peptides Mechanisms, Simple and Complex. Chem. Biol. Interact. 2022, 368, 110194. [Google Scholar] [CrossRef]
- Bidwell, G.L.; Raucher, D. Therapeutic Peptides for Cancer Therapy. Part I—Peptide Inhibitors of Signal Transduction Cascades. Expert Opin. Drug Deliv. 2009, 6, 1033–1047. [Google Scholar] [CrossRef]
- Raucher, D.; Moktan, S.; Massodi, I.; Bidwell, G.L. Therapeutic Peptides for Cancer Therapy. Part II—Cell Cycle Inhibitory Peptides and Apoptosis-Inducing Peptides. Expert Opin. Drug Deliv. 2009, 6, 1049–1064. [Google Scholar] [CrossRef]
- Dutoit, V.; Migliorini, D.; Ranzanici, G.; Marinari, E.; Widmer, V.; Lobrinus, J.A.; Momjian, S.; Costello, J.; Walker, P.R.; Okada, H.; et al. Antigenic Expression and Spontaneous Immune Responses Support the Use of a Selected Peptide Set from the IMA950 Glioblastoma Vaccine for Immunotherapy of Grade II and III Glioma. Oncoimmunology 2018, 7, e1391972. [Google Scholar] [CrossRef] [PubMed]
- Thundimadathil, J. Cancer Treatment Using Peptides: Current Therapies and Future Prospects. J. Amino Acids 2012, 2012, 1–13. [Google Scholar] [CrossRef]
- Miller, W.R.; Scott, W.N.; Morris, R.; Fraser, H.M.; Sharpe, R.M. Growth of Human Breast Cancer Cells Inhibited by a Luteinizing Hormone-Releasing Hormone Agonist. Nature 1985, 313, 231–233. [Google Scholar] [CrossRef] [PubMed]
- Schally, A.V.; Zhang, X.; Cai, R.; Hare, J.M.; Granata, R.; Bartoli, M. Actions and Potential Therapeutic Applications of Growth Hormone–Releasing Hormone Agonists. Endocrinology 2019, 160, 1600–1612. [Google Scholar] [CrossRef]
- Tornesello, A.L.; Borrelli, A.; Buonaguro, L.; Buonaguro, F.M.; Tornesello, M.L. Antimicrobial Peptides as Anticancer Agents: Functional Properties and Biological Activities. Molecules 2020, 25, 2850. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Peinado, C.; Valle, J.; Freire, J.M.; Andreu, D. Tumor Cell Attack by Crotalicidin (Ctn) and Its Fragment Ctn[15-34]: Insights into Their Dual Membranolytic and Intracellular Targeting Mechanism. ACS Chem. Biol. 2020, 15, 2945–2957. [Google Scholar] [CrossRef] [PubMed]
- Pazgier, M.; Liu, M.; Zou, G.; Yuan, W.; Li, C.; Li, C.; Li, J.; Monbo, J.; Zella, D.; Tarasov, S.G.; et al. Structural Basis for High-Affinity Peptide Inhibition of P53 Interactions with MDM2 and MDMX. Proc. Natl. Acad. Sci. USA 2009, 106, 4665–4670. [Google Scholar] [CrossRef]
- Liu, M.; Pazgier, M.; Li, C.; Yuan, W.; Li, C.; Lu, W. A Left-Handed Solution to Peptide Inhibition of the P53–MDM2 Interaction. Angew. Chem. Int. Ed. 2010, 49, 3649–3652. [Google Scholar] [CrossRef]
- Giordano, R.J.; Cardó-Vila, M.; Lahdenranta, J.; Pasqualini, R.; Arap, W. Biopanning and Rapid Analysis of Selective Interactive Ligands. Nat. Med. 2001, 7, 1249–1253. [Google Scholar] [CrossRef]
- Giordano, R.J.; Cardó-Vila, M.; Salameh, A.; Anobom, C.D.; Zeitlin, B.D.; Hawke, D.H.; Valente, A.P.; Almeida, F.C.L.; Nör, J.E.; Sidman, R.L.; et al. From Combinatorial Peptide Selection to Drug Prototype (I): Targeting the Vascular Endothelial Growth Factor Receptor Pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 5112–5117. [Google Scholar] [CrossRef]
- Rezazadeh, F.; Sadeghzadeh, N.; Abedi, S.M.; Abediankenari, S. 99mTc Labeled D(LPR): A Novel Retro-Inverso Peptide for VEGF Receptor-1 Targeted Tumor Imaging. Nucl. Med. Biol. 2018, 62–63, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Mei, L.; Yu, Q.; Zhang, Q.; Gao, H.; Zhang, Z.; He, Q. Integrin Avβ3 Targeting Activity Study of Different Retro-Inverso Sequences of RGD and Their Potentiality in the Designing of Tumor Targeting Peptides. Amino Acids 2015, 47, 2533–2539. [Google Scholar] [CrossRef]
- Ramezanizadeh, M.; Masterifarahani, A.; Sadeghzadeh, N.; Abediankenari, S.; Mardanshahi, A.; Maleki, F. 99mTc-D(RGD): Molecular Imaging Probe for Diagnosis of α β -Positive Tumors. Nucl. Med. Commun. 2020, 41, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Mandelin, J.; Cardó-Vila, M.; Driessen, W.H.P.; Mathew, P.; Navonea, N.M.; Lin, S.H.; Logothetis, C.J.; Rietz, A.C.; Dobroff, A.S.; Proneth, B.; et al. Selection and Identification of Ligand Peptides Targeting a Model of Castrate-Resistant Osteogenic Prostate Cancer and Their Receptors. Proc. Natl. Acad. Sci. USA 2015, 112, 3776–3781. [Google Scholar] [CrossRef] [PubMed]
- Ran, D.; Mao, J.; Shen, Q.; Xie, C.; Zhan, C.; Wang, R.; Lu, W. GRP78 Enabled Micelle-Based Glioma Targeted Drug Delivery. J. Control. Release 2017, 255, 120–131. [Google Scholar] [CrossRef]
- Teesalu, T.; Sugahara, K.N.; Kotamraju, V.R.; Ruoslahti, E. C-End Rule Peptides Mediate Neuropilin-1-Dependent Cell, Vascular, and Tissue Penetration. Proc. Natl. Acad. Sci. USA 2009, 106, 16157–16162. [Google Scholar] [CrossRef]
- Tomita, T.; Kato, M.; Hiratsuka, S. Regulation of Vascular Permeability in Cancer Metastasis. Cancer Sci. 2021, 112, 2966–2974. [Google Scholar] [CrossRef]
- Wang, R.; Shen, Q.; Li, X.; Xie, C.; Lu, W.; Wang, S.; Wang, J.; Wang, D.; Liu, M. Efficacy of Inverso Isomer of CendR Peptide on Tumor Tissue Penetration. Acta Pharm. Sin. B 2018, 8, 825–832. [Google Scholar] [CrossRef]
- Papadimitriou, K.; Kountourakis, P.; Kottorou, A.E.; Antonacopoulou, A.G.; Rolfo, C.; Peeters, M.; Kalofonos, H.P. Follicle-Stimulating Hormone Receptor (FSHR): A Promising Tool in Oncology? Mol. Diagn. Ther. 2016, 20, 523–530. [Google Scholar] [CrossRef]
- Perales-Puchalt, A.; Svoronos, N.; Rutkowski, M.R.; Allegrezza, M.J.; Tesone, A.J.; Payne, K.K.; Wickramasinghe, J.; Nguyen, J.M.; O’Brien, S.W.; Gumireddy, K.; et al. Follicle-Stimulating Hormone Receptor Is Expressed by Most Ovarian Cancer Subtypes and Is a Safe and Effective Immunotherapeutic Target. Clin. Cancer Res. 2017, 23, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Y.; Chen, J.; Zheng, Y.F.; Gao, X.L.; Kang, Y.; Liu, J.C.; Cheng, M.J.; Sun, H.; Xu, C.J. Follicle-Stimulating Hormone Peptide Can Facilitate Paclitaxel Nanoparticles to Target Ovarian Carcinoma In Vivo. Cancer Res. 2009, 69, 6506–6514. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.M.; Zhang, M.M.; Wang, J.; Cai, Q.; Zhao, R.; Yu, Y.; Tai, H.; Zhang, X.; Xu, C. Retro-Inverso Follicle-Stimulating Hormone Peptide-Mediated Polyethylenimine Complexes for Targeted Ovarian Cancer Gene Therapy. Drug Deliv. 2018, 25, 995–1003. [Google Scholar] [CrossRef]
- Liu, M.; Li, C.; Pazgier, M.; Li, C.; Mao, Y.; Lv, Y.; Gu, B.; Wei, G.; Yuan, W.; Zhan, C.; et al. D-Peptide Inhibitors of the P53-MDM2 Interaction for Targeted Molecular Therapy of Malignant Neoplasms. Proc. Natl. Acad. Sci. USA 2010, 107, 14321–14326. [Google Scholar] [CrossRef]
- Calvanese, L.; Caporale, A.; Focà, G.; Iaccarino, E.; Sandomenico, A.; Doti, N.; Apicella, I.; Incisivo, G.M.; De Falco, S.; Falcigno, L.; et al. Targeting VEGF Receptors with Non-Neutralizing Cyclopeptides for Imaging Applications. Amino Acids 2018, 50, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xie, Z.; Xie, C.; Lu, W.; Gao, C.; Ren, H.; Ying, M.; Wei, X.; Gao, J.; Su, B.; et al. D-SP5 Peptide-Modified Highly Branched Polyethylenimine for Gene Therapy of Gastric Adenocarcinoma. Bioconjug. Chem. 2015, 26, 1494–1503. [Google Scholar] [CrossRef]
- Januchta, W.; Serocki, M.; Dzierzbicka, K.; Cholewiński, G.; Skladanowski, A. Synthesis of Functionalized New Conjugates of Batracylin with Tuftsin/Retro-Tuftsin Derivatives and Their Biological Evaluation. Eur. J. Med. Chem. 2015, 106, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Torabizadeh, S.A.; Abedi, S.M.; Noaparast, Z.; Hosseinimehr, S.J. Comparative Assessment of a 99mTc Labeled H1299.2-HYNIC Peptide Bearing Two Different Co-Ligands for Tumor-Targeted Imaging. Bioorg. Med. Chem. 2017, 25, 2583–2592. [Google Scholar] [CrossRef]
- Xie, Z.; Shen, Q.; Xie, C.; Lu, W.; Peng, C.; Wei, X.; Li, X.; Su, B.; Gao, C.; Liu, M. Retro-Inverso Bradykinin Opens the Door of Blood–Brain Tumor Barrier for Nanocarriers in Glioma Treatment. Cancer Lett. 2015, 369, 144–151. [Google Scholar] [CrossRef]
- Ran, D.; Mao, J.; Zhan, C.; Xie, C.; Ruan, H.; Ying, M.; Zhou, J.; Lu, W.-L.; Lu, W. D-Retroenantiomer of Quorum-Sensing Peptide-Modified Polymeric Micelles for Brain Tumor-Targeted Drug Delivery. ACS Appl. Mater. Interfaces 2017, 9, 25672–25682. [Google Scholar] [CrossRef]
- Carriero, M.V.; Bifulco, K.; Ingangi, V.; Costantini, S.; Botti, G.; Ragone, C.; Minopoli, M.; Motti, M.L.; Rea, D.; Scognamiglio, G.; et al. Retro-Inverso Urokinase Receptor Antagonists for the Treatment of Metastatic Sarcomas. Sci. Rep. 2017, 7, 1312. [Google Scholar] [CrossRef]
- Tang, J.; Wang, Q.; Yu, Q.; Qiu, Y.; Mei, L.; Wan, D.; Wang, X.; Li, M.; He, Q. A Stabilized Retro-Inverso Peptide Ligand of Transferrin Receptor for Enhanced Liposome-Based Hepatocellular Carcinoma-Targeted Drug Delivery. Acta Biomater. 2019, 83, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Du, M.; Qi, X.; Wang, Y.; Li, G.; Xu, C.; Zhang, X. Retro-Inversion Follicle-Stimulating Hormone Peptide-Modified Nanoparticles for Delivery of PDK2 ShRNA against Chemoresistant Ovarian Cancer by Switching Glycolysis to Oxidative Phosphorylation. Cancer Nanotechnol. 2022, 13, 23. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, Q.; Zhang, M.; Cao, C.; Liu, X.; Zhang, M.; Li, G.; Xu, C.; Zhang, X. Enhanced Antitumor Effects of Follicle-Stimulating Hormone Receptor-Mediated Hexokinase-2 Depletion on Ovarian Cancer Mediated by a Shift in Glucose Metabolism. J. Nanobiotechnol. 2020, 18, 161. [Google Scholar] [CrossRef] [PubMed]
- Hilchie, A.L.; Haney, E.F.; Pinto, D.M.; Hancock, R.E.W.; Hoskin, D.W. Enhanced Killing of Breast Cancer Cells by a D-Amino Acid Analog of the Winter Flounder-Derived Pleurocidin NRC-03. Exp. Mol. Pathol. 2015, 99, 426–434. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Fan, Z.; Chen, Y.; Fang, X.; Sha, X. Retro-Inverso Carbohydrate Mimetic Peptides with Annexin1-Binding Selectivity, Are Stable In Vivo, and Target Tumor Vasculature. PLoS ONE 2013, 8, e80390. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic Peptides: Current Applications and Future Directions. Signal Transduct. Target. Ther. 2022, 7, 48. [Google Scholar] [CrossRef]
- Pizzolato-Cezar, L.R.; Okuda-Shinagawa, N.M.; Teresa Machini, M. Combinatory Therapy Antimicrobial Peptide-Antibiotic to Minimize the Ongoing Rise of Resistance. Front. Microbiol. 2019, 10, 1703. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carrera-Aubesart, A.; Gallo, M.; Defaus, S.; Todorovski, T.; Andreu, D. Topoisomeric Membrane-Active Peptides: A Review of the Last Two Decades. Pharmaceutics 2023, 15, 2451. https://doi.org/10.3390/pharmaceutics15102451
Carrera-Aubesart A, Gallo M, Defaus S, Todorovski T, Andreu D. Topoisomeric Membrane-Active Peptides: A Review of the Last Two Decades. Pharmaceutics. 2023; 15(10):2451. https://doi.org/10.3390/pharmaceutics15102451
Chicago/Turabian StyleCarrera-Aubesart, Adam, Maria Gallo, Sira Defaus, Toni Todorovski, and David Andreu. 2023. "Topoisomeric Membrane-Active Peptides: A Review of the Last Two Decades" Pharmaceutics 15, no. 10: 2451. https://doi.org/10.3390/pharmaceutics15102451
APA StyleCarrera-Aubesart, A., Gallo, M., Defaus, S., Todorovski, T., & Andreu, D. (2023). Topoisomeric Membrane-Active Peptides: A Review of the Last Two Decades. Pharmaceutics, 15(10), 2451. https://doi.org/10.3390/pharmaceutics15102451