

Dendritic Glycerol-Cholesterol Amphiphiles as Drug Delivery Systems: A Comparison between Monomeric and Polymeric Structures
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis of Polymeric Amphiphiles
2.1.1. Synthesis of Acetal-Protected G1-Methacrylate Monomer
2.1.2. Synthesis of Cholesterol Methacrylate (CMA) Monomer
2.1.3. First Step: Synthesis of pG1MA Homopolymer
2.1.4. Second Step: Synthesis of pG1MA-b-pCMA Block Copolymer
2.1.5. Synthesis of pG1MAOH-b-pCMA Polymeric Amphiphiles by Deprotection of Dendritic Polyglycerol Units
2.2. Synthesis of Monomeric Amphiphiles
2.2.1. Synthesis of Acetal-Protected G2-CHOL
2.2.2. Synthesis of G2OH-CHOL Monomeric Amphiphiles by Deprotection of Dendritic Polyglycerol Units
2.3. Preparation of Doxorubicin (Free Base)
2.4. Micelle Formation and Characterization
2.5. Cryo-Tem Imaging
2.6. Encapsulation Studies
2.7. Cytotoxicity Studies
2.8. Cellular Uptake Studies
3. Results
3.1. Synthesis of Polymeric Amphiphiles
3.2. Synthesis of Monomeric Amphiphiles
3.3. Formation and Characterization of Micelles
3.4. Cell Viability
3.5. Characterization and In Vitro Studies of DOX-Loaded Polymeric Micelles
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CMC | Critical Micelle Concentration |
| DDS | Drug Delivery System |
| DOX | Doxorubicin |
| EE | Encapsulation Efficiency |
| LC | Loading Capacity |
| MW | Molecular Weight |
| RAFT | Reversible Addition–Fragmentation Chain Transfer |
Appendix A. Chemicals, Materials, and Methods
Appendix A.1. Chemicals and Materials
Appendix A.2. Methods
Appendix B. Synthesis of Acetal-Protected G2-NH2 Precursor

Appendix C. Calculation of Monomer Conversion

Appendix D. Calculation of the Degree of Polymerization, Molecular Weights, and HLB Values

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | M (g/mol) | p | p | MW (g/mol) | HLB 1 |
|---|---|---|---|---|---|
| pG1MA | 5000 | 12 | − | − | − |
| pG1MA-b-pCMA | 6900 | 12 | 4 | − | − |
| pG1MAOH-b-pCMA | 5940 2 | 12 | 4 | 4040 | 13.6 |
| pG1MAOH | 4040 2 | 12 | − | 4040 | − |
| G2OH-CHOL | 948 | − | − | 536 | 11.3 |
Appendix E. Loading Capacity (LC) and Encapsulation Efficiency (EE) Calculation
References
- Pucci, C.; Martinelli, C.; Ciofani, G. Innovative approaches for cancer treatment: Current perspectives and new challenges. Ecancer 2019, 13, 961. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Votruba, A.R.; Farokhzad, O.C.; Langer, R. Nanotechnology in drug delivery and tissue engineering: From discovery to applications. Nano Lett. 2010, 10, 3223–3230. [Google Scholar] [CrossRef] [PubMed]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Qin, X.; Kong, F.; Chen, P.; Pan, G. Improving cellular uptake of therapeutic entities through interaction with components of cell membrane. Drug Deliv. 2019, 26, 328–342. [Google Scholar] [CrossRef] [PubMed]
- Liechty, W.B.; Kryscio, D.R.; Slaughter, B.V.; Peppas, N.A. Polymers for drug delivery systems. Annu. Rev. Chem. Biomol. Eng. 2010, 1, 149–173. [Google Scholar] [CrossRef] [PubMed]
- Kumari, P.; Ghosh, B.; Biswas, S. Nanocarriers for cancer-targeted drug delivery. J. Drug Target. 2016, 24, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Ling, P.; Zhang, T. Polymeric Micelles, a Promising Drug Delivery System to Enhance Bioavailability of Poorly Water-Soluble Drugs. J. Drug Deliv. 2013, 2013, 340315. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.y.; Zhang, P.y. Polymersomes in Nanomedicine—A Review. Curr. Nanosci. 2016, 13, 124–129. [Google Scholar] [CrossRef]
- Nagarajan, R.; Ganesh, K. Block copolymer self-assembly in selective solvents: Spherical micelles with segregated cores. J. Chem. Phys. 1989, 90, 5843–5856. [Google Scholar] [CrossRef]
- Martin, C.; Marino, N.; Curran, C.; McHale, A.P.; Callan, J.F.; Callan, B. Cholesteryl to improve the cellular uptake of polymersomes within HeLa cells. Int. J. Pharm. 2016, 511, 570–578. [Google Scholar] [CrossRef]
- Kumari, P.; Muddineti, O.S.; Rompicharla, S.V.K.; Ghanta, P.; Adithya, K.B.; Ghosh, B.; Biswas, S. Cholesterol-conjugated poly(D, L-lactide)-based micelles as a nanocarrier system for effective delivery of curcumin in cancer therapy. Drug Deliv. 2017, 24, 209–223. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Tian, Q.; Huang, Z.; Fan, D.; She, Z.; Liu, X.; Cheng, X.; Yu, B.; Deng, Y. Self-assembled micelles of novel amphiphilic copolymer cholesterol-coupled F68 containing cabazitaxel as a drug delivery system. Int. J. Nanomed. 2014, 9, 2307–2317. [Google Scholar] [CrossRef]
- Lee, A.L.; Venkataraman, S.; Sirat, S.B.; Gao, S.; Hedrick, J.L.; Yang, Y.Y. The use of cholesterol-containing biodegradable block copolymers to exploit hydrophobic interactions for the delivery of anticancer drugs. Biomaterials 2012, 33, 1921–1928. [Google Scholar] [CrossRef]
- Misiak, P.; Markiewicz, K.H.; Szymczuk, D.; Wilczewska, A.Z. Polymeric drug delivery systems bearing cholesterol moieties: A review. Polymers 2020, 12, 2620. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.H.; Nguyen, C.T.; Gonzalez-Fajardo, L.; Hargrove, D.; Song, D.; Deshmukh, P.; Mahajan, L.; Ndaya, D.; Lai, L.; Kasi, R.M.; et al. Long Circulating Self-Assembled Nanoparticles from Cholesterol-Containing Brush-Like Block Copolymers for Improved Drug Delivery to Tumors. Biomacromolecules 2014, 15, 4363–4375. [Google Scholar] [CrossRef] [PubMed]
- Thunemann, A.F.; Gruber, A.; Klinger, D. Amphiphilic Nanogels: Fuzzy Spheres with a Pseudo-Periodic Internal Structure. Langmuir 2020, 36, 10979–10988. [Google Scholar] [CrossRef] [PubMed]
- Wan, Q.; Huang, B.; Li, T.; Xiao, Y.; He, Y.; Du, W.; Wang, B.Z.; Dakin, G.F.; Rosenbaum, M.; Goncalves, M.D.; et al. Selective targeting of visceral adiposity by polycation nanomedicine. Nat. Nanotechnol. 2022, 17, 1311–1321. [Google Scholar] [CrossRef] [PubMed]
- Perrier, S. 50th Anniversary Perspective: RAFT Polymerization—A User Guide. Macromolecules 2017, 50, 7433–7447. [Google Scholar] [CrossRef]
- He, S.J.; Zhang, Y.; Cui, Z.H.; Tao, Y.Z.; Zhang, B.L. Controlled radical polymerization of cholesteryl acrylate and its block copolymer with styrene via the RAFT process. Eur. Polym. J. 2009, 45, 2395–2401. [Google Scholar] [CrossRef]
- Keddie, D.J. A guide to the synthesis of block copolymers using reversible-addition fragmentation chain transfer (RAFT) polymerization. Chem. Soc. Rev. 2014, 43, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Magami, S.M.; Abdulganiyyu, U. Raft approach to the copolymerisation of methyl methacrylate based polymeric micelles. Bayero J. Pure Appl. Sci. 2017, 10, 197. [Google Scholar] [CrossRef]
- Semsarilar, M.; Perrier, S. ’Green’ reversible addition-fragmentation chain-transfer (RAFT) polymerization. Nat. Chem. 2010, 2, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Boyer, C.; Bulmus, V.; Davis, T.P.; Ladmiral, V.; Liu, J.; Perrier, S. Bioapplications of RAFT polymerization. Chem. Rev. 2009, 109, 5402–5436. [Google Scholar] [CrossRef] [PubMed]
- Moad, G.; Rizzardo, E.; Thang, S.H. Living radical polymerization by the RAFT process—A second update. Aust. J. Chem. 2009, 62, 1402–1472. [Google Scholar] [CrossRef]
- Misiak, P.; Niemirowicz- Laskowska, K.; Markiewicz, K.H.; Misztalewska-Turkowicz, I.; Wielgat, P.; Kurowska, I.; Siemiaszko, G.; Destarac, M.; Car, H.; Wilczewska, A.Z. Evaluation of cytotoxic effect of cholesterol end-capped poly(N-isopropylacrylamide)s on selected normal and neoplastic cells. Int. J. Nanomed. 2020, 15, 7263–7278. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.L.; Lavasanifar, A.; Kwon, G.S. Amphiphilic block copolymers for drug delivery. J. Pharm. Sci. 2003, 92, 1343–1355. [Google Scholar] [CrossRef]
- Yoncheva, K.; Calleja, P.; Agüeros, M.; Petrov, P.; Miladinova, I.; Tsvetanov, C.; Irache, J.M. Stabilized micelles as delivery vehicles for paclitaxel. Int. J. Pharm. 2012, 436, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Achazi, K.; Böttcher, C.; Haag, R.; Sharma, S.K. Fabrication of nanostructures through self-assembly of non-ionic amphiphiles for biomedical applications. RSC Adv. 2017, 7, 22121–22132. [Google Scholar] [CrossRef]
- Rodrigo, A.C.; Malhotra, S.; Böttcher, C.; Adeli, M.; Haag, R. Dendritic polyglycerol cyclodextrin amphiphiles and their self-assembled architectures to transport hydrophobic guest molecules. RSC Adv. 2014, 4, 61656–61659. [Google Scholar] [CrossRef]
- Haag, R.; Kratz, F. Polymer therapeutics: Concepts and applications. Angew. Chem.-Int. Ed. 2006, 45, 1198–1215. [Google Scholar] [CrossRef]
- Trappmann, B.; Ludwig, K.; Radowski, M.R.; Shukla, A.; Mohr, A.; Rehage, H.; Böttcher, C.; Haag, R. A new family of nonionic dendritic amphiphiles displaying unexpected packing parameters in micellar assemblies. J. Am. Chem. Soc. 2010, 132, 11119–11124. [Google Scholar] [CrossRef]
- Richter, A.; Wiedekind, A.; Krause, M.; Kissel, T.; Haag, R.; Olbrich, C. Non-ionic dendritic glycerol-based amphiphiles: Novel excipients for the solubilization of poorly water-soluble anticancer drug Sagopilone. Eur. J. Pharm. Sci. 2010, 40, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Kasi, R.M. Synthesis and Characterization of Polycholesteryl Methacrylate–Polyhydroxyethyl Methyacrylate Block Copolymers. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 6801–6809. [Google Scholar] [CrossRef]
- Prasad, S.; Achazi, K.; Schade, B.; Haag, R.; Sharma, S.K. Nonionic Dendritic and Carbohydrate Based Amphiphiles: Self-Assembly and Transport Behavior. Macromol. Biosci. 2018, 18, 1800019. [Google Scholar] [CrossRef] [PubMed]
- Wyszogrodzka, M.; Haag, R. A convergent approach to biocompatible polyglycerol "click" dendrons for the synthesis of modular core-shell architectures and their transport behavior. Chem.-Eur. J. 2008, 14, 9202–9214. [Google Scholar] [CrossRef]
- Patravale, V.B.; Upadhaya, P.G.; Jain, R.D. Preparation and Characterization of Micelles; Humana Press Inc.: Totowa, NJ, USA, 2019; Volume 2000, pp. 19–29. [Google Scholar] [CrossRef]
- Rashmi; Singh, A.K.; Achazi, K.; Schade, B.; Böttcher, C.; Haag, R.; Sharma, S.K. Synthesis of non-ionic bolaamphiphiles and study of their self-assembly and transport behaviour for drug delivery applications. RSC Adv. 2018, 8, 31777–31782. [Google Scholar] [CrossRef] [PubMed]
- Tschiche, A.; Staedtler, A.M.; Malhotra, S.; Bauer, H.; Böttcher, C.; Sharbati, S.; Calderón, M.; Koch, M.; Zollner, T.M.; Barnard, A.; et al. Polyglycerol-based amphiphilic dendrons as potential siRNA carriers for in vivo applications. J. Mater. Chem. B 2014, 2, 2153–2167. [Google Scholar] [CrossRef]
- Tabas, I. Consequences of cellular cholesterol accumulation: Basic concepts and physiological implications. J. Clin. Investig. 2002, 110, 905–911. [Google Scholar] [CrossRef]
- Krause, M.R.; Regen, S.L. The Structural Role of Cholesterol in Cell Membranes: From Condensed Bilayers to Lipid Rafts. Accounts Chem. Res. 2014, 47, 3512–3521. [Google Scholar] [CrossRef] [PubMed]
- Alpugan, S.; Garcia, G.; Poyer, F.; Durmuş, M.; Maillard, P.; Ahsen, V.; Dumoulin, F. Dendrimeric-like hexadecahydroxylated zinc phthalocyanine. J. Porphyrins Phthalocyanines 2013, 17, 596–603. [Google Scholar] [CrossRef]
- Griffin, W.C. Calculation of HLB values of non-ionic surfactants. J. Soc. Cosmet. Chem. 1954, 5, 249–256. [Google Scholar]




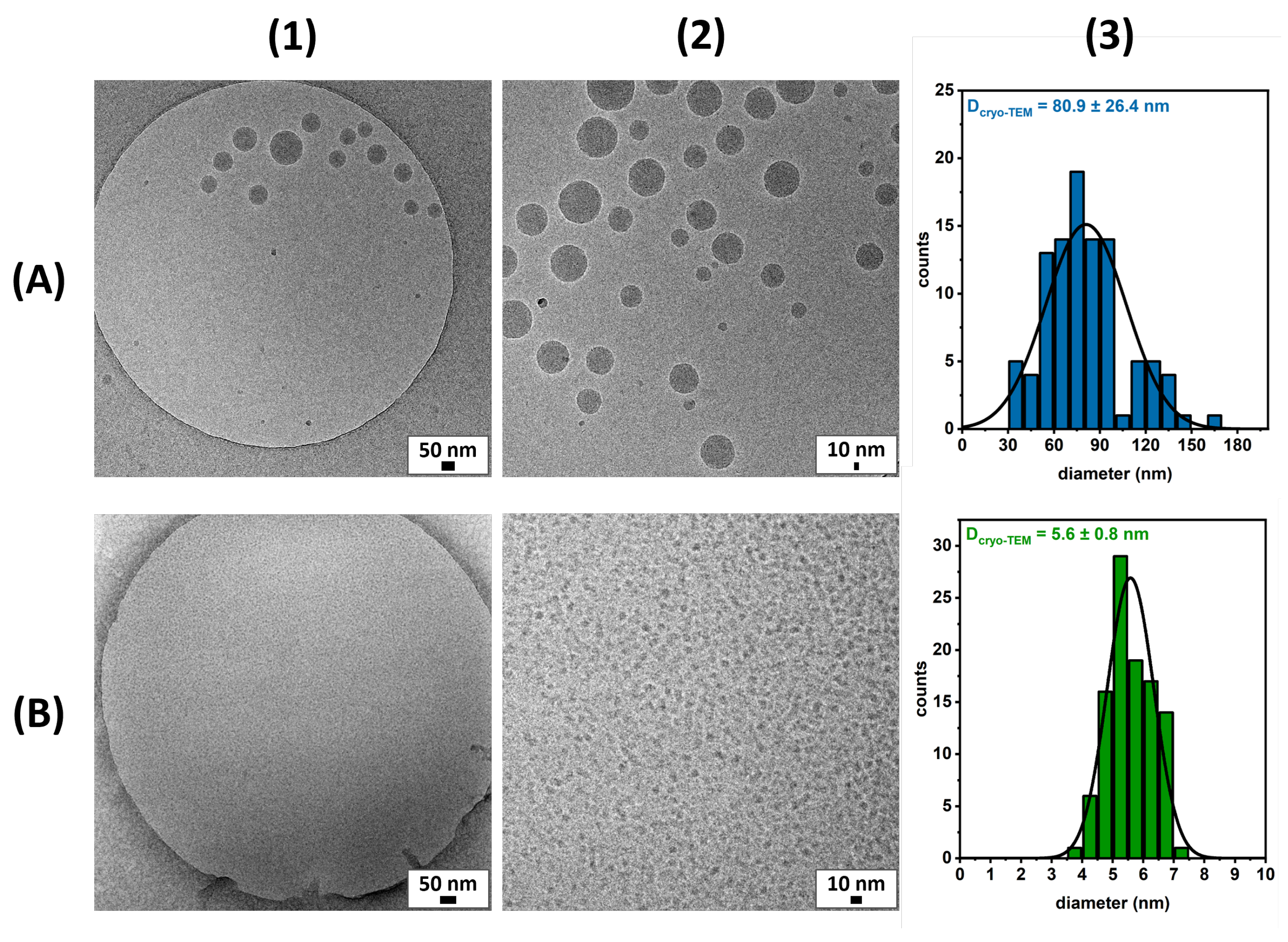



| Micelles System | MW (g/mol) | CMC (μg/mL) | (nm) | HLB 1 |
|---|---|---|---|---|
| polymeric | 5940 | 0.2 2 | 145 ± 7 | 13.6 |
| monomeric | 948 | 17.0 3 | 10 ± 1 | 11.3 |
| IC (μg/mL Polymeric Micelles) | IC (μg/mL DOX) | |
|---|---|---|
| empty micelles | 1555.0 | − |
| DOX-loaded micelles | 264.5 | 5.3 |
| free DOX | − | 9.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romero, J.F.; Herziger, S.; Cherri, M.; Dimde, M.; Achazi, K.; Mohammadifar, E.; Haag, R. Dendritic Glycerol-Cholesterol Amphiphiles as Drug Delivery Systems: A Comparison between Monomeric and Polymeric Structures. Pharmaceutics 2023, 15, 2452. https://doi.org/10.3390/pharmaceutics15102452
Romero JF, Herziger S, Cherri M, Dimde M, Achazi K, Mohammadifar E, Haag R. Dendritic Glycerol-Cholesterol Amphiphiles as Drug Delivery Systems: A Comparison between Monomeric and Polymeric Structures. Pharmaceutics. 2023; 15(10):2452. https://doi.org/10.3390/pharmaceutics15102452
Chicago/Turabian StyleRomero, Jocelyn Fernanda, Svenja Herziger, Mariam Cherri, Mathias Dimde, Katharina Achazi, Ehsan Mohammadifar, and Rainer Haag. 2023. "Dendritic Glycerol-Cholesterol Amphiphiles as Drug Delivery Systems: A Comparison between Monomeric and Polymeric Structures" Pharmaceutics 15, no. 10: 2452. https://doi.org/10.3390/pharmaceutics15102452
APA StyleRomero, J. F., Herziger, S., Cherri, M., Dimde, M., Achazi, K., Mohammadifar, E., & Haag, R. (2023). Dendritic Glycerol-Cholesterol Amphiphiles as Drug Delivery Systems: A Comparison between Monomeric and Polymeric Structures. Pharmaceutics, 15(10), 2452. https://doi.org/10.3390/pharmaceutics15102452