
The Role of Five-Membered Heterocycles in the Molecular Structure of Antibacterial Drugs Used in Therapy





Abstract
:1. Introduction
1.1. Antibacterials Shortage
1.2. Bacterial Resistance Phenomenon
1.3. FDA-Approved Antibiotics Whose Structure Includes Five-Members Heterocycles
1.4. Aim of the Work
2. Materials and Methods
3. Five-Membered Heterocycles Used in the Design of Antibacterial Drugs
- One heteroatom (nitrogen, oxygen, or sulfur);
- Two heteroatoms (oxygen and nitrogen; sulfur and nitrogen atoms);
- Three heteroatoms (three nitrogen atoms, e.g., triazoles, and one sulfur and two nitrogen atoms, e.g., thiadiazoles);
- Four heteroatoms (tetrazoles).
4. Five-Membered Heterocycles Containing Nitrogen Atoms
4.1. Pyrrolidine
4.1.1. Beta-Lactam Antibiotics
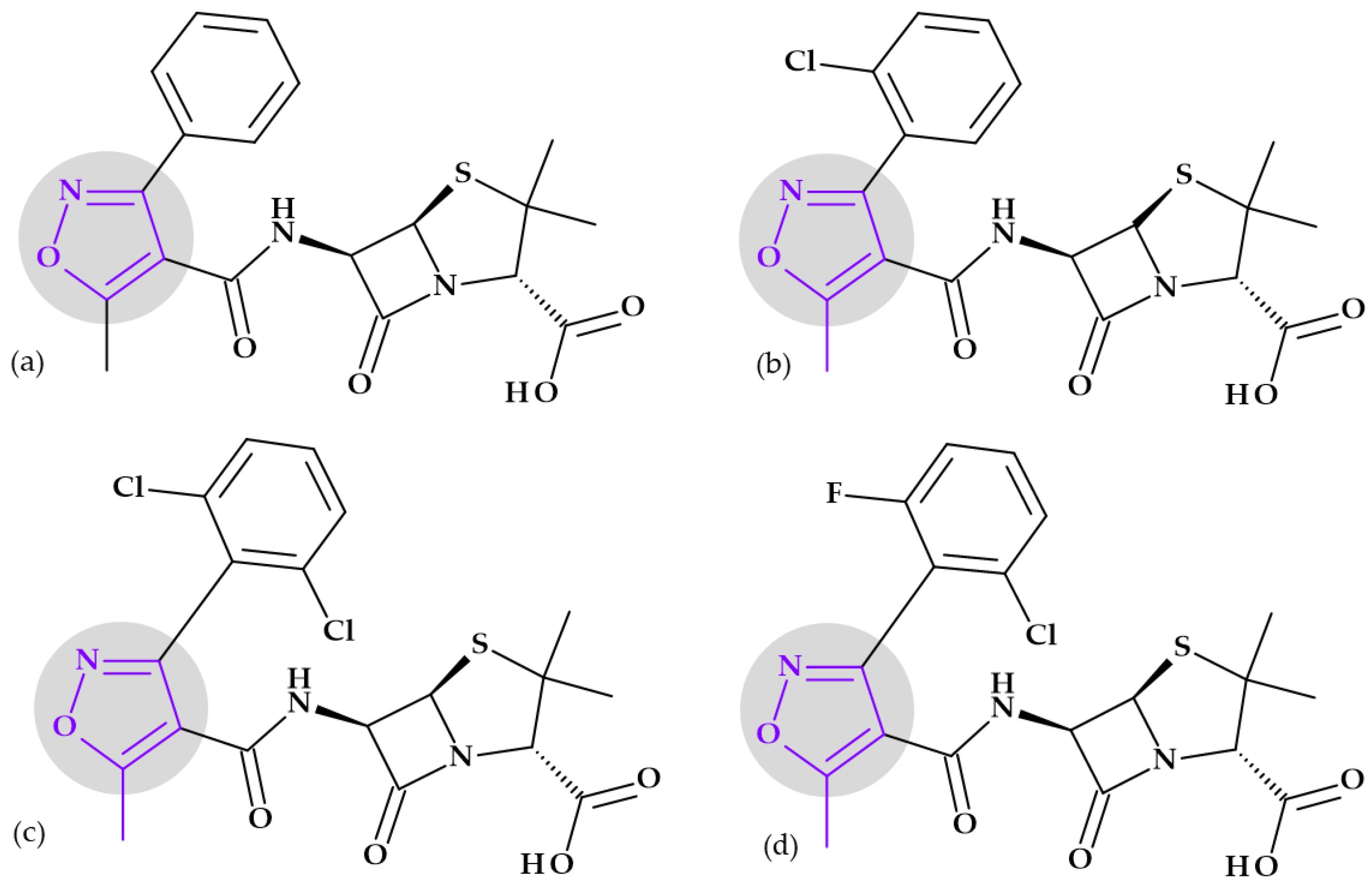
4.1.2. Fluoroquinolones
4.1.3. Lincosamides (Lincomycin and Clindamycin)
4.1.4. Streptogramins
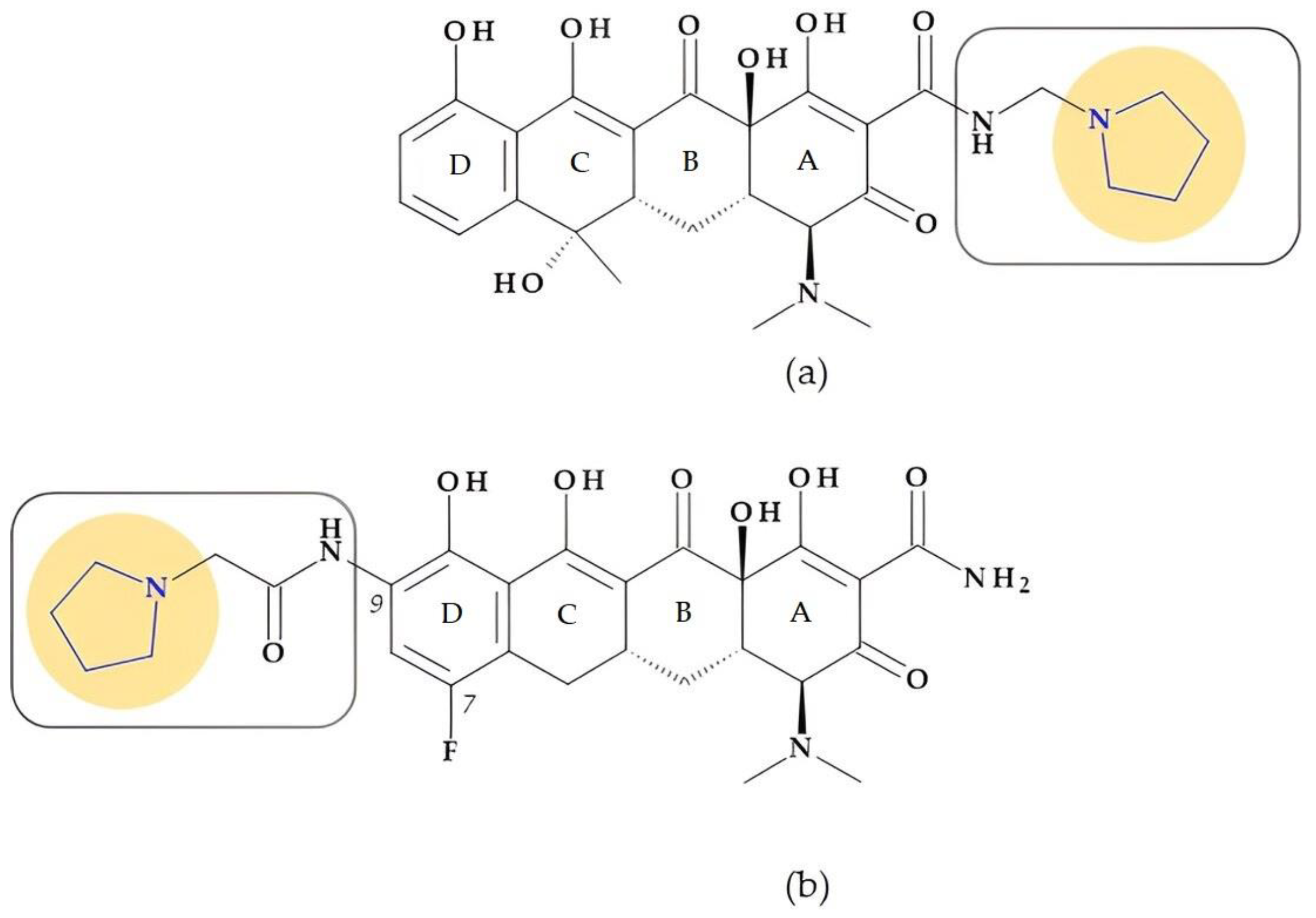
4.1.5. Tetracyclines
4.1.6. Other Antibacterials
4.2. Imidazole
4.2.1. Macrolides (Ketolides Subclass)
4.2.2. Nitroimidazoles
4.3. 2-Imidazolidinone
Beta-Lactamase Inhibitors
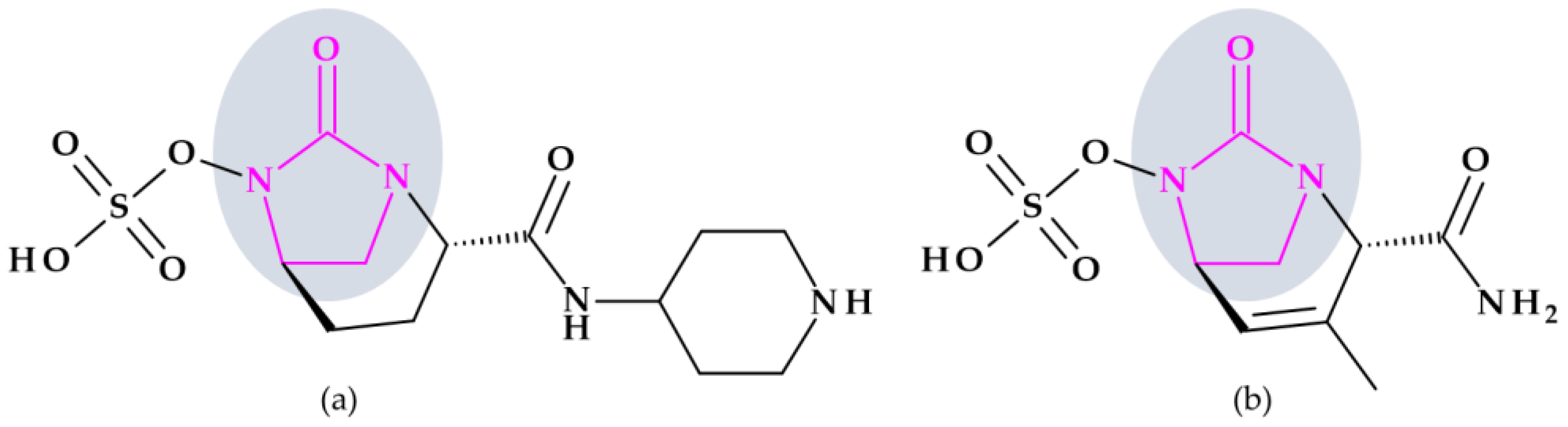
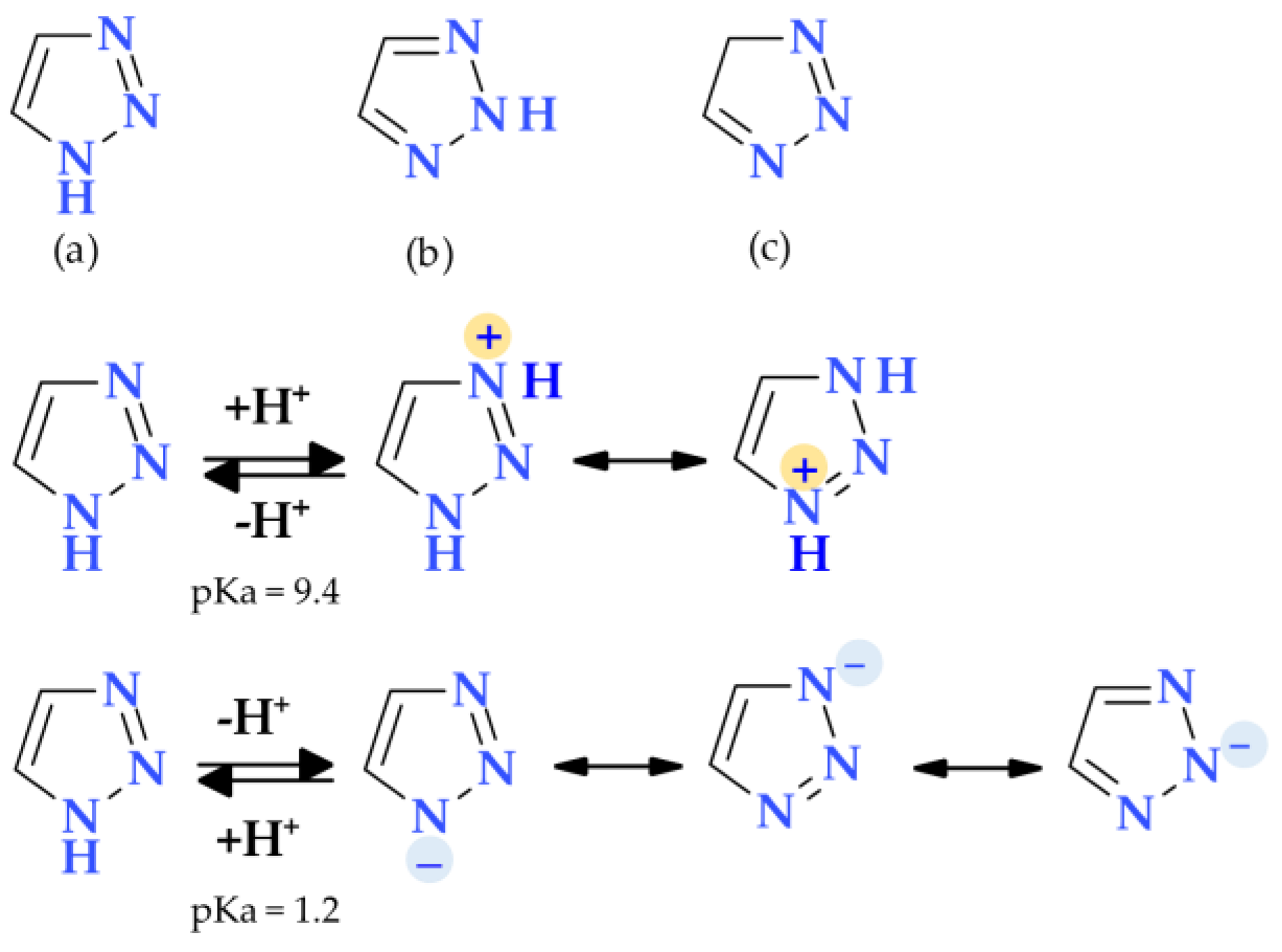
4.4. 1,2,3-Triazole
4.4.1. Beta-Lactamase Inhibitors
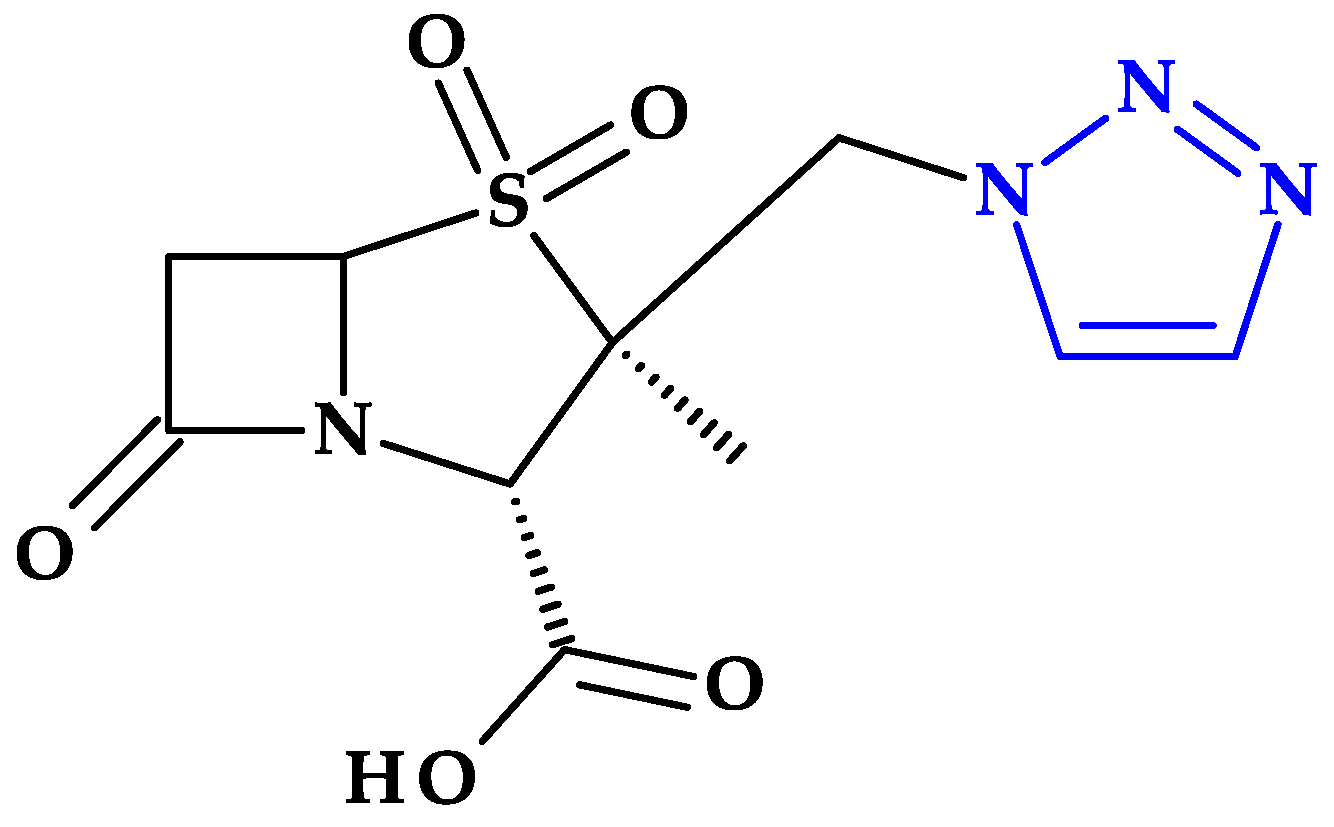
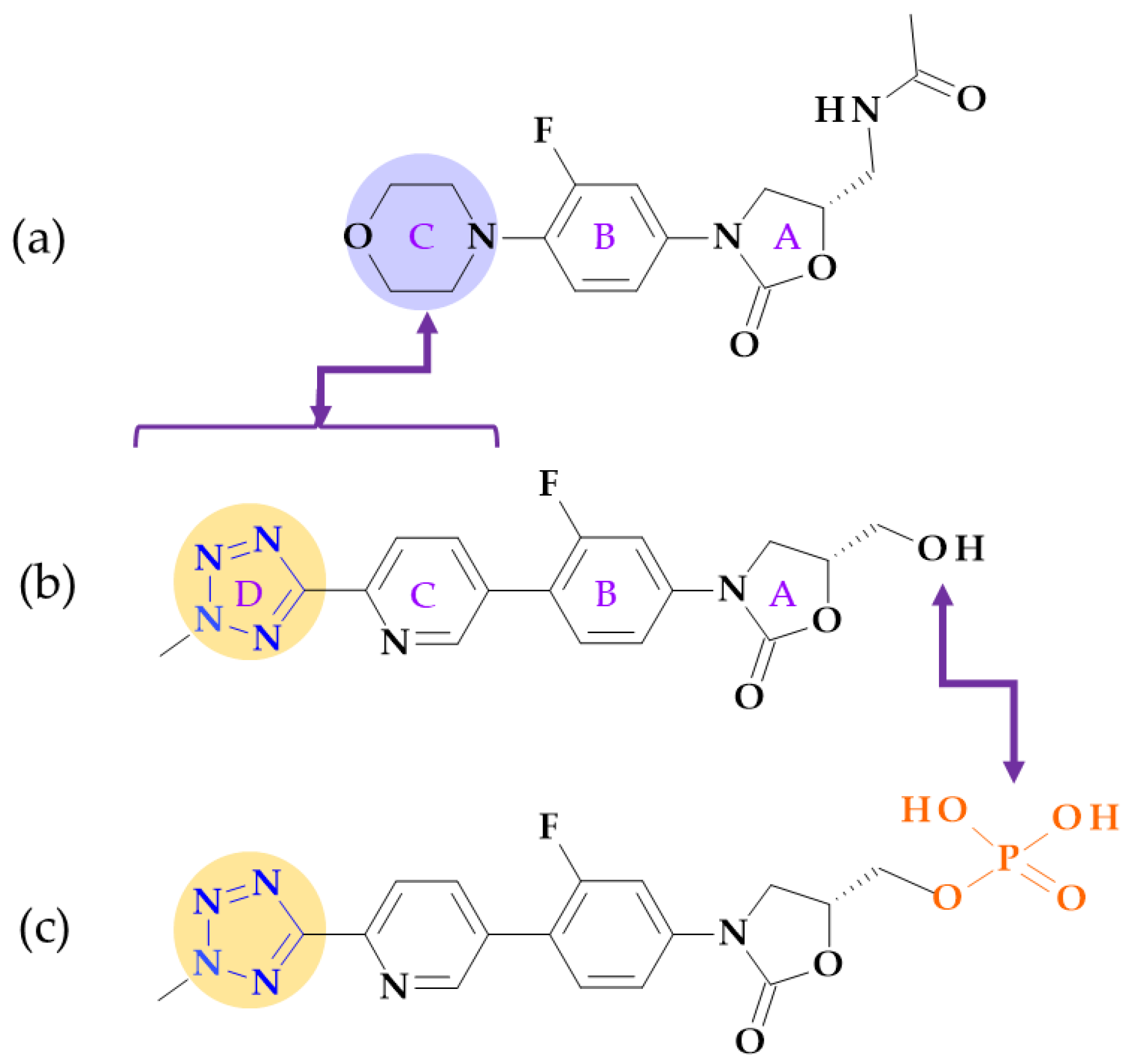
4.5. Tetrazole
4.5.1. Beta-Lactam Antibiotics

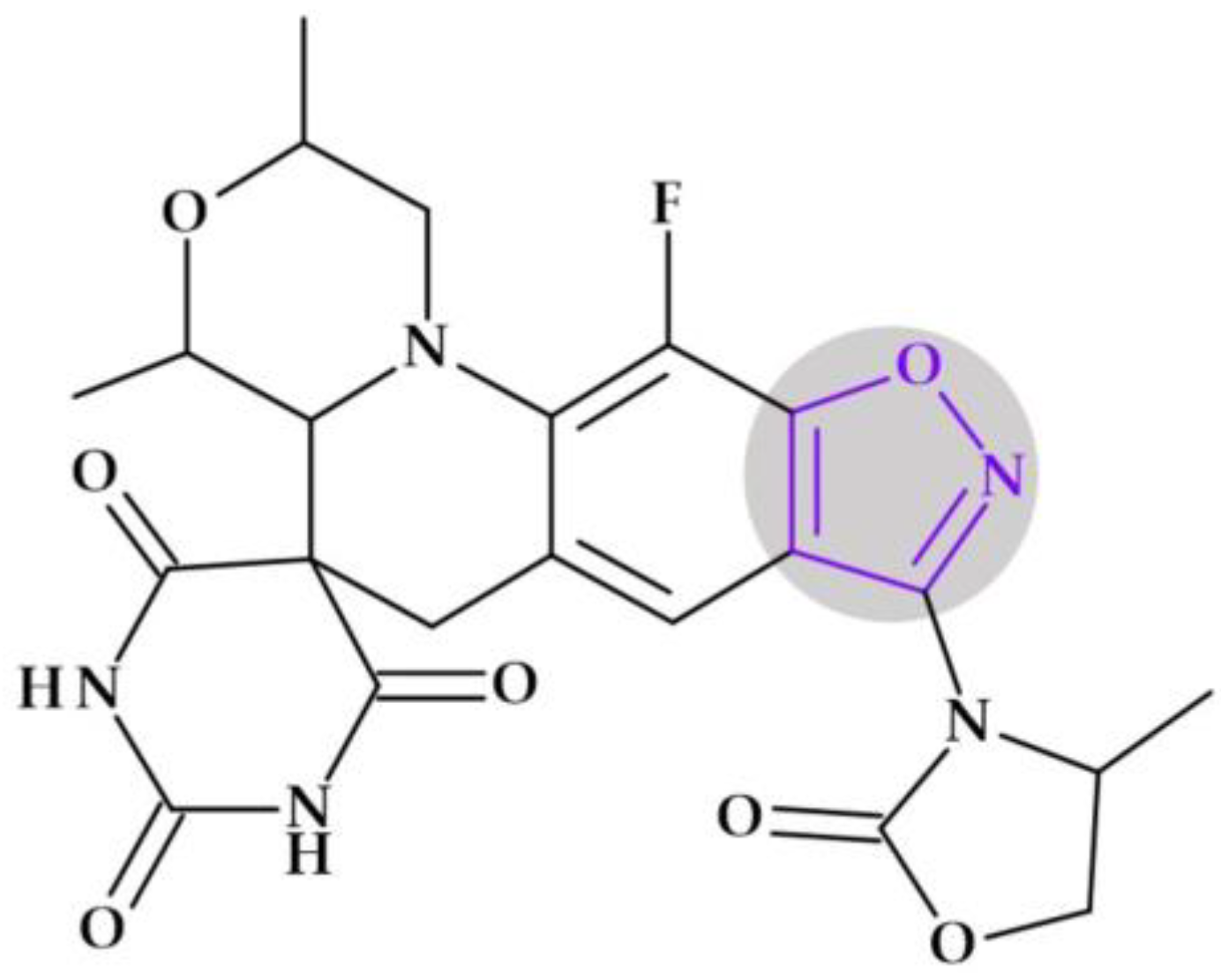
4.5.2. Oxazolidinones
5. Five-Membered Heterocycles Containing Oxygen Atoms
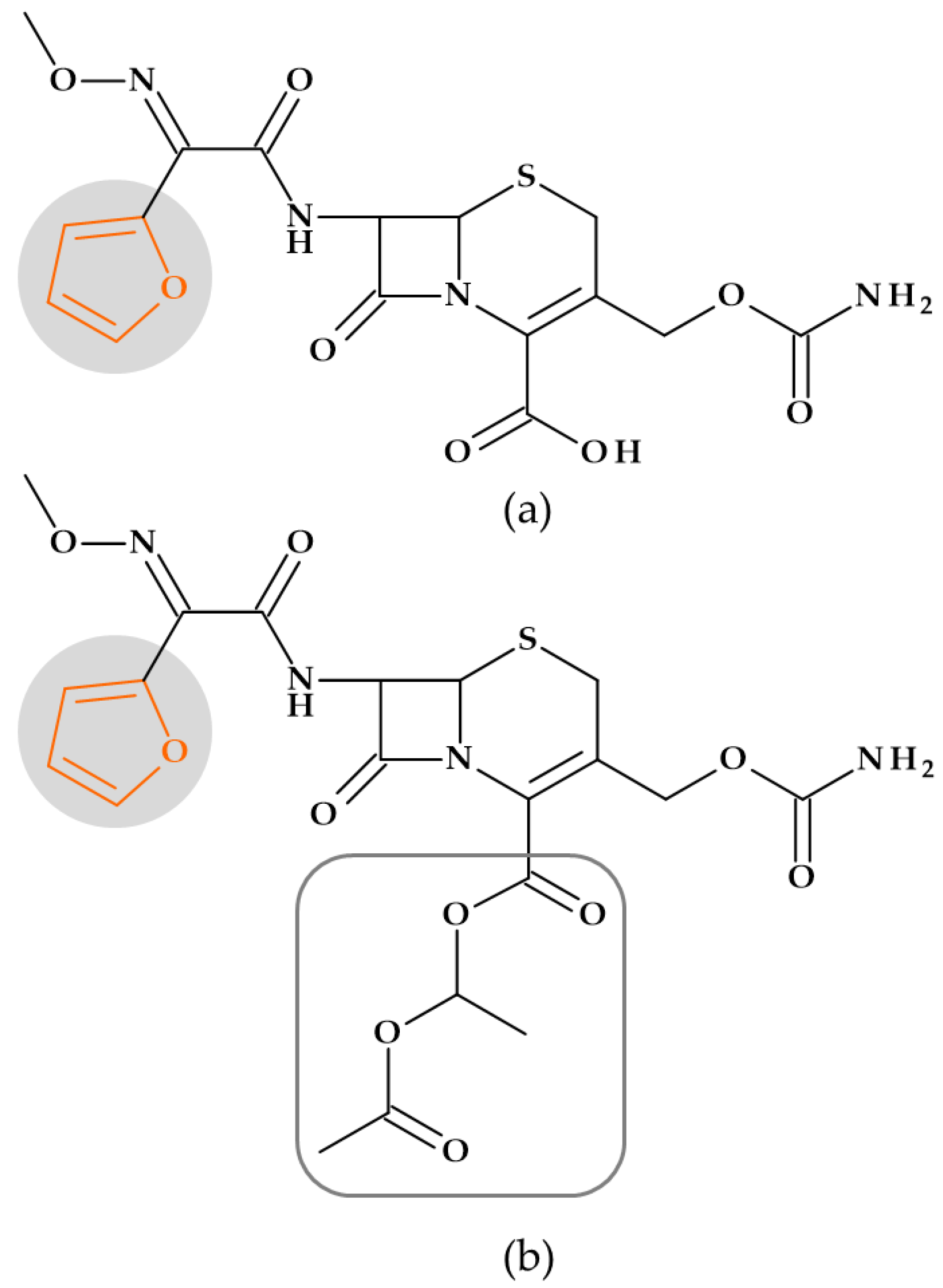
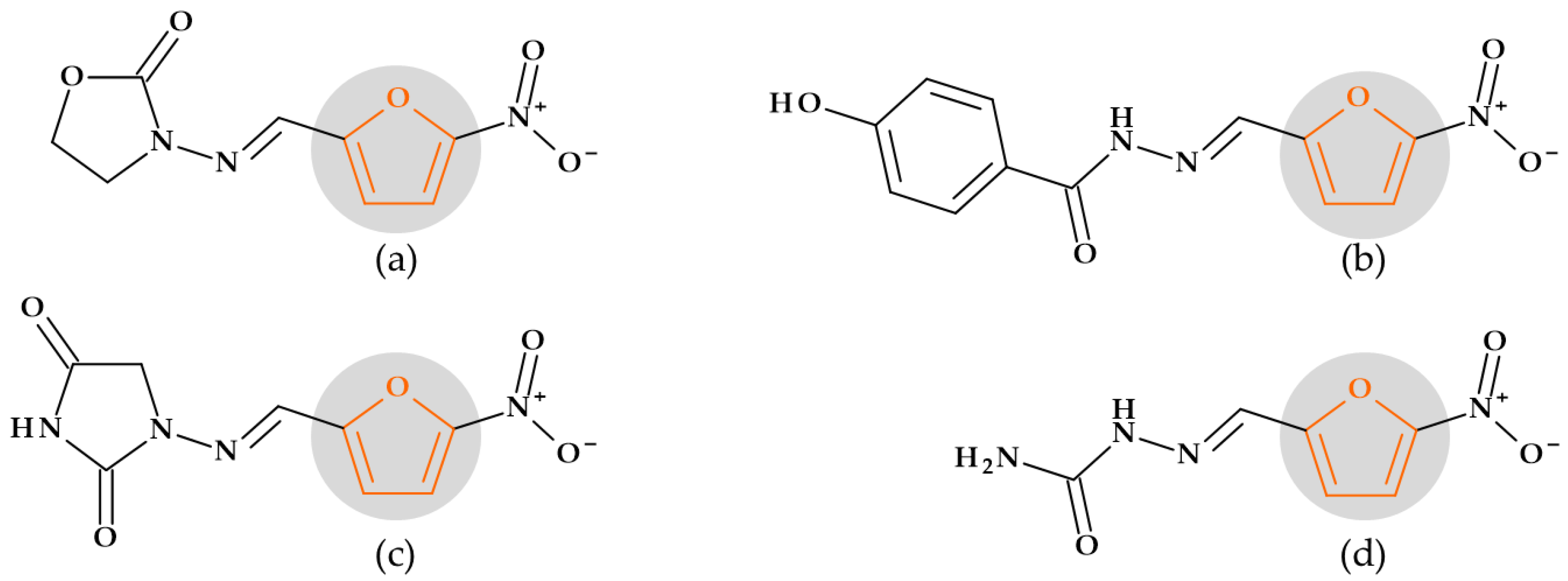
5.1. Furan
5.1.1. Beta-Lactam Antibiotics
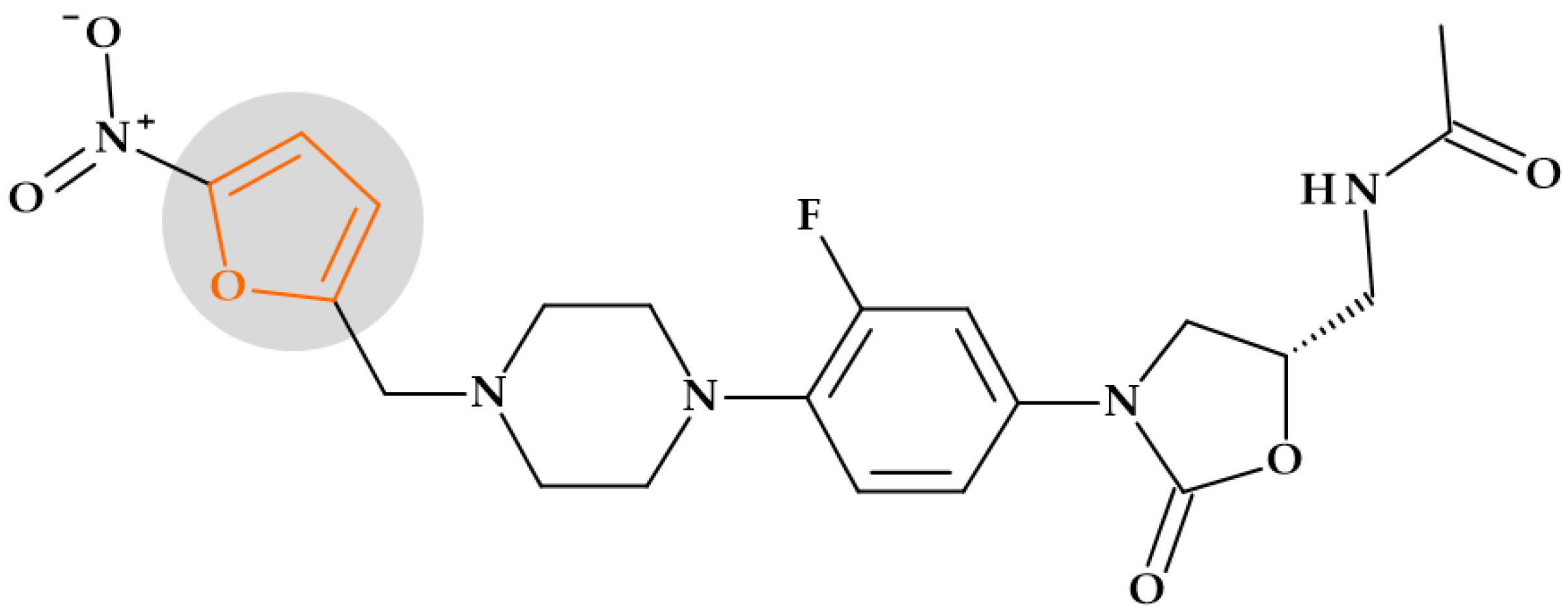
5.1.2. Nitrofurans
5.1.3. Oxazolidinones
6. Five-Membered Heterocycles Containing Oxygen and Nitrogen Atoms
6.1. 1,3-Oxazolidine
Oxazolidinones
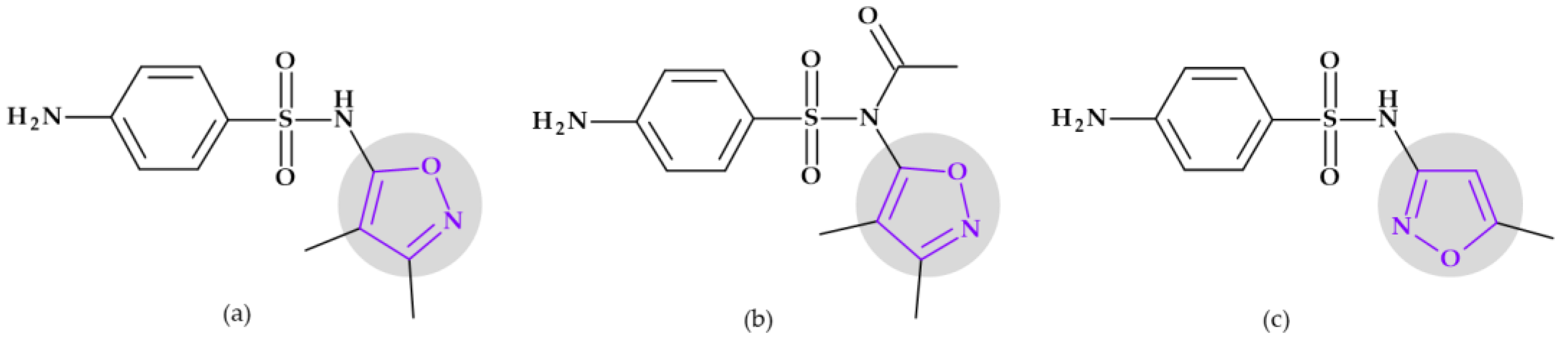
6.2. Oxazoles and Isoxazoles
6.2.1. Beta-Lactam Antibiotics
6.2.2. Izoxazolidinones
6.2.3. Sulfonamides
7. Five-Membered Heterocycles Containing One Sulfur Atom
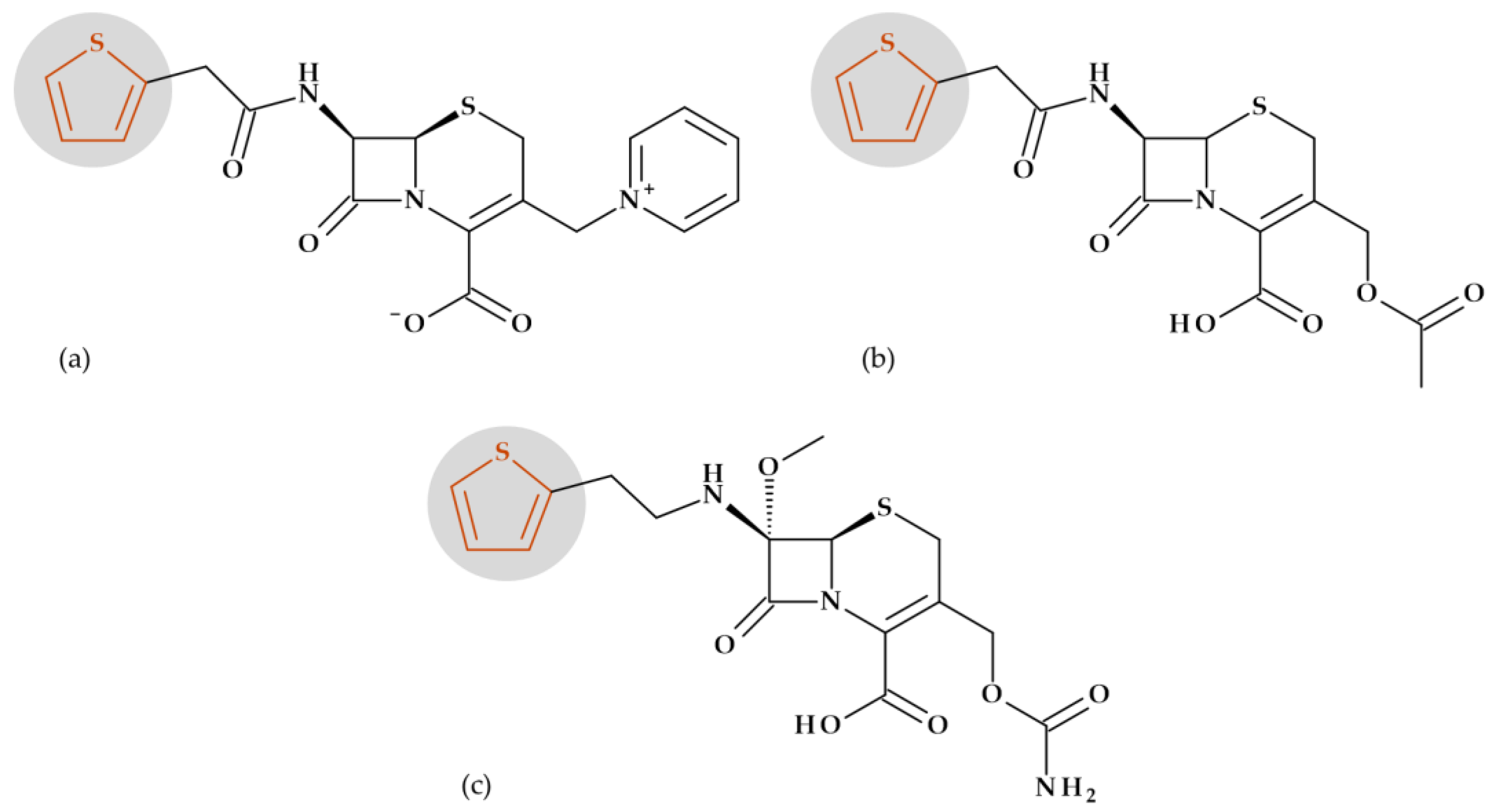
7.1. Tiophene
Beta-Lactam Antibiotics
8. Five-Membered Heterocycles Containing Sulfur and Nitrogen Atoms
8.1. 1,3-Thiazolidine
Beta-Lactam Antibiotics: Natural and Semisynthetic Penicillins
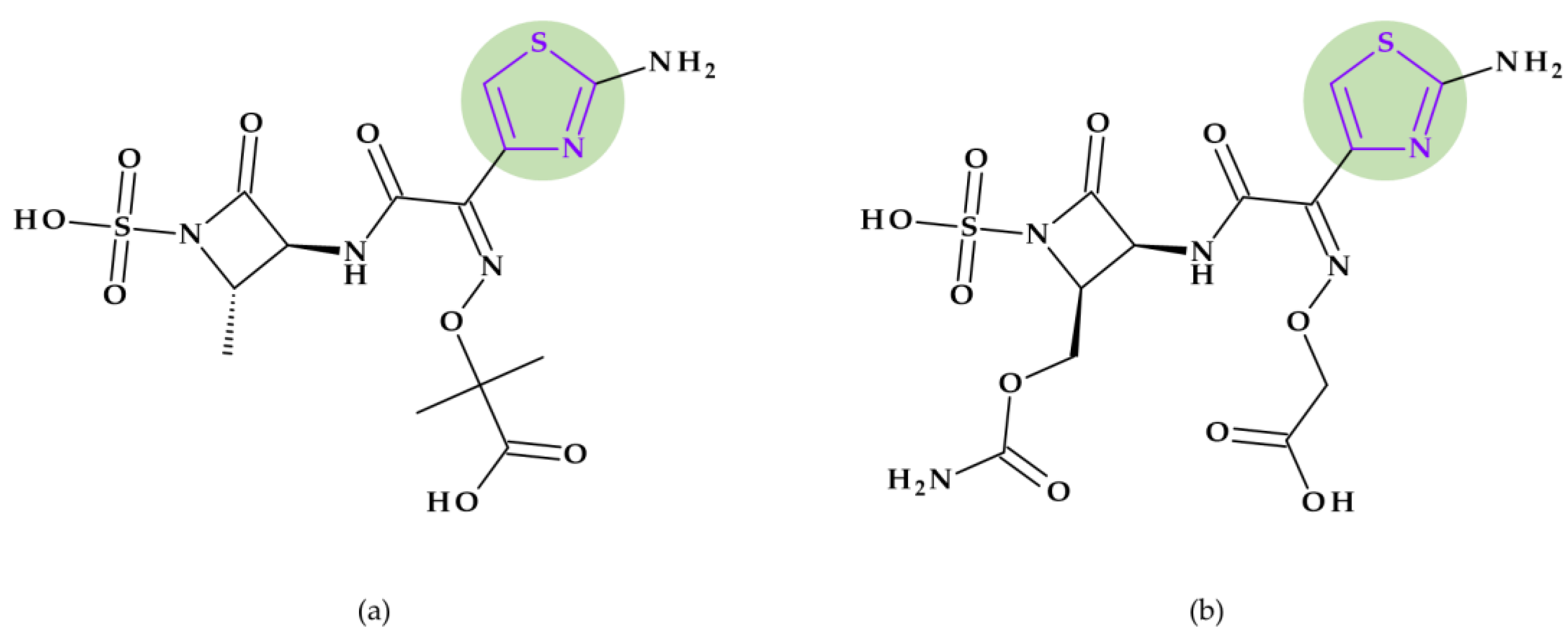
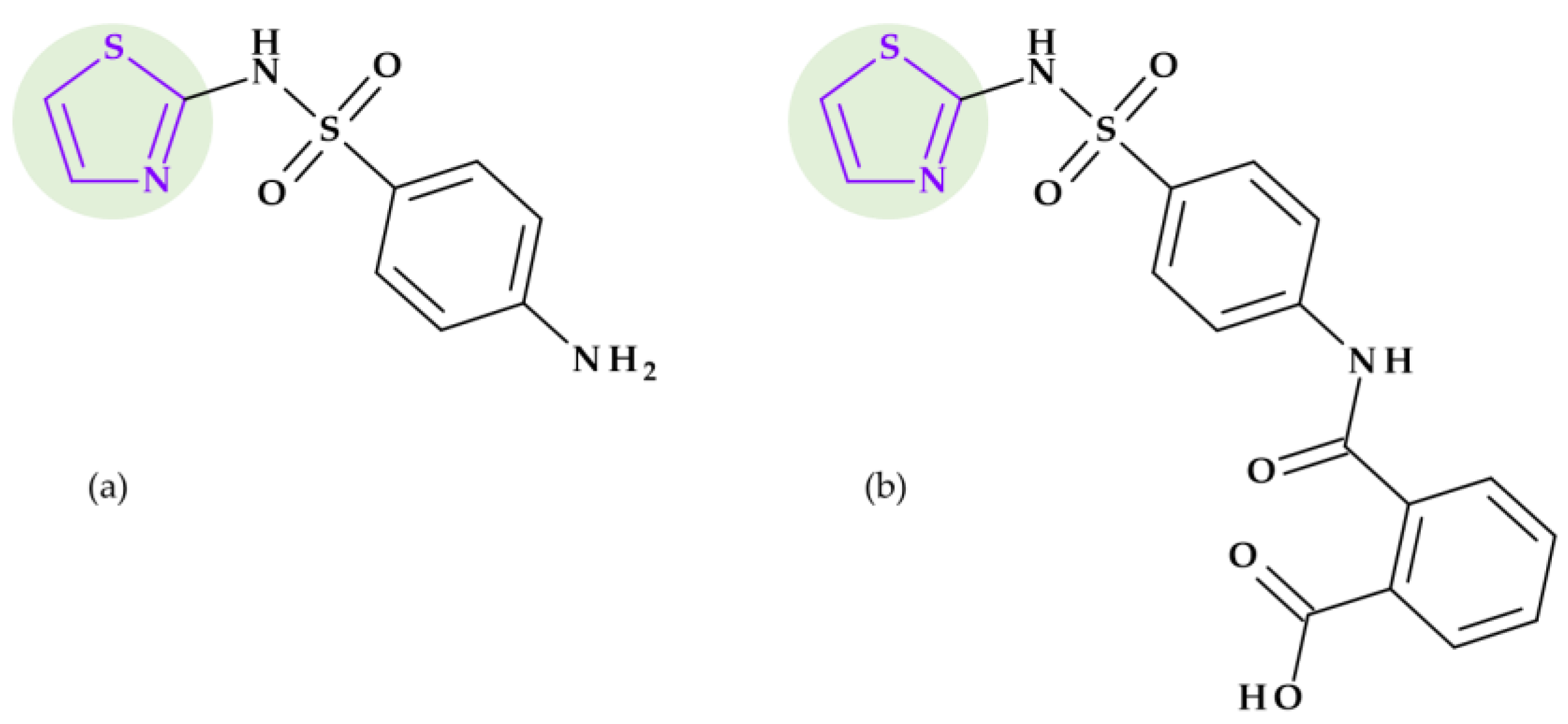
8.2. 1,3-Thiazole
8.2.1. Beta-Lactam Antibiotics
8.2.2. Sulphonamides
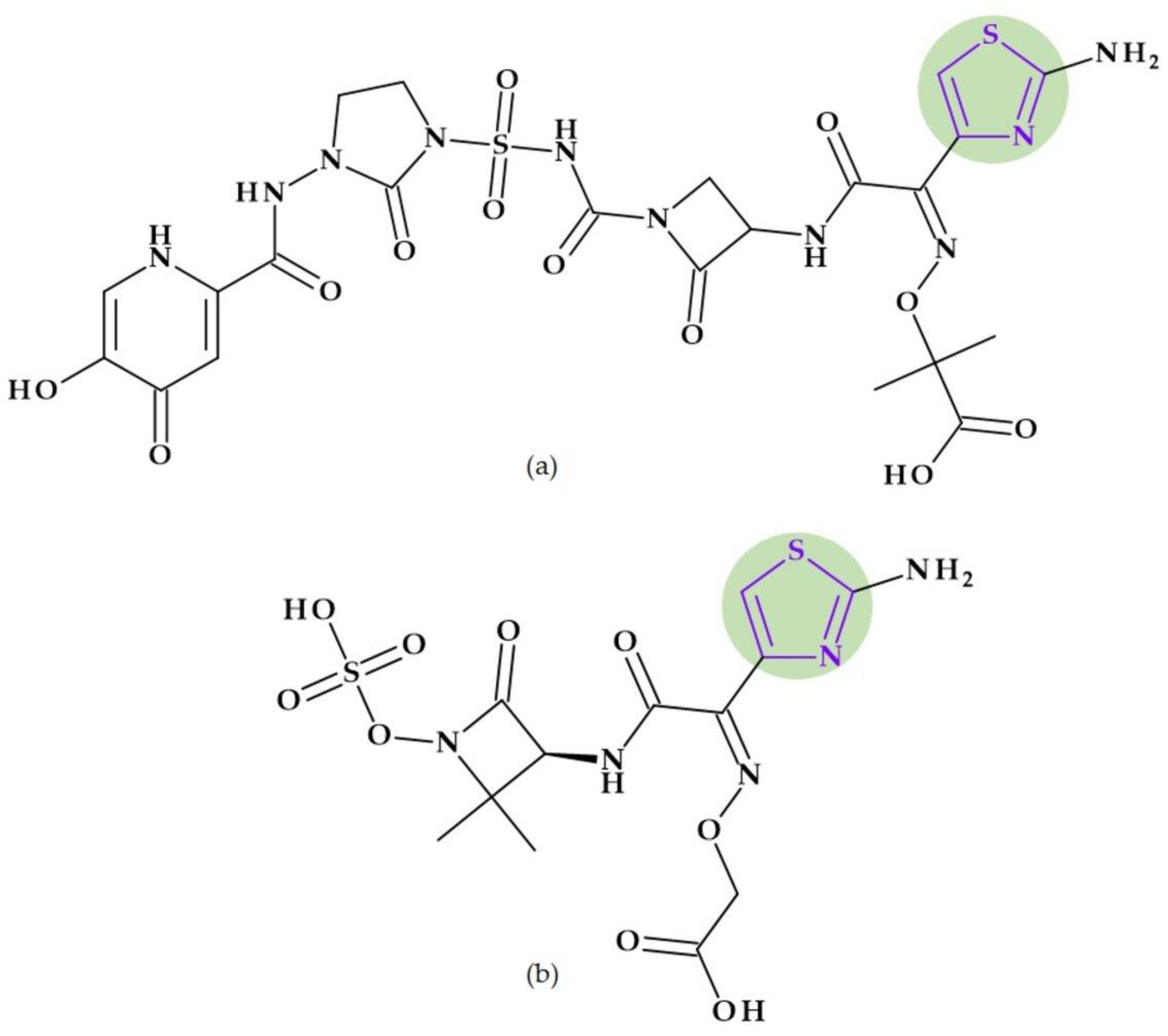
8.3. Thiadiazoles
- 1,2,4-Thiadiazoles. A five-membered, unsaturated, conjugated heteroaromatic with one sulfur atom and two nitrogen atoms, one of which is next to the sulfur and the other of which is one carbon distinct from it, is known as 1,2,4-thiadiazole. It is a π-excessive ring though relatively π deficient at the two carbon atoms. The nucleophilic substitution is easy at the C5 position due to the lowest π-electron density [24]. In nucleophilic substitution reactions, the C5 position of 1,2,4-thiadiazoles is the most reactive. The 1,2,4-thiadiazoles exhibit extremely few electrophilic reactions. Considering acid-base properties, 1,2,4-thiadiazoles are weak bases. They produce salts with mineral acids, and with heavy metal salts generate additional compounds [186]. Derivatives of 1,2,4-thiadiazoles are useful as antibiotics, cysteine protease inhibitors (cysteine protease cathepsin K), melanocortin-4-receptor agonists, and modulators of adenosine A3 receptors. Also, agrochemicals such as pesticides, soil fungicides, lubricating greases and vulcanization agents are found as derivatives of 1,2,4-thiadiazoles [24,186,187].
- 1,3,4-Thiadiazoles. 1,3,4-Thiadiazoles are a five-membered, aromatic, weakly basic, planar, electron-deficient heterocyclic ring system composed of two carbon atoms, one sulfur atom, and two nitrogen atoms that resemble pyridine in the N3- and N4-positions of the ring. The 1,3,4-thiadiazole’s dipole moment indicates that it is a polar, symmetric molecule with pseudo-aromatic properties. Due to the inductive influence of nitrogen and sulfur, the carbon atoms at the C2- and C5-positions are electron deficient, so they’re inert to electrophilic substitution but reactive to nucleophilic attack. The high aromaticity of the ring and the inductive action of sulfur are responsible for the 1,3,4-thiadiazole’s weak basicity. In aqueous acidic media, it is relatively stable. However, in an aqueous base, it does not undergo ring cleavage [24]. Numerous medications, including antibiotic, anti-inflammatory, anti-hypertensive, anti-HIV, anti-depressant, local anesthetic, and anti-convulsant drugs, have been discovered based on the 1,3,4-thiadiazole ring [24,29,184].
8.3.1. Beta-Lactam Antibiotics
8.3.2. Sulfonamides
9. Conclusions
- One or more nitrogen atoms: pyrrolidine, imidazole, 2-imidazolidinone, 1,2,3-triazole, and tetrazole;
- One oxygen atom: furan;
- Oxygen and nitrogen atoms: 1,3-oxazolidine, oxazole, and isoxazole;
- One sulfur atom: thiophene;
- One sulfur and nitrogen atoms: 1,3-thiazolidine, 1,3-thiazole, and thiadiazoles.
Author Contributions
Funding
Conflicts of Interest
References
- Gupta, R.R.; Kumar, M.; Gupta, V. Heterocyclic Chemistry: Volume II: Five-Membered Heterocycles; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2013; ISBN 978-3-662-07757-3. [Google Scholar]
- Gomtsyan, A. Heterocycles in Drugs and Drug Discovery. Chem. Heterocycl. Comp. 2012, 48, 7–10. [Google Scholar] [CrossRef]
- Barreca, M.; Spanò, V.; Rocca, R.; Bivacqua, R.; Gualtieri, G.; Raimondi, M.V.; Gaudio, E.; Bortolozzi, R.; Manfreda, L.; Bai, R.; et al. Identification of Pyrrolo [3′,4′:3,4]Cyclohepta[1,2-d][1,2]Oxazoles as Promising New Candidates for the Treatment of Lymphomas. Eur. J. Med. Chem. 2023, 254, 115372. [Google Scholar] [CrossRef]
- Grillone, K.; Riillo, C.; Rocca, R.; Ascrizzi, S.; Spanò, V.; Scionti, F.; Polerà, N.; Maruca, A.; Barreca, M.; Juli, G.; et al. The New Microtubule-Targeting Agent SIX2G Induces Immunogenic Cell Death in Multiple Myeloma. Int. J. Mol. Sci. 2022, 23, 10222. [Google Scholar] [CrossRef]
- Lee, B.; Kim, D.G.; Lee, A.; Kim, Y.M.; Cui, L.; Kim, S.; Choi, I. Synthesis and Discovery of the First Potent Proteolysis Targeting Chimaera (PROTAC) Degrader of AIMP2-DX2 as a Lung Cancer Drug. J. Enzym. Inhib. Med. Chem. 2023, 38, 51–66. [Google Scholar] [CrossRef]
- Bivacqua, R.; Barreca, M.; Spanò, V.; Raimondi, M.V.; Romeo, I.; Alcaro, S.; Andrei, G.; Barraja, P.; Montalbano, A. Insight into Non-Nucleoside Triazole-Based Systems as Viral Polymerases Inhibitors. Eur. J. Med. Chem. 2023, 249, 115136. [Google Scholar] [CrossRef]
- Fesatidou, M.; Petrou, A.; Athina, G. Heterocycle Compounds with Antimicrobial Activity. Curr. Pharm. Des. 2020, 26, 867–904. [Google Scholar] [CrossRef] [PubMed]
- Chinemerem Nwobodo, D.; Ugwu, M.C.; Oliseloke Anie, C.; Al-Ouqaili, M.T.S.; Chinedu Ikem, J.; Victor Chigozie, U.; Saki, M. Antibiotic Resistance: The Challenges and Some Emerging Strategies for Tackling a Global Menace. J. Clin. Lab. Anal. 2022, 36, e24655. [Google Scholar] [CrossRef]
- Chin, K.W.; Michelle Tiong, H.L.; Luang-In, V.; Ma, N.L. An Overview of Antibiotic and Antibiotic Resistance. Environ. Adv. 2023, 11, 100331. [Google Scholar] [CrossRef]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, Research, and Development of New Antibiotics: The WHO Priority List of Antibiotic-Resistant Bacteria and Tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Dutescu, I.A.; Hillier, S.A. Encouraging the Development of New Antibiotics: Are Financial Incentives the Right Way Forward? A Systematic Review and Case Study. Infect. Drug Resist. 2021, 14, 415–434. [Google Scholar] [CrossRef]
- Livermore, D.M.; Tulkens, P.M. Temocillin Revived. J. Antimicrob. Chemother. 2008, 63, 243–245. [Google Scholar] [CrossRef] [PubMed]
- Pulcini, C.; Bush, K.; Craig, W.A.; Frimodt-Møller, N.; Grayson, M.L.; Mouton, J.W.; Turnidge, J.; Harbarth, S.; Gyssens, I.C.; the ESCMID Study Group for Antibiotic Policies. Forgotten Antibiotics: An Inventory in Europe, the United States, Canada, and Australia. Clin. Infect. Dis. 2012, 54, 268–274. [Google Scholar] [CrossRef]
- Miljković, N.; Polidori, P.; Kohl, S. Managing Antibiotic Shortages: Lessons from EAHP and ECDC Surveys. Eur. J. Hosp. Pharm. 2022, 29, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Urban-Chmiel, R.; Marek, A.; Stępień-Pyśniak, D.; Wieczorek, K.; Dec, M.; Nowaczek, A.; Osek, J. Antibiotic Resistance in Bacteria—A Review. Antibiotics 2022, 11, 1079. [Google Scholar] [CrossRef]
- WHO Antibiotic Resistance. Available online: https://www.who.int/news-room/fact-sheets/detail/antibiotic-resistance (accessed on 20 July 2023).
- Sun, G.; Zhang, Q.; Dong, Z.; Dong, D.; Fang, H.; Wang, C.; Dong, Y.; Wu, J.; Tan, X.; Zhu, P.; et al. Antibiotic Resistant Bacteria: A Bibliometric Review of Literature. Front. Public Health 2022, 10, 1002015. [Google Scholar] [CrossRef]
- Research Center for Drug Evaluation and Drug Approvals and Databases. Available online: https://www.fda.gov/drugs/development-approval-process-drugs/drug-approvals-and-databases (accessed on 22 August 2023).
- Research Center for Drug Evaluation and New Drugs at FDA: CDER’s New Molecular Entities and New Therapeutic Biological Products. Available online: https://www.fda.gov/drugs/development-approval-process-drugs/new-drugs-fda-cders-new-molecular-entities-and-new-therapeutic-biological-products (accessed on 22 August 2023).
- BIOVIA Draw for Academics. Available online: https://discover.3ds.com/biovia-draw-academic (accessed on 9 August 2023).
- NIH PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/ (accessed on 12 July 2023).
- Eicher, T.; Hauptmann, S. The Chemistry of Heterocycles: Structure, Reactions, Syntheses and Applications; Wiley: Hoboken, NJ, USA, 2002; ISBN 978-3-527-30887-3. [Google Scholar]
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef] [PubMed]
- Ji Ram, V.; Sethi, A.; Nath, M.; Pratap, R. Chapter 5—Five-Membered Heterocycles. In The Chemistry of Heterocycles; Ji Ram, V., Sethi, A., Nath, M., Pratap, R., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 149–478. ISBN 978-0-08-101033-4. [Google Scholar]
- Joule, J.A.; Mills, K. Heterocyclic Chemistry; Wiley: Hoboken, NJ, USA, 2010; ISBN 978-1-4051-3300-5. [Google Scholar]
- Islam, M.T.; Mubarak, M.S. Pyrrolidine Alkaloids and Their Promises in Pharmacotherapy. Adv. Tradit. Med. 2020, 20, 13–22. [Google Scholar] [CrossRef]
- Johari, S.A.; Mohtar, M.; Syed Mohammad, S.A.; Sahdan, R.; Shaameri, Z.; Hamzah, A.S.; Mohammat, M.F. In Vitro Inhibitory and Cytotoxic Activity of MFM 501, a Novel Codonopsinine Derivative, against Methicillin-Resistant Staphylococcus Aureus Clinical Isolates. Biomed. Res. Int. 2015, 2015, 823829. [Google Scholar] [CrossRef]
- Łowicki, D.; Przybylski, P. Tandem Construction of Biological Relevant Aliphatic 5-Membered N-Heterocycles. Eur. J. Med. Chem. 2022, 235, 114303. [Google Scholar] [CrossRef]
- Wilson and Gisvold’s Textbook of Organic Medicinal and Pharmaceutical Chemistry, 12th ed.; Beale, J.M., Jr.; Block, J.H. (Eds.) Wolters Kluwer Health: Baltimore, MD, USA, 2010; ISBN 978-0-7817-7929-6. [Google Scholar]
- Papp-Wallace, K.M.; Endimiani, A.; Taracila, M.A.; Bonomo, R.A. Carbapenems: Past, Present, and Future. Antimicrob. Agents Chemother. 2011, 55, 4943–4960. [Google Scholar] [CrossRef]
- Foye, W.O. Foye’s Principles of Medicinal Chemistry; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; ISBN 978-1-4511-7572-1. [Google Scholar]
- El-Lababidi, R.M.; Rizk, J.G. Cefiderocol: A Siderophore Cephalosporin. Ann. Pharmacother. 2020, 54, 1215–1231. [Google Scholar] [CrossRef] [PubMed]
- Rusu, A.; Lungu, I.-A. The New Fifth-Generation Cephalosporins—A Balance between Safety and Efficacy. Rom. J. Pharm. Pract. 2020, 13, 121–126. [Google Scholar] [CrossRef]
- Rusu, A.; Lungu, I.-A.; Moldovan, O.-L.; Tanase, C.; Hancu, G. Structural Characterization of the Millennial Antibacterial (Fluoro)Quinolones—Shaping the Fifth Generation. Pharmaceutics 2021, 13, 1289. [Google Scholar] [CrossRef] [PubMed]
- Schinzer, W.C.; Bergren, M.S.; Aldrich, D.S.; Chao, R.S.; Dunn, M.J.; Jeganathan, A.; Madden, L.M. Characterization and Interconversion of Polymorphs of Premafloxacin, a New Quinolone Antibiotic. J. Pharm. Sci. 1997, 86, 1426–1431. [Google Scholar] [CrossRef] [PubMed]
- Quinupristin-Dalfopristin. In LiverTox: Clinical and Research Information on Drug-Induced Liver Injury; National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD, USA, 2012.
- Rusu, A.; Buta, E.L. The Development of Third-Generation Tetracycline Antibiotics and New Perspectives. Pharmaceutics 2021, 13, 2085. [Google Scholar] [CrossRef]
- Smith, J.R.; Rybak, J.M.; Claeys, K.C. Imipenem-Cilastatin-Relebactam: A Novel β-Lactam–β-Lactamase Inhibitor Combination for the Treatment of Multidrug-Resistant Gram-Negative Infections. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2020, 40, 343–356. [Google Scholar] [CrossRef]
- Armstrong, T.; Fenn, S.J.; Hardie, K.R. JMM Profile: Carbapenems: A Broad-Spectrum Antibiotic. J. Med. Microbiol. 2021, 70, 001462. [Google Scholar] [CrossRef]
- O’Connor, A.; Lopez, M.J.; Eranki, A.P. Cefepime. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Chapman, T.M.; Perry, C.M. Cefepime: A Review of Its Use in the Management of Hospitalized Patients with Pneumonia. Am. J. Respir. Med. 2003, 2, 75–107. [Google Scholar] [CrossRef]
- Sato, T.; Yamawaki, K. Cefiderocol: Discovery, Chemistry, and In Vivo Profiles of a Novel Siderophore Cephalosporin. Clin. Infect. Dis. 2019, 69, S538–S543. [Google Scholar] [CrossRef]
- FDA FETROJA (Cefiderocol) for Injection, for Intravenous Use. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/209445s000lbl.pdf (accessed on 23 June 2023).
- Wu, J.Y.; Srinivas, P.; Pogue, J.M. Cefiderocol: A Novel Agent for the Management of Multidrug-Resistant Gram-Negative Organisms. Infect. Dis. Ther. 2020, 9, 17–40. [Google Scholar] [CrossRef]
- Yamano, Y. In Vitro Activity of Cefiderocol Against a Broad Range of Clinically Important Gram-Negative Bacteria. Clin. Infect. Dis. 2019, 69, S544–S551. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Sato, T.; Ota, M.; Takemura, M.; Nishikawa, T.; Toba, S.; Kohira, N.; Miyagawa, S.; Ishibashi, N.; Matsumoto, S.; et al. In Vitro Antibacterial Properties of Cefiderocol, a Novel Siderophore Cephalosporin, against Gram-Negative Bacteria. Antimicrob. Agents Chemother. 2017, 62, e01454-17. [Google Scholar] [CrossRef] [PubMed]
- Shionogi Use of Cefiderocol in the Management of Gram-Negative Infections (PERSEUS). Available online: https://www.clinicaltrials.gov/study/NCT05789199?term=cefiderocol&rank=2 (accessed on 27 June 2023).
- El Solh, A. Ceftobiprole: A New Broad Spectrum Cephalosporin. Expert. Opin. Pharmacother. 2009, 10, 1675–1686. [Google Scholar] [CrossRef] [PubMed]
- Giacobbe, D.R.; Rosa, F.G.D.; Bono, V.D.; Grossi, P.A.; Pea, F.; Petrosillo, N.; Rossolini, G.M.; Tascini, C.; Tumbarello, M.; Viale, P.; et al. Ceftobiprole: Drug Evaluation and Place in Therapy. Expert Rev. Anti-Infect. Ther. 2019, 17, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Morosini, M.-I.; Díez-Aguilar, M.; Cantón, R. Mechanisms of Action and Antimicrobial Activity of Ceftobiprole. Rev. Esp. Quim. 2019, 32, 3–10. [Google Scholar]
- Shalaby, M.-A.W.; Dokla, E.M.E.; Serya, R.A.T.; Abouzid, K.A.M. Penicillin Binding Protein 2a: An Overview and a Medicinal Chemistry Perspective. Eur. J. Med. Chem. 2020, 199, 112312. [Google Scholar] [CrossRef]
- Pham, T.D.M.; Ziora, Z.M.; Blaskovich, M.A.T. Quinolone Antibiotics. Medchemcomm 2019, 10, 1719–1739. [Google Scholar] [CrossRef]
- Zhang, G.-F.; Zhang, S.; Pan, B.; Liu, X.; Feng, L.-S. 4-Quinolone Derivatives and Their Activities against Gram Positive Pathogens. Eur. J. Med. Chem. 2018, 143, 710–723. [Google Scholar] [CrossRef]
- Rusu, A.; Munteanu, A.-C.; Arbănași, E.-M.; Uivarosi, V. Overview of Side-Effects of Antibacterial Fluoroquinolones: New Drugs versus Old Drugs, a Step Forward in the Safety Profile? Pharmaceutics 2023, 15, 804. [Google Scholar] [CrossRef]
- Liu, J.; Ren, Z.; Fan, L.; Wei, J.; Tang, X.; Xu, X.; Yang, D. Design, Synthesis, Biological Evaluation, Structure-Activity Relationship, and Toxicity of Clinafloxacin-Azole Conjugates as Novel Antitubercular Agents. Bioorganic Med. Chem. 2019, 27, 175–187. [Google Scholar] [CrossRef]
- Zhanel, G.G.; Ennis, K.; Vercaigne, L.; Walkty, A.; Gin, A.S.; Embil, J.; Smith, H.; Hoban, D.J. A Critical Review of the Fluoroquinolones. Drugs 2002, 62, 13–59. [Google Scholar] [CrossRef]
- W-L Withdraws Clinafloxacin NDA—Pharmaceutical Industry News. Available online: https://www.thepharmaletter.com/article/w-l-withdraws-clinafloxacin-nda (accessed on 4 July 2023).
- Kocsis, B.; Domokos, J.; Szabo, D. Chemical Structure and Pharmacokinetics of Novel Quinolone Agents Represented by Avarofloxacin, Delafloxacin, Finafloxacin, Zabofloxacin and Nemonoxacin. Ann. Clin. Microbiol. Antimicrob. 2016, 15, 34. [Google Scholar] [CrossRef] [PubMed]
- Kocsis, B.; Gulyás, D.; Szabó, D. Delafloxacin, Finafloxacin, and Zabofloxacin: Novel Fluoroquinolones in the Antibiotic Pipeline. Antibiotics 2021, 10, 1506. [Google Scholar] [CrossRef] [PubMed]
- Gemifloxacin. In LiverTox: Clinical and Research Information on Drug-Induced Liver Injury; National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD, USA, 2012.
- Saravolatz, L.D.; Leggett, J. Gatifloxacin, Gemifloxacin, and Moxifloxacin: The Role of 3 Newer Fluoroquinolones. Clin. Infect. Dis. 2003, 37, 1210–1215. [Google Scholar] [CrossRef] [PubMed]
- EMA Factive: Withdrawn Application. Available online: https://www.ema.europa.eu/en/medicines/human/withdrawn-applications/factive (accessed on 9 August 2023).
- WHO Gemifloxacin: Withdrawal of Marketing Authorization Application. Available online: https://apps.who.int/iris/handle/10665/74447 (accessed on 9 August 2023).
- Tanaka, K.; Vu, H.; Hayashi, M. In Vitro Activities and Spectrum of lascufloxacin (KRP-AM1977) against Anaerobes. J. Infect. Chemother. 2021, 27, 1265–1269. [Google Scholar] [CrossRef]
- Limberakis, C. Quinolone Antibiotics: Levofloxacin (Levaquin®), Moxifloxacin (Avelox®), Gemifloxacin (Factive®), and Garenoxacin (T-3811). In The Art of Drug Synthesis; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2007; pp. 39–69. ISBN 978-0-470-13497-9. [Google Scholar]
- Wise, R. A Review of the Clinical Pharmacology of Moxifloxacin, a New 8-Methoxyquinolone, and Its Potential Relation to Therapeutic Efficacy. Clin. Drug Investig. 1999, 17, 365–387. [Google Scholar] [CrossRef]
- Miravitlles, M. Moxifloxacin in the Management of Exacerbations of Chronic Bronchitis and COPD. Int. J. Chron. Obs. Pulmon. Dis. 2007, 2, 191–204. [Google Scholar]
- Keating, G.M. Sitafloxacin: In Bacterial Infections. Drugs 2011, 71, 731–744. [Google Scholar] [CrossRef]
- Chen, C.-K.; Cheng, I.-L.; Chen, Y.-H.; Lai, C.-C. Efficacy and Safety of Sitafloxacin in the Treatment of Acute Bacterial Infection: A Meta-Analysis of Randomized Controlled Trials. Antibiotics 2020, 9, 106. [Google Scholar] [CrossRef]
- Guo, S.; Li, X.; Li, Y.; Tong, H.; Wei, M.; Yan, B.; Tian, M.; Xu, B.; Shao, J. Sitafloxacin Pharmacokinetics/Pharmacodynamics against Multidrug-Resistant Bacteria in a Dynamic Urinary Tract Infection in Vitro Model. J. Antimicrob. Chemother. 2023, 78, 141–149. [Google Scholar] [CrossRef]
- Spížek, J.; Řezanka, T. Lincosamides: Chemical Structure, Biosynthesis, Mechanism of Action, Resistance, and Applications. Biochem. Pharmacol. 2017, 133, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Mukhtar, T.A.; Wright, G.D. Streptogramins, Oxazolidinones, and Other Inhibitors of Bacterial Protein Synthesis. Chem. Rev. 2005, 105, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Johnston, N.J.; Mukhtar, T.A.; Wright, G.D. Streptogramin Antibiotics: Mode of Action and Resistance. Curr. Drug Targets 2002, 3, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Manzella, J.P. Quinupristin-Dalfopristin: A New Antibiotic for Severe Gram-Positive Infections. Am. Fam. Physician 2001, 64, 1863–1867. [Google Scholar]
- Scott, L.J. Eravacycline: A Review in Complicated Intra-Abdominal Infections. Drugs 2019, 79, 315–324. [Google Scholar] [CrossRef]
- FDA XERAVA (Eravacycline) for Injection. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/211109lbl.pdf (accessed on 1 August 2023).
- European Medicines Agency Xerava. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/xerava (accessed on 3 August 2021).
- Clark, R.B.; Hunt, D.K.; He, M.; Achorn, C.; Chen, C.-L.; Deng, Y.; Fyfe, C.; Grossman, T.H.; Hogan, P.C.; O’Brien, W.J.; et al. Fluorocyclines. 2. Optimization of the C-9 Side-Chain for Antibacterial Activity and Oral Efficacy. J. Med. Chem. 2012, 55, 606–622. [Google Scholar] [CrossRef]
- Zhanel, G.G.; Cheung, D.; Adam, H.; Zelenitsky, S.; Golden, A.; Schweizer, F.; Gorityala, B.; Lagacé-Wiens, P.R.S.; Walkty, A.; Gin, A.S.; et al. Review of Eravacycline, a Novel Fluorocycline Antibacterial Agent. Drugs 2016, 76, 567–588. [Google Scholar] [CrossRef]
- Lee, Y.R.; Burton, C.E. Eravacycline, a Newly Approved Fluorocycline. Eur. J. Clin. Microbiol. Infect. Dis. 2019, 38, 1787–1794. [Google Scholar] [CrossRef]
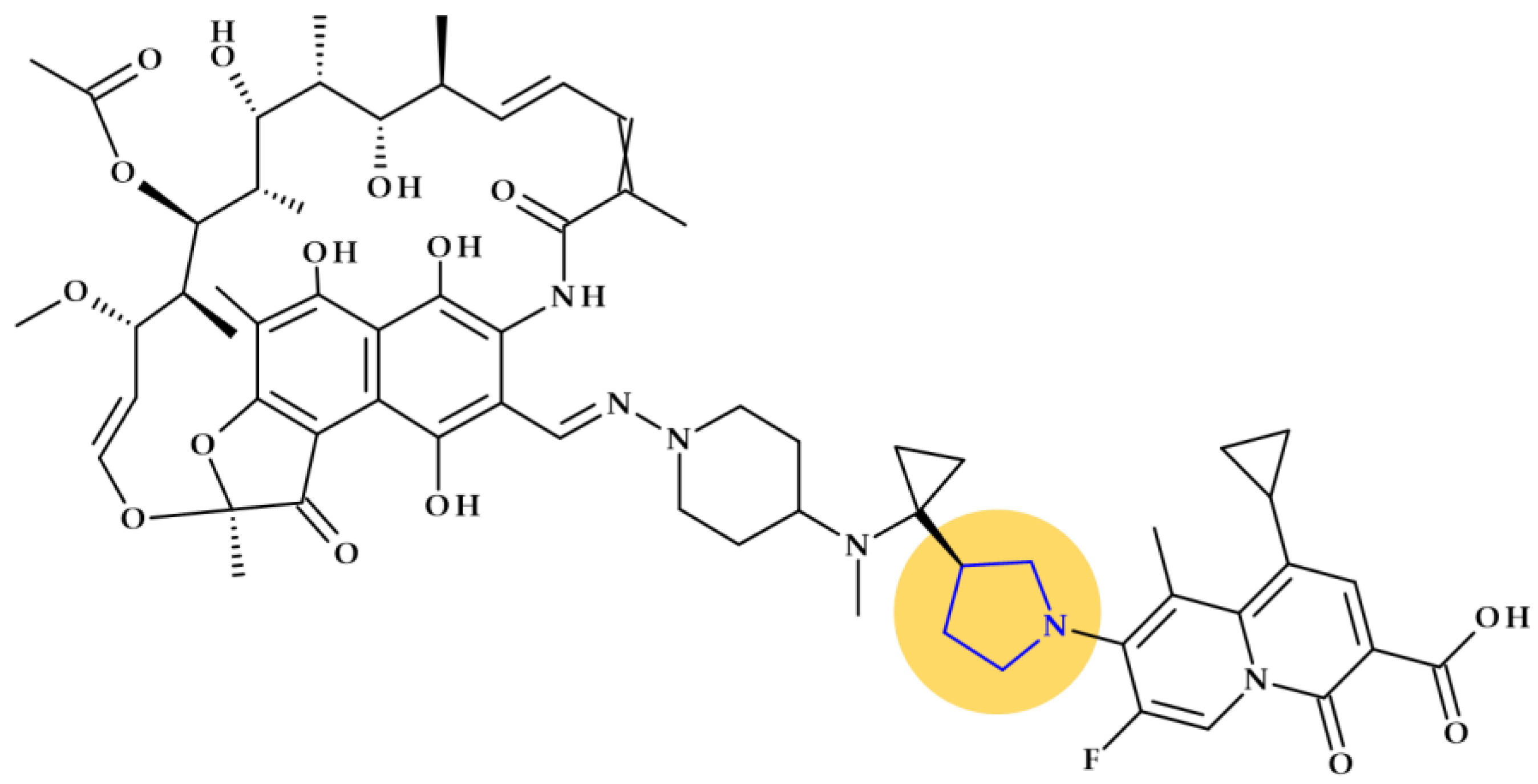
- Ma, Z.; Lynch, A.S. Development of a Dual-Acting Antibacterial Agent (TNP-2092) for the Treatment of Persistent Bacterial Infections. J. Med. Chem. 2016, 59, 6645–6657. [Google Scholar] [CrossRef]
- NCATS Inxight Drugs Rifaquizinone. Available online: https://drugs.ncats.io/drug/W2P7EF7O6O (accessed on 29 August 2023).
- DrugBank TNP-2092. Available online: https://go.drugbank.com/drugs/DB16312 (accessed on 29 August 2023).
- Meulbroek, J.A.; Oleksijew, A.; Tanaka, S.K.; Alder, J.D. Efficacy of ABT-719, a 2-Pyridone Antimicrobial, against Enterococci, Escherichia Coli, and Pseudomonas Aeruginosa in Experimental Murine Pyelonephritis. J. Antimicrob. Chemother. 1996, 38, 641–653. [Google Scholar] [CrossRef]
- Butler, M.S.; Henderson, I.R.; Capon, R.J.; Blaskovich, M.A.T. Antibiotics in the Clinical Pipeline as of December 2022. J. Antibiot. 2023, 76, 431–473. [Google Scholar] [CrossRef]
- GlobalData TNP-2092 by TenNor Therapeutics for Bacterial Infections: Likelihood of Approval. Pharmaceutical Technology 2023. Available online: https://www.pharmaceutical-technology.com/data-insights/tnp-2092-tennor-therapeutics-bacterial-infections-likelihood-of-approval/ (accessed on 8 August 2023).
- TenNor Therapeutics Limited Phase 2, Double-Blind, Randomized, Multicenter, Parallel, Controlled Study to Evaluate the Safety, Tolerability, Pharmacokinetics, and Efficacy of TNP-2092 to Treat Acute Bacterial Skin and Skin Structure Infection in Adults; clinicaltrials.gov. 2020. Available online: https://clinicaltrials.gov/study/NCT03964493 (accessed on 24 October 2023).
- Siwach, A.; Verma, P.K. Synthesis and Therapeutic Potential of Imidazole Containing Compounds. BMC Chem. 2021, 15, 12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Peng, X.-M.; Damu, G.L.V.; Geng, R.-X.; Zhou, C.-H. Comprehensive Review in Current Developments of Imidazole-Based Medicinal Chemistry. Med. Res. Rev. 2014, 34, 340–437. [Google Scholar] [CrossRef] [PubMed]
- Molina, P.; Tárraga, A.; Otón, F. Imidazole Derivatives: A Comprehensive Survey of Their Recognition Properties. Org. Biomol. Chem. 2012, 10, 1711–1724. [Google Scholar] [CrossRef]
- Tran, M.P. Telithromycin: A Novel Agent for the Treatment of Community-Acquired Upper Respiratory Infections. Bayl. Univ. Med. Cent. Proc. 2004, 17, 475–479. [Google Scholar] [CrossRef] [PubMed]
- FDA Ketek (Telithromycin) Label NDA 21-144/S-012. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2007/021144s012lbl.pdf (accessed on 15 July 2023).
- Douthwaite, S.; Champney, W.S. Structures of Ketolides and Macrolides Determine Their Mode of Interaction with the Ribosomal Target Site. J. Antimicrob. Chemother. 2001, 48, 1–8. [Google Scholar] [CrossRef]
- Wu, Y.-J. Chapter 1—Heterocycles and Medicine: A Survey of the Heterocyclic Drugs Approved by the U.S. FDA from 2000 to Present. In Progress in Heterocyclic Chemistry; Gribble, G.W., Joule, J.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2012; Volume 24, pp. 1–53. [Google Scholar]
- Ross, D.B. The FDA and the Case of Ketek. N. Engl. J. Med. 2007, 356, 1601–1604. [Google Scholar] [CrossRef]
- European Medicines Agency Ketek. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/ketek (accessed on 15 July 2023).
- Wetzel, D.M.; Phillips, M.A. Chemotherapy of Protozoal Infections: Amebiasis, Giardiasis, Trichomoniasis, Trypanosomiasis, Leishmaniasis, and Other Protozoal Infections. In Goodman & Gilman’s: The Pharmacological Basis of Therapeutics; Brunton, L.L., Hilal-Dandan, R., Knollmann, B.C., Eds.; McGraw-Hill Education: New York, NY, USA, 2017. [Google Scholar]
- Zhanel, G.G.; Lawrence, C.K.; Adam, H.; Schweizer, F.; Zelenitsky, S.; Zhanel, M.; Lagacé-Wiens, P.R.S.; Walkty, A.; Denisuik, A.; Golden, A.; et al. Imipenem–Relebactam and Meropenem–Vaborbactam: Two Novel Carbapenem-β-Lactamase Inhibitor Combinations. Drugs 2018, 78, 65–98. [Google Scholar] [CrossRef]
- FDA Drugs@FDA: FDA-Approved Drugs, New Drug Application (NDA): 212819, RECARBRIO. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&varApplNo=212819 (accessed on 22 August 2023).
- Research Center for Drug Evaluation and FDA Approves New Treatment for Pneumonia Caused by Certain Difficult-to-Treat Bacteria. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-new-treatment-pneumonia-caused-certain-difficult-treat-bacteria (accessed on 22 August 2023).
- Granata, G.; Taglietti, F.; Schiavone, F.; Petrosillo, N. Durlobactam in the Treatment of Multidrug-Resistant Acinetobacter Baumannii Infections: A Systematic Review. J. Clin. Med. 2022, 11, 3258. [Google Scholar] [CrossRef]
- Kolb, H.C.; Sharpless, K.B. The Growing Impact of Click Chemistry on Drug Discovery. Drug Discov. Today 2003, 8, 1128–1137. [Google Scholar] [CrossRef]
- Whiting, M.; Muldoon, J.; Lin, Y.-C.; Silverman, S.M.; Lindstrom, W.; Olson, A.J.; Kolb, H.C.; Finn, M.G.; Sharpless, K.B.; Elder, J.H.; et al. Inhibitors of HIV-1 Protease by Using In Situ Click Chemistry. Angew. Chem. 2006, 118, 1463–1467. [Google Scholar] [CrossRef]
- Masood, M.M. Progress in Synthetic Trends Followed for the Development of 1,2,3-Triazole Derivatives: A Review. Polycycl. Aromat. Compd. 2023. [CrossRef]
- Phetsang, W.; Blaskovich, M.A.T.; Butler, M.S.; Huang, J.X.; Zuegg, J.; Mamidyala, S.K.; Ramu, S.; Kavanagh, A.M.; Cooper, M.A. An Azido-Oxazolidinone Antibiotic for Live Bacterial Cell Imaging and Generation of Antibiotic Variants. Bioorganic Med. Chem. 2014, 22, 4490–4498. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.-H.; Wang, Y. Recent Researches in Triazole Compounds as Medicinal Drugs. Curr. Med. Chem. 2012, 19, 239–280. [Google Scholar] [CrossRef] [PubMed]
- Eiamphungporn, W.; Schaduangrat, N.; Malik, A.A.; Nantasenamat, C. Tackling the Antibiotic Resistance Caused by Class A β-Lactamases through the Use of β-Lactamase Inhibitory Protein. Int. J. Mol. Sci. 2018, 19, 2222. [Google Scholar] [CrossRef] [PubMed]
- Patowary, P.; Deka, B.; Bharali, D. Tetrazole Moiety as a Pharmacophore in Medicinal Chemistry: A Review. Malar. Contr. Elimination 2021, 10, 1–11. [Google Scholar]
- Ostrovskii, V.A.; Trifonov, R.E.; Popova, E.A. Medicinal Chemistry of Tetrazoles. Russ. Chem. Bull. 2012, 61, 768–780. [Google Scholar] [CrossRef]
- Zou, Y.; Liu, L.; Liu, J.; Liu, G. Bioisosteres in Drug Discovery: Focus on Tetrazole. Future Med. Chem. 2020, 12, 91–93. [Google Scholar] [CrossRef]
- Ballatore, C.; Huryn, D.M.; Smith, A.B. Carboxylic Acid (Bio)Isosteres in Drug Design. ChemMedChem 2013, 8, 385–395. [Google Scholar] [CrossRef]
- Brown, N. Bioisosteres in Medicinal Chemistry; John Wiley & Sons: Hoboken, NJ, USA, 2012; ISBN 978-3-527-65432-1. [Google Scholar]
- Stevens, E. Medicinal Chemistry: The Modern Drug Discovery Process; Pearson: London, UK, 2014; ISBN 978-0-321-71048-2. [Google Scholar]
- Kaushik, N.; Kumar, N.; Kumar, A.; Singh, U.K. Tetrazoles: Synthesis and Biological Activity. Immunol. Endocr. Metab. Agents Med. Chem. 2018, 18, 3–21. [Google Scholar] [CrossRef]
- Lipsky, J.J. N-Methyl-Thio-Tetrazole Inhibition of the Gamma Carboxylation of Glutamic Acid: Possible Mechanism for Antibiotic-Associated Hypoprothrombinaemia. Lancet 1983, 2, 192–193. [Google Scholar] [CrossRef]
- Fekety, F.R. Safety of Parenteral Third-Generation Cephalosporins. Am. J. Med. 1990, 88, S38–S44. [Google Scholar] [CrossRef] [PubMed]
- Vardanyan, R.S.; Hruby, V.J. 32—Antibiotics. In Synthesis of Essential Drugs; Vardanyan, R.S., Hruby, V.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2006; pp. 425–498. ISBN 978-0-444-52166-8. [Google Scholar]
- Goodman and Gilman’s The Pharmacological Basis of Therapeutics, 13th ed.; McGraw-Hill Education: New York, NY, USA, 2018; ISBN 978-1-259-58474-9.
- Castle, S.S. Cefamandole. In xPharm: The Comprehensive Pharmacology Reference; Enna, S.J., Bylund, D.B., Eds.; Elsevier: New York, NY, USA, 2007; pp. 1–4. ISBN 978-0-08-055232-3. [Google Scholar]
- Saltiel, E.; Brogden, R.N. Cefonicid. Drugs 1986, 32, 222–259. [Google Scholar] [CrossRef] [PubMed]
- McLeod, D.C.; Tartaglione, T.A.; Polk, R.E. Review of the New Second-Generation Cephalosporins: Cefonicid, Ceforanide, and Cefuroxime. Drug Intell. Clin. Pharm. 1985, 19, 188–198. [Google Scholar] [CrossRef]
- Jones, R.N. Cefmetazole (CS-1170), a “New” Cephamycin with a Decade of Clinical Experience. Diagn. Microbiol. Infect. Dis. 1989, 12, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Martindale: The Complete Drug Reference, 36th ed.; Pharmaceutical Press: London, UK; Chicago, IL, USA, 2009; ISBN 978-0-85369-840-1.
- Darlow, C.A.; da Costa, R.M.A.; Ellis, S.; Franceschi, F.; Sharland, M.; Piddock, L.; Das, S.; Hope, W. Potential Antibiotics for the Treatment of Neonatal Sepsis Caused by Multidrug-Resistant Bacteria. Paediatr. Drugs 2021, 23, 465–484. [Google Scholar] [CrossRef]
- DrugBank Latamoxef. Available online: https://go.drugbank.com/drugs/DB04570 (accessed on 31 July 2023).
- DrugBank Flomoxef. Available online: https://go.drugbank.com/drugs/DB11935 (accessed on 16 September 2023).
- DrugBank Tedizolid. Available online: https://go.drugbank.com/drugs/DB14569 (accessed on 16 September 2023).
- Zhanel, G.G.; Love, R.; Adam, H.; Golden, A.; Zelenitsky, S.; Schweizer, F.; Gorityala, B.; Lagacé-Wiens, P.R.S.; Rubinstein, E.; Walkty, A.; et al. Tedizolid: A Novel Oxazolidinone with Potent Activity Against Multidrug-Resistant Gram-Positive Pathogens. Drugs 2015, 75, 253–270. [Google Scholar] [CrossRef]
- Michalska, K.; Karpiuk, I.; Król, M.; Tyski, S. Recent Development of Potent Analogues of Oxazolidinone Antibacterial Agents. Bioorganic Med. Chem. 2013, 21, 577–591. [Google Scholar] [CrossRef] [PubMed]
- Rybak, J.M.; Roberts, K. Tedizolid Phosphate: A Next-Generation Oxazolidinone. Infect. Dis. Ther. 2015, 4, 1–14. [Google Scholar] [CrossRef]
- Flanagan, S.D.; Bien, P.A.; Muñoz, K.A.; Minassian, S.L.; Prokocimer, P.G. Pharmacokinetics of Tedizolid Following Oral Administration: Single and Multiple Dose, Effect of Food, and Comparison of Two Solid Forms of the Prodrug. Pharmacotherapy 2013, 34, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Brier, M.E.; Stalker, D.J.; Aronoff, G.R.; Batts, D.H.; Ryan, K.K.; O’Grady, M.; Hopkins, N.K.; Jungbluth, G.L. Pharmacokinetics of Linezolid in Subjects with Renal Dysfunction. Antimicrob. Agents Chemother. 2003, 47, 2775–2780. [Google Scholar] [CrossRef] [PubMed]
- DrugBank Radezolid. Available online: https://go.drugbank.com/drugs/DB12339 (accessed on 19 July 2023).
- Michalska, K.; Bednarek, E.; Gruba, E.; Lewandowska, K.; Mizera, M.; Cielecka-Piontek, J. Comprehensive Spectral Identification of Key Intermediates to the Final Product of the Chiral Pool Synthesis of Radezolid. Chem. Cent. J. 2017, 11, 82. [Google Scholar] [CrossRef]
- Melinta Therapeutics, Inc. A Phase 2, Multicenter, Randomized, Open-Label, Comparative Study to Evaluate the Safety and Efficacy of RX-1741 Versus Linezolid in the Outpatient Treatment of Adult Patients With Uncomplicated Skin and Skin Structure Infection; Melinta Therapeutics, Inc.: Troy Hills, NJ, USA, 2014. [Google Scholar]
- Melinta Therapeutics, Inc. A Phase 2, Multicenter, Randomized, Double-Blind Study to Evaluate the Safety and Efficacy of RX-1741 in the Treatment of Adult Patients With Mild to Moderate Severity of Community-Acquired Pneumonia (CAP); Melinta Therapeutics, Inc.: Troy Hills, NJ, USA, 2016. [Google Scholar]
- Wang, L.; Zhang, Y.; Liu, S.; Huang, N.; Zeng, W.; Xu, W.; Zhou, T.; Shen, M. Comparison of Anti-Microbic and Anti-Biofilm Activity Among Tedizolid and Radezolid Against Linezolid-Resistant Enterococcus Faecalis Isolates. Infect. Drug Resist. 2021, 14, 4619–4627. [Google Scholar] [CrossRef]
- DrugBank Furans | DrugBank Online. Available online: https://go.drugbank.com/categories/DBCAT000766 (accessed on 1 August 2023).
- Le Dang, N.; Hughes, T.B.; Miller, G.P.; Swamidass, S.J. Computational Approach to Structural Alerts: Furans, Phenols, Nitroaromatics, and Thiophenes. Chem. Res. Toxicol. 2017, 30, 1046–1059. [Google Scholar] [CrossRef] [PubMed]
- PubChem Cefuroxime. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/5479529 (accessed on 4 August 2023).
- Das, B.; Rudra, S.; Yadav, A.; Ray, A.; Rao, A.V.S.R.; Srinivas, A.S.S.V.; Soni, A.; Saini, S.; Shukla, S.; Pandya, M.; et al. Synthesis and SAR of Novel Oxazolidinones: Discovery of Ranbezolid. Bioorganic Med. Chem. Lett. 2005, 15, 4261–4267. [Google Scholar] [CrossRef] [PubMed]
- Mathur, T.; Kalia, V.; Barman, T.K.; Singhal, S.; Khan, S.; Upadhyay, D.J.; Rattan, A.; Raj, V.S. Anti-Anaerobic Potential of Ranbezolid: Insight into Its Mechanism of Action against Bacteroides Fragilis. Int. J. Antimicrob. Agents 2013, 41, 36–40. [Google Scholar] [CrossRef]
- Karaman, N.; Adil Zainel, R.; Kapkaç, H.A.; Karaca Gençer, H.; Ilgın, S.; Karaduman, A.B.; Karaküçük-İyidoğan, A.; Oruç-Emre, E.E.; Koçyiğit-Kaymakçıoğlu, B. Design and Evaluation of Biological Activities of 1,3-Oxazolidinone Derivatives Bearing Amide, Sulfonamide, and Thiourea Moieties. Arch. Pharm. 2018, 351, e1800057. [Google Scholar] [CrossRef]
- Neu, H.C.; Novelli, A.; Saha, G.; Chin, N.-X. In Vitro Activities of Two Oxazolidinone Antimicrobial Agents DuP 721 and DuP 105. Antimicrob. Agents Chemother. 1988, 32, 580–583. [Google Scholar] [CrossRef]
- Matsingos, C.; Al-Adhami, T.; Jamshidi, S.; Hind, C.; Clifford, M.; Mark Sutton, J.; Rahman, K.M. Synthesis, Microbiological Evaluation and Structure Activity Relationship Analysis of Linezolid Analogues with Different C5-Acylamino Substituents. Bioorganic Med. Chem. 2021, 49, 116397. [Google Scholar] [CrossRef]
- Foti, C.; Piperno, A.; Scala, A.; Giuffrè, O. Oxazolidinone Antibiotics: Chemical, Biological and Analytical Aspects. Molecules 2021, 26, 4280. [Google Scholar] [CrossRef]
- Chellat, M.F.; Raguž, L.; Riedl, R. Targeting Antibiotic Resistance. Angew. Chem. Int. Ed. 2016, 55, 6600–6626. [Google Scholar] [CrossRef] [PubMed]
- Polacek, N.; Mankin, A.S. The Ribosomal Peptidyl Transferase Center: Structure, Function, Evolution, Inhibition. Crit. Rev. Biochem. Mol. Biol. 2005, 40, 285–311. [Google Scholar] [CrossRef] [PubMed]
- Pathania, S.; Petrova-Szczasiuk, K.; Pentikäinen, O.; Singh, P.K. Oxazolidinones: Are They Only Good for the Discovery of Antibiotics? A Worm’s Eye View. J. Mol. Struct. 2023, 1286, 135630. [Google Scholar] [CrossRef]
- Hashemian, S.M.R.; Farhadi, T.; Ganjparvar, M. Linezolid: A Review of Its Properties, Function, and Use in Critical Care. Drug Des. Dev. Ther. 2018, 12, 1759. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-Z.; Zhao, Z.-L.; Zhou, C.-H. Recent Advance in Oxazole-Based Medicinal Chemistry. Eur. J. Med. Chem. 2018, 144, 444–492. [Google Scholar] [CrossRef]
- Bush, K. CHAPTER 14-β-Lactam Antibiotics: Penicillins. In Antibiotic and Chemotherapy, 9th ed.; Finch, R.G., Greenwood, D., Norrby, S.R., Whitley, R.J., Eds.; W.B. Saunders: London, UK, 2010; pp. 200–225. ISBN 978-0-7020-4064-1. [Google Scholar]
- Alagarsamy, V. Textbook of Medicinal Chemistry Vol II—E-Book; Elsevier: Amsterdam, The Netherlands, 2012; ISBN 978-81-312-3259-0. [Google Scholar]
- Thomas, G. Fundamentals of Medicinal Chemistry; John Wiley & Sons: Hoboken, NJ, USA, 2004; ISBN 978-0-470-87169-0. [Google Scholar]
- Cotrina-Santome, A.; Ulloa-Esquivel, L.; Vásquez-Quispe, S.; Arevalo-Flores, M.; Pedraz-Petrozzi, B. Cycloserine-Induced Psychosis in Patients with Drug-Resistant Tuberculosis: A Systematic Review of Case Reports. Egypt. J. Neurol. Psychiatry Neurosurg. 2023, 59, 37. [Google Scholar] [CrossRef]
- Deshpande, D.; Alffenaar, J.-W.C.; Köser, C.U.; Dheda, K.; Chapagain, M.L.; Simbar, N.; Schön, T.; Sturkenboom, M.G.G.; McIlleron, H.; Lee, P.S.; et al. D-Cycloserine Pharmacokinetics/Pharmacodynamics, Susceptibility, and Dosing Implications in Multidrug-Resistant Tuberculosis: A Faustian Deal. Clin. Infect. Dis. 2018, 67, S308–S316. [Google Scholar] [CrossRef]
- Bradford, P.A.; Miller, A.A.; O’Donnell, J.; Mueller, J.P. Zoliflodacin: An Oral Spiropyrimidinetrione Antibiotic for the Treatment of Neisseria Gonorrheae, Including Multi-Drug-Resistant Isolates. ACS Infect. Dis. 2020, 6, 1332–1345. [Google Scholar] [CrossRef]
- Golparian, D.; Jacobsson, S.; Ohnishi, M.; Unemo, M. Complete Reference Genome Sequence of the Clinical Neisseria Gonorrhoeae Strain H035, with Resistance to the Novel Antimicrobial Zoliflodacin, Identified in Japan in 2000. Microbiol. Resour. Announc. 2023, 12, e0113022. [Google Scholar] [CrossRef]
- Kemnic, T.R.; Coleman, M. Trimethoprim Sulfamethoxazole. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Shah, R.; Verma, P.K. Therapeutic Importance of Synthetic Thiophene. Chem. Cent. J. 2018, 12, 137. [Google Scholar] [CrossRef]
- Pathania, S.; Narang, R.K.; Rawal, R.K. Role of Sulphur-Heterocycles in Medicinal Chemistry: An Update. Eur. J. Med. Chem. 2019, 180, 486–508. [Google Scholar] [CrossRef] [PubMed]
- Scott, K.A.; Njardarson, J.T. Analysis of US FDA-Approved Drugs Containing Sulfur Atoms. Top. Curr. Chem. 2018, 376, 5. [Google Scholar] [CrossRef] [PubMed]
- Ticarcillin. In LiverTox: Clinical and Research Information on Drug-Induced Liver Injury; National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD, USA, 2012.
- Lupia, T.; De Benedetto, I.; Stroffolini, G.; Di Bella, S.; Mornese Pinna, S.; Zerbato, V.; Rizzello, B.; Bosio, R.; Shbaklo, N.; Corcione, S.; et al. Temocillin: Applications in Antimicrobial Stewardship as a Potential Carbapenem-Sparing Antibiotic. Antibiotics 2022, 11, 493. [Google Scholar] [CrossRef]
- Sköld, O. Penicillins and Other Betalactams. In Antibiotics and Antibiotic Resistance; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2011; pp. 69–94. ISBN 978-1-118-07560-9. [Google Scholar]
- Norrby, S.R. CHAPTER 5—Antimicrobial Agents and the Kidneys. In Antibiotic and Chemotherapy, 9th ed.; Finch, R.G., Greenwood, D., Norrby, S.R., Whitley, R.J., Eds.; W.B. Saunders: London, UK, 2010; pp. 60–67. ISBN 978-0-7020-4064-1. [Google Scholar]
- Godzeski, C.W.; Brier, G.; Pavey, D.E. Cephalothin, a New Cephalosporin with a Broad Antibacterial Spectrum. I. In Vitro Studies Employing the Gradient Plate Technique. I. In Vitro Studies Employing the Gradient Plate Technique. Appl. Microbiol. 1963, 11, 122–127. [Google Scholar] [CrossRef]
- DrugBank Cefalotin. Available online: https://go.drugbank.com/drugs/DB00456 (accessed on 16 August 2023).
- Dondoni, A.; Merino, P. 3.06—Thiazoles. In Comprehensive Heterocyclic Chemistry II; Katritzky, A.R., Rees, C.W., Scriven, E.F.V., Eds.; Pergamon: Oxford, UK, 1996; pp. 373–474. ISBN 978-0-08-096518-5. [Google Scholar]
- Li, J.J. Heterocyclic Chemistry in Drug Discovery; Wiley: Hoboken, NJ, USA, 2013; ISBN 978-1-118-35442-1. [Google Scholar]
- Ayati, A.; Emami, S.; Asadipour, A.; Shafiee, A.; Foroumadi, A. Recent Applications of 1,3-Thiazole Core Structure in the Identification of New Lead Compounds and Drug Discovery. Eur. J. Med. Chem. 2015, 97, 699–718. [Google Scholar] [CrossRef]
- Sharma, P.C.; Bansal, K.K.; Sharma, A.; Sharma, D.; Deep, A. Thiazole-Containing Compounds as Therapeutic Targets for Cancer Therapy. Eur. J. Med. Chem. 2020, 188, 112016. [Google Scholar] [CrossRef] [PubMed]
- Borcea, A.-M.; Ionuț, I.; Crișan, O.; Oniga, O. An Overview of the Synthesis and Antimicrobial, Antiprotozoal, and Antitumor Activity of Thiazole and Bisthiazole Derivatives. Molecules 2021, 26, 624. [Google Scholar] [CrossRef]
- Ayati, A.; Emami, S.; Moghimi, S.; Foroumadi, A. Thiazole in the Targeted Anticancer Drug Discovery. Future Med. Chem. 2019, 11, 1929–1952. [Google Scholar] [CrossRef] [PubMed]
- Arshad, M.F.; Alam, A.; Alshammari, A.A.; Alhazza, M.B.; Alzimam, I.M.; Alam, M.A.; Mustafa, G.; Ansari, M.S.; Alotaibi, A.M.; Alotaibi, A.A.; et al. Thiazole: A Versatile Standalone Moiety Contributing to the Development of Various Drugs and Biologically Active Agents. Molecules 2022, 27, 3994. [Google Scholar] [CrossRef]
- Singh, A.; Malhotra, D.; Singh, K.; Chadha, R.; Bedi, P.M.S. Thiazole Derivatives in Medicinal Chemistry: Recent Advancements in Synthetic Strategies, Structure Activity Relationship and Pharmacological Outcomes. J. Mol. Struct. 2022, 1266, 133479. [Google Scholar] [CrossRef]
- Decuyper, L.; Jukič, M.; Sosič, I.; Žula, A.; D’hooghe, M.; Gobec, S. Antibacterial and β-Lactamase Inhibitory Activity of Monocyclic β-Lactams. Med. Res. Rev. 2018, 38, 426–503. [Google Scholar] [CrossRef]
- Neu, H.C. Aztreonam Activity, Pharmacology, and Clinical Uses. Am. J. Med. 1990, 88, S2–S6. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, C.; MacGowan, A.P. A Review of the Pharmacokinetics and Pharmacodynamics of Aztreonam. J. Antimicrob. Chemother. 2016, 71, 2704–2712. [Google Scholar] [CrossRef] [PubMed]
- Bulitta, J.B.; Duffull, S.B.; Landersdorfer, C.B.; Kinzig, M.; Holzgrabe, U.; Stephan, U.; Drusano, G.L.; Sörgel, F. Comparison of the Pharmacokinetics and Pharmacodynamic Profile of Carumonam in Cystic Fibrosis Patients and Healthy Volunteers. Diagn. Microbiol. Infect. Dis. 2009, 65, 130–141. [Google Scholar] [CrossRef] [PubMed]
- Chatwal, G.R. Medicinal Chemistry; Himalaya Publishing House: Mumbai, India, 2010; ISBN 978-1-4416-6248-4. [Google Scholar]
- Anand, N. Sulfonamides: Structure-Activity Relationships and Mechanism of Action. In Inhibition of Folate Metabolism in Chemotherapy: The Origins and Uses of Co-trimoxazole; Hitchings, G.H., Ed.; Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 1983; pp. 25–54. ISBN 978-3-642-81890-5. [Google Scholar]
- Testa, B.; Mayer, J.M. Hydrolysis in Drug and Prodrug Metabolism; John Wiley & Sons: Hoboken, NJ, USA, 2003; ISBN 978-3-906390-25-3. [Google Scholar]
- Ameta, K.L.; Kant, R.; Penoni, A.; Maspero, A.; Scapinello, L. N-Heterocycles: Synthesis and Biological Evaluation; Springer: Berlin/Heidelberg, Germany, 2022; ISBN 978-981-19083-2-3. [Google Scholar]
- Hu, Y.; Li, C.-Y.; Wang, X.-M.; Yang, Y.-H.; Zhu, H.-L. 1,3,4-Thiadiazole: Synthesis, Reactions, and Applications in Medicinal, Agricultural, and Materials Chemistry. Chem. Rev. 2014, 114, 5572–5610. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, D.J. 5.08—1,2,4-Thiadiazoles. In Comprehensive Heterocyclic Chemistry III; Katritzky, A.R., Ramsden, C.A., Scriven, E.F.V., Taylor, R.J.K., Eds.; Elsevier: Oxford, UK, 2008; pp. 487–513. ISBN 978-0-08-044992-0. [Google Scholar]
- Delgado, C.P.; Rocha, J.B.T.; Orian, L.; Bortoli, M.; Nogara, P.A. In Silico Studies of Mpro and PLpro from SARS-CoV-2 and a New Class of Cephalosporin Drugs Containing 1,2,4-Thiadiazole. Struct. Chem. 2022, 33, 2205–2220. [Google Scholar] [CrossRef]
- Laudano, J.B. Ceftaroline Fosamil: A New Broad-Spectrum Cephalosporin. J. Antimicrob. Chemother. 2011, 66, iii11–iii18. [Google Scholar] [CrossRef] [PubMed]
- Zhanel, G.G.; Sniezek, G.; Schweizer, F.; Zelenitsky, S.; Lagacé-Wiens, P.R.S.; Rubinstein, E.; Gin, A.S.; Hoban, D.J.; Karlowsky, J.A. Ceftaroline. Drugs 2009, 69, 809–831. [Google Scholar] [CrossRef]
- Iizawa, Y.; Okonogi, K.; Hayashi, R.; Iwahi, T.; Yamazaki, T.; Imada, A. Therapeutic Effect of Cefozopran (SCE-2787), a New Parenteral Cephalosporin, against Experimental Infections in Mice. Antimicrob. Agents Chemother. 1993, 37, 100–105. [Google Scholar] [CrossRef]
- Wu, G.L.; Shentu, J.Z.; Zhou, H.L.; Zhu, M.X.; Hu, X.J.; Liu, J.; Wu, L.H. Pharmacokinetics of Cefozopran by Single and Multiple Intravenous Infusions in Healthy Chinese Volunteers. Drugs R&D 2015, 15, 63–70. [Google Scholar] [CrossRef]
- Bui, T.; Preuss, C.V. Cephalosporins. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Duplessis, C.; Crum-Cianflone, N.F. Ceftaroline: A New Cephalosporin with Activity against Methicillin-Resistant Staphylococcus Aureus (MRSA). Clin. Med. Rev. Ther. 2011, 3, a2466. [Google Scholar] [CrossRef] [PubMed]
- FDA TEFLARO® (Ceftaroline Fosamil) for Injection, for Intravenous Use. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/200327s015lbl.pdf (accessed on 22 August 2023).
- Kouki, I.; Montagner, C.; Mauhin, W.; London, J.; Lazard, T.; Grimbert, S.; Zeller, V.; Lidove, O. Coagulation Disorders during Treatment with Cefazolin and Rifampicin: Rare but Dangerous. J. Bone Jt. Infect. 2021, 6, 131–134. [Google Scholar] [CrossRef] [PubMed]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FDA Approval Year | Antibiotic Compound | Antibiotic Class (Generation) | Five-Member Heterocycle in the Structure |
|---|---|---|---|
| 1980 | Cefotaxime | Beta-lactam cephalosporin (2nd generation) | 1,3-Thiazole |
| 1981 | Cefoperazone | Beta-lactam cephalosporin (3rd generation) | Tetrazole |
| 1981 | Cefotiam | Beta-lactam cephalosporin (2nd generation) | 1,3-Thiazole, Tetrazole |
| 1982 | Ceftriaxone | Beta-lactam cephalosporin (3rd generation) | 1,3-Thiazole |
| 1982 | Latamoxef/Moxalactam | Beta-lactam, oxacephem cephalosporin (1st generation) | Tetrazole |
| 1983 | Cefonicid | Beta-lactam cephalosporin (2nd generation) | Tetrazole |
| 1983 | Cefuroxime | Beta-lactam cephalosporin (2nd generation) | Furan |
| 1984 | Ceftazidime | Beta-lactam cephalosporin (2nd generation | 1,3-Thiazole |
| 1986 | Aztreonam | Beta-lactam monobactam | 1,3-Thiazole |
| 1987 | Cefotetan | Beta-lactam cephalosporin (3rd generation) | Tetrazole |
| 1992 | Cefpodoxime proxetil | Beta-lactam cephalosporin (3rd generation) | 1,3-Thiazole |
| 1991 | Cefuroxime axetil | Beta-lactam cephalosporin (3rd generation) | Furan |
| 1992 | Ceftibuten | Beta-lactam cephalosporin (3rd generation) | 1,3-Thiazole |
| 1992 | Tazobactam | Beta-lactamase inhibitor | 1,2,3-Triazole |
| 1993 | Cefixime | Beta-lactam cephalosporin (3rd generation) | 1,3-Thiazole |
| 1996 | Cefepime | Beta-lactam cephalosporin (4th generation) | Pyrrolidine, 1,3-Thiazole |
| 1996 | Meropenem | Beta-lactam carbapenem | Pyrrolidine |
| 1997 | Cefdinir | Beta-lactam cephalosporin (4th generation) | 1,3-Thiazole |
| 2000 | Linezolid | Oxazolidinone | 1,3-Oxazolidine |
| 2001 | Ertapenem | Beta-lactam carbapenem | Pyrrolidine |
| 2001 | Telithromycin | Ketolide macrolide | Imidazole |
| 2003 | Gemifloxacin | Fluoroquinolone | Pyrrolidine |
| 2009 | Ceftobiprole | Beta-lactam cephalosporin (5th generation) | Pyrrolidine |
| 2014 | Doripenem | Beta-lactam carbapenem | Pyrrolidine |
| 2014 | Finafloxacin | Fluoroquinolone | Pyrrole (in a bicycle) |
| 2014 | Tedizolid | Oxazolidinone | 1,3-Oxazolidin-2-one, Tetrazole |
| 2018 | Eravacycline | Tetracycline | Pyrrolidine |
| 2018 | Gemifloxacin | Fluoroquinolone | Pyrrolidine |
| 2019 | Imipenem + Cilastatin + Relebactam | Relebactam: beta-lactamase inhibitor | 2-Imidazolidinone (in an azabicycle) |
| 2019 | Cefidorocol | Beta-lactam cephalosporin (5th generation) | Pyrrolidine, thiazole |
| 2021 | Ceftidoren pivoxil | Beta-lactam cephalosporin (5th generation) | Thiazole (2 groups) |
| 2023 | Sulbactam + durlobactam | Sulbactam: beta-lactam antibacterial and beta-lactamase inhibitor Durlobactam: beta-lactamase inhibitor | Sulbactam: 1,3-Thiazolidine 1,1-dioxide Durlobactam: 2-Imidazolidinone (in an azabicycle) |
| Five-Membered Heterocycles | Heteroatom (s) | Chemical Structure | MW (g/mol) | HBA | HBD |
|---|---|---|---|---|---|
| Pyrrolidine | N | 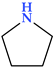 | 71.12 | 1 | 1 |
| Imidazole | N(2) | 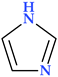 | 68.08 | 1 | 1 |
| 1,2,3-Triazole | N(3) |  | 69.07 | 2 | 1 |
| Tetrazole | N(4) | 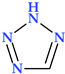 | 70.05 | 3 | 1 |
| Furan | O(1) |  | 68.07 | 1 | 0 |
| 1,3-Oxazolidine | N(1),O(1) | 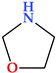 | 73.09 | 2 | 1 |
| 1,3-Oxazole | N(1),O(1) | 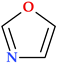 | 69.06 | 2 | 0 |
| 1,2-Oxazole (Isoxazole) | N(1),O(1) |  | 69.06 | 2 | 0 |
| Thiophene | S(1) |  | 84.14 | 1 | 0 |
| 1,3-Thiazolidine | N(1),S(1) |  | 89.6 | 2 | 1 |
| 1,3-Thiazole | N(1),S(1) |  | 89.16 | 2 | 1 |
| 1,2,4-Thiadiazole | N(2),S(1) |  | 86.12 | 3 | 0 |
| 1,3,4-Thiadiazole | N(2),S(1) | 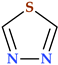 | 86.12 | 3 | 0 |
| No. | Therapeutic Class | Subclass | Reprezentatives | Ref. |
|---|---|---|---|---|
| 1 | Beta-lactam antibiotics | Carbapenemes | Doripenem | [30] |
| Ertapenem | [30] | |||
| Meropenem | [30] | |||
| Cephalosporins | Cefepime | [31] | ||
| Cefiderocol | [32] | |||
| Ceftobiprole | [33] | |||
| 2 | Fluoroquinolones | - | Clinafloxacin | [34] |
| Finafloxacin | [34] | |||
| Gemifloxacin | [34] | |||
| Lascufloxacin | [34] | |||
| Premafloxacin | [35] | |||
| Sitafloxacin | [34] | |||
| Trovafloxacin | [34] | |||
| 3 | Lincosamides | - | Lincomycin | [31] |
| Clindamycin | [31] | |||
| 4 | Streptogramins | - | Quinupristin/Dalfopristin | [36] |
| 5 | Tetracyclines | - | Rolitetracycline | [29] |
| Glycylcyclines | Eravacycline | [37] |
| Associated Side-Effects | Comparison with Other Substituents in the C7 Position |
|---|---|
| Genotoxicity | Pyrrolidine > Piperazine > Alkyl |
| Pyrrolidine (unsubstituted) > Piperazine (unsubstituted) > Pyrrolidine (substituted) > Piperazine (substituted) | |
| Neuropsychiatric toxicity, seizures (GABA receptor binding) | Alkyl > Piperazine (unsubstituted) > Pyrrolidine (unsubstituted) > Piperazine (substituted) or Pyrrolidine (substituted) |
| Some NSAIDs interactions | Piperazine (unsubstituted) > Pyrrolidine (unsubstituted) > Piperazine (substituted) or Pyrrolidine (substituted) |
| Theophylline interactions | Pyrrolidine (unsubstituted) > Piperazine (unsubstituted) > Piperazine (substituted) or Pyrrolidine (substituted) |
| No. | Cephalosporin | Generation | 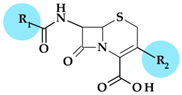 | Administration | t1/2 (Hours) | Acid Resistant | Resistance to β-lactamases | Antibacterial Spectrum | Activity against Pseudomonas sp. | |
|---|---|---|---|---|---|---|---|---|---|---|
| R1 | R2 | |||||||||
| 1 | Cefpodoxime (proxetil) | 3rd |  |  | Oral | 2.2 | Yes | Good | Extended | No |
| 2 | Cefotaxime | 3rd | 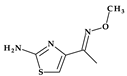 |  | Parenteral | 1 | No | Good | Extended | Yes |
| 3 | Ceftazidime | 3rd |  |  | Parenteral | 2 | No | Good | Extended | Yes |
| 4 | Ceftriaxone | 3rd | 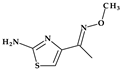 | 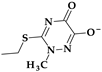 | Parenteral | 6–9 | No | Good | Extended | Yes |
| 5 | Cefixime | 3rd | 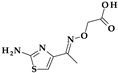 |  | Oral | 3–4 | Yes | Good | Extended | No |
| 6 | Cefdinir | 3rd | 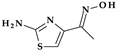 | 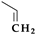 | Oral | 1.7 | Yes | Good | Extended | No |
| 7 | Cefditoren (pivoxil) | 3rd |  | 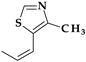 | Oral | 1.6 | Yes | Good | Extended | No |
| 8 | Ceftibuten | 3rd |  | -H | Oral | 2–2.3 | Yes | Good | Extended | No |
| 9 | Ceftizoxime | 3rd | 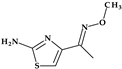 | -H | Parenteral | 1.7 | No | Good | Extended | Yes |
| 10 | Cefepime | 4th |  |  | Parenteral | 2 | No | Good | Extended | Yes |
| 11 | Cefpirome | 4th | 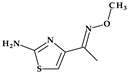 | 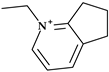 | Parenteral | 2 | No | Good | Extended | Yes |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rusu, A.; Moga, I.-M.; Uncu, L.; Hancu, G. The Role of Five-Membered Heterocycles in the Molecular Structure of Antibacterial Drugs Used in Therapy. Pharmaceutics 2023, 15, 2554. https://doi.org/10.3390/pharmaceutics15112554
Rusu A, Moga I-M, Uncu L, Hancu G. The Role of Five-Membered Heterocycles in the Molecular Structure of Antibacterial Drugs Used in Therapy. Pharmaceutics. 2023; 15(11):2554. https://doi.org/10.3390/pharmaceutics15112554
Chicago/Turabian StyleRusu, Aura, Ioana-Maria Moga, Livia Uncu, and Gabriel Hancu. 2023. "The Role of Five-Membered Heterocycles in the Molecular Structure of Antibacterial Drugs Used in Therapy" Pharmaceutics 15, no. 11: 2554. https://doi.org/10.3390/pharmaceutics15112554
APA StyleRusu, A., Moga, I.-M., Uncu, L., & Hancu, G. (2023). The Role of Five-Membered Heterocycles in the Molecular Structure of Antibacterial Drugs Used in Therapy. Pharmaceutics, 15(11), 2554. https://doi.org/10.3390/pharmaceutics15112554