
Podophyllotoxin: Recent Advances in the Development of Hybridization Strategies to Enhance Its Antitumoral Profile


 , ,
, ,  , and
, and 
Abstract
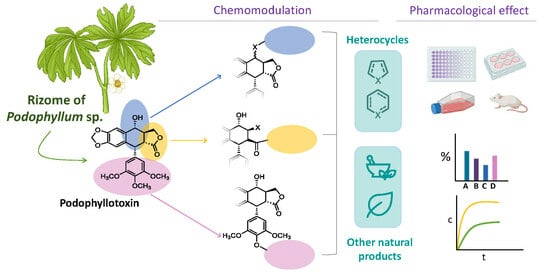
:
{kind=link}
{kind=link}
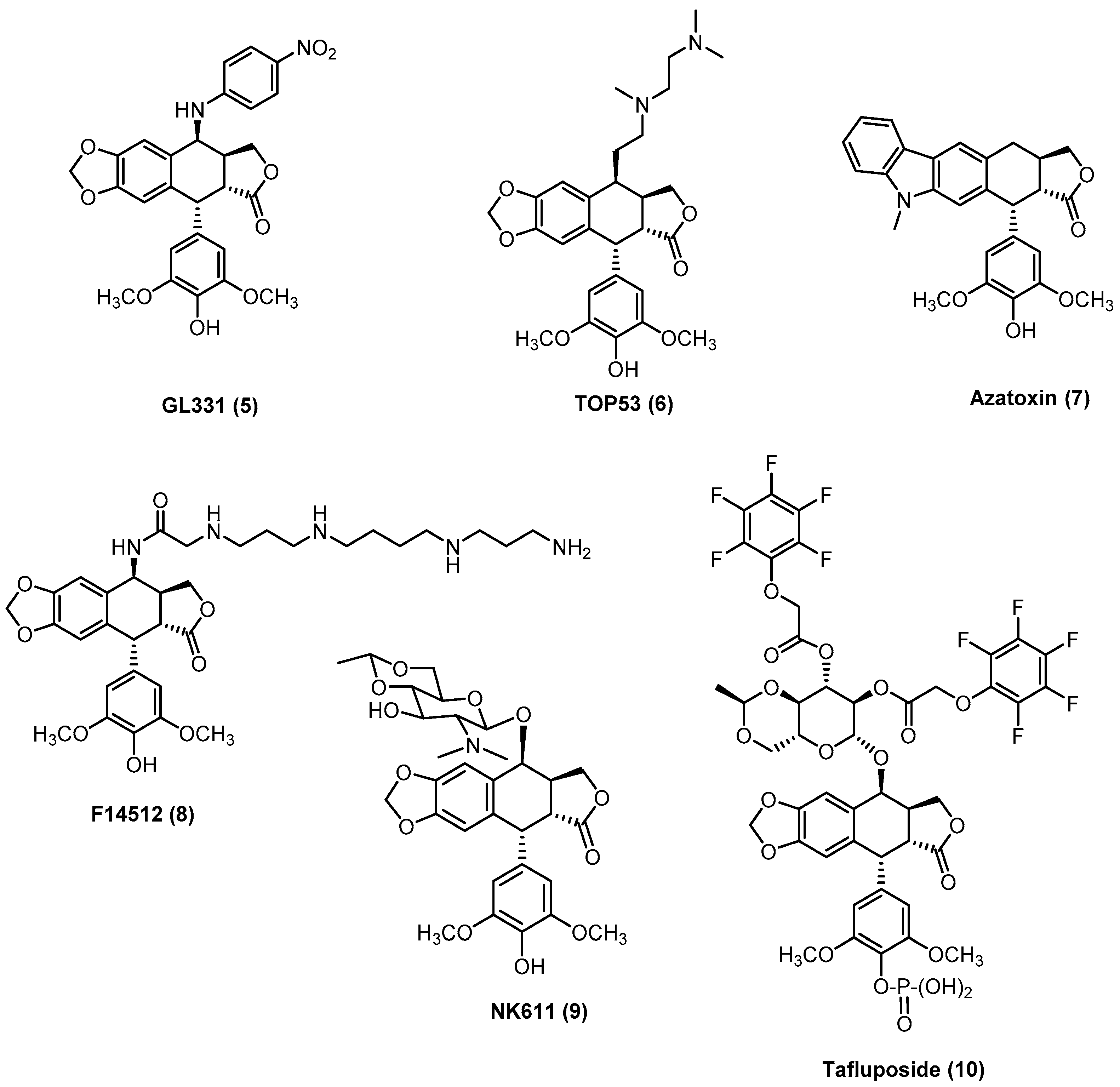{kind=link}
{kind=link}
{kind=link}
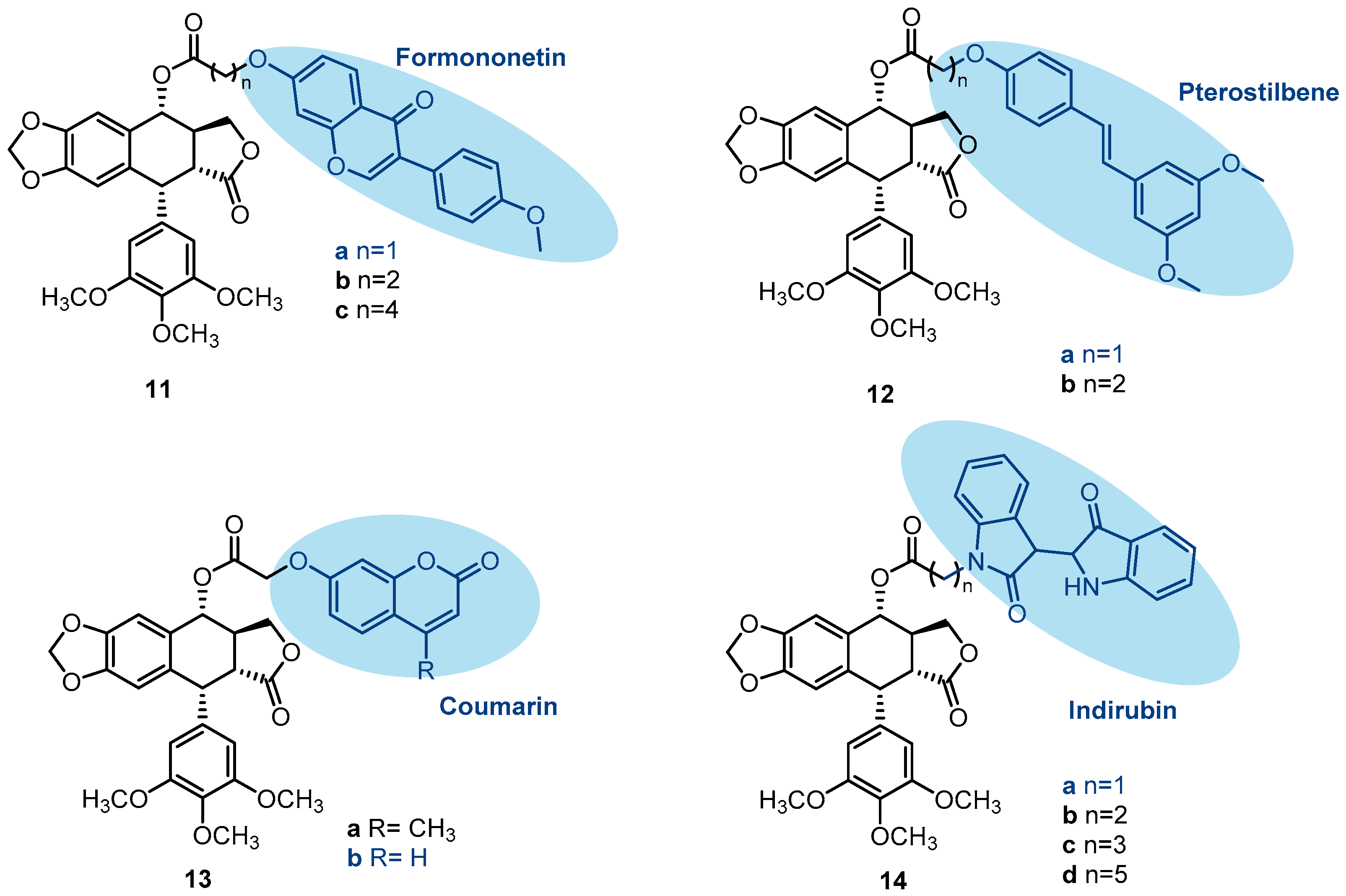{kind=link}
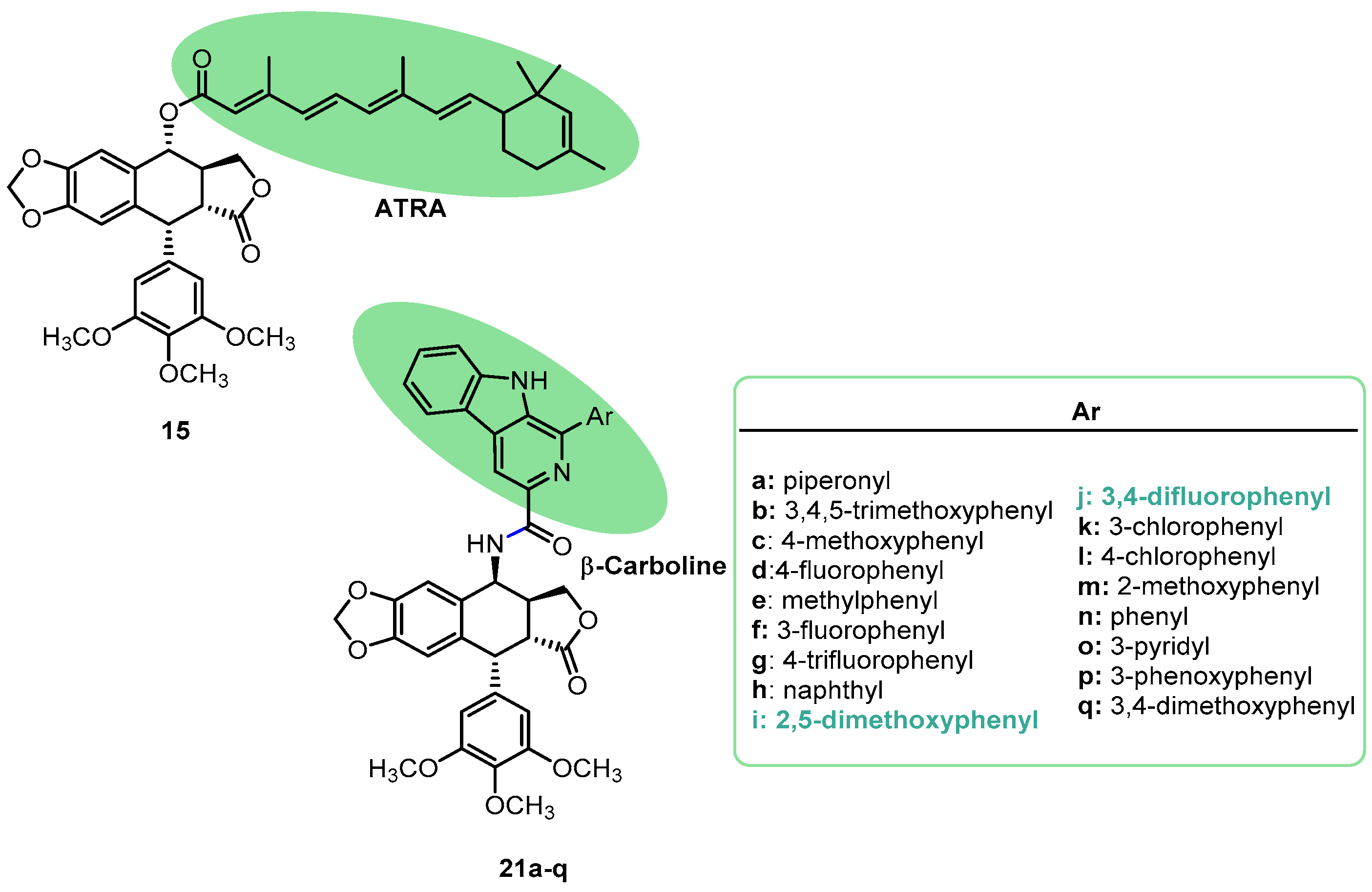{kind=link}
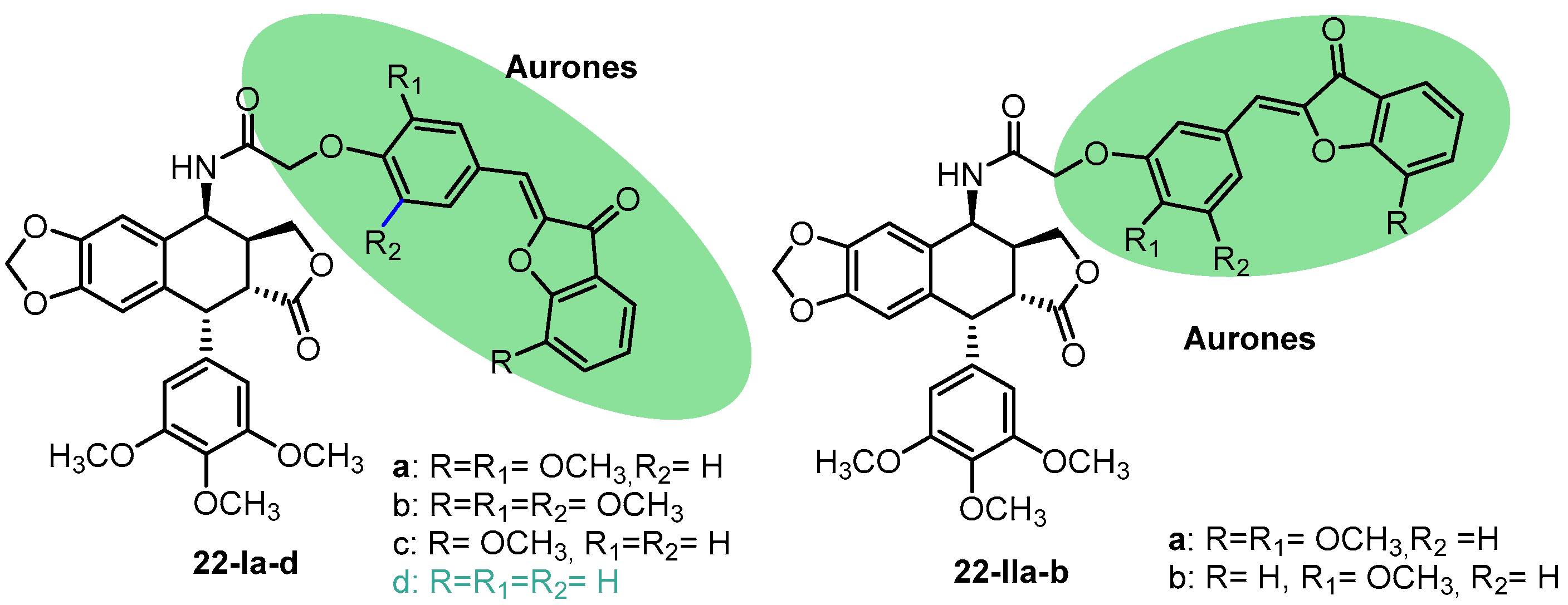{kind=link}
{kind=link}
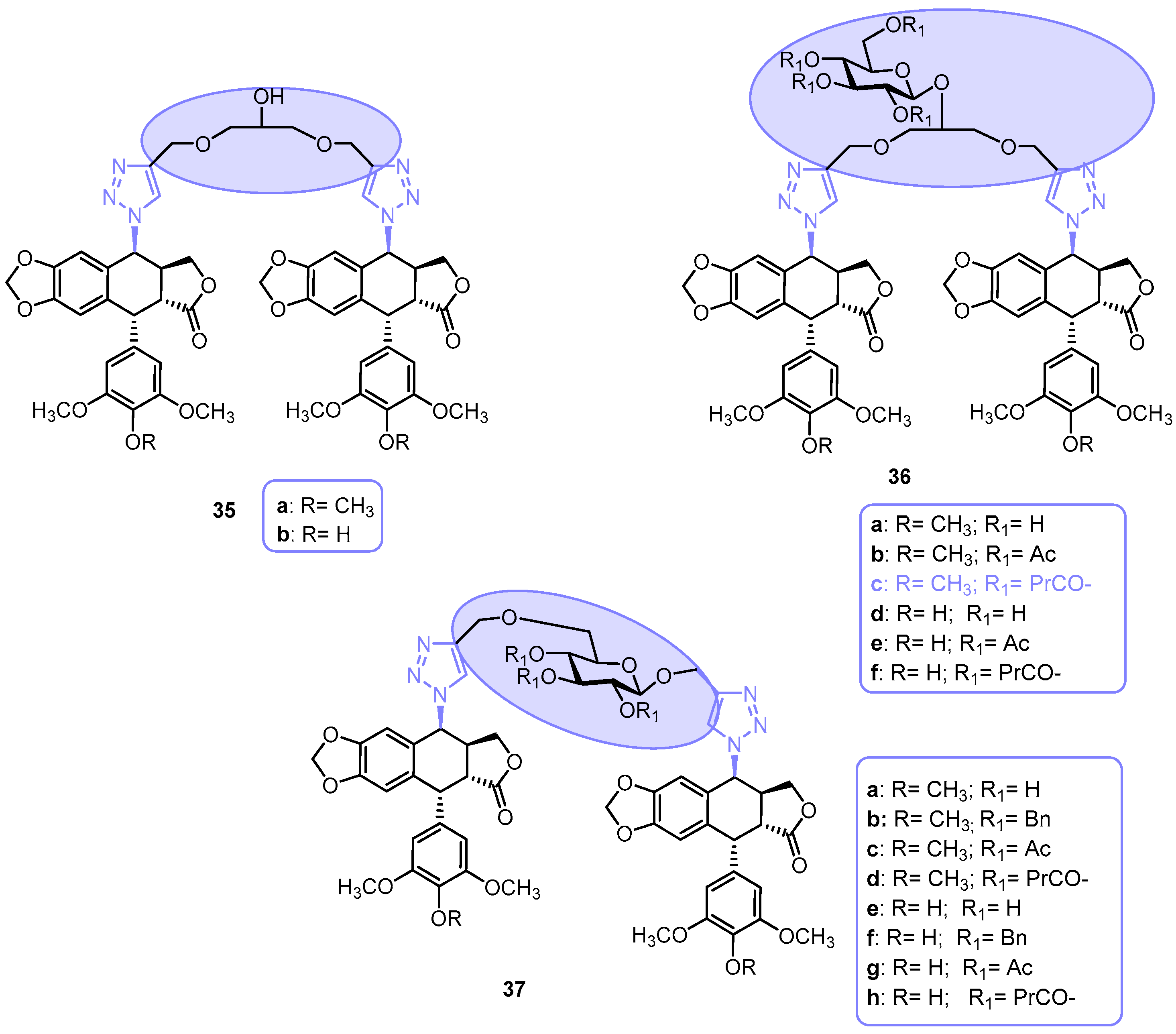{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
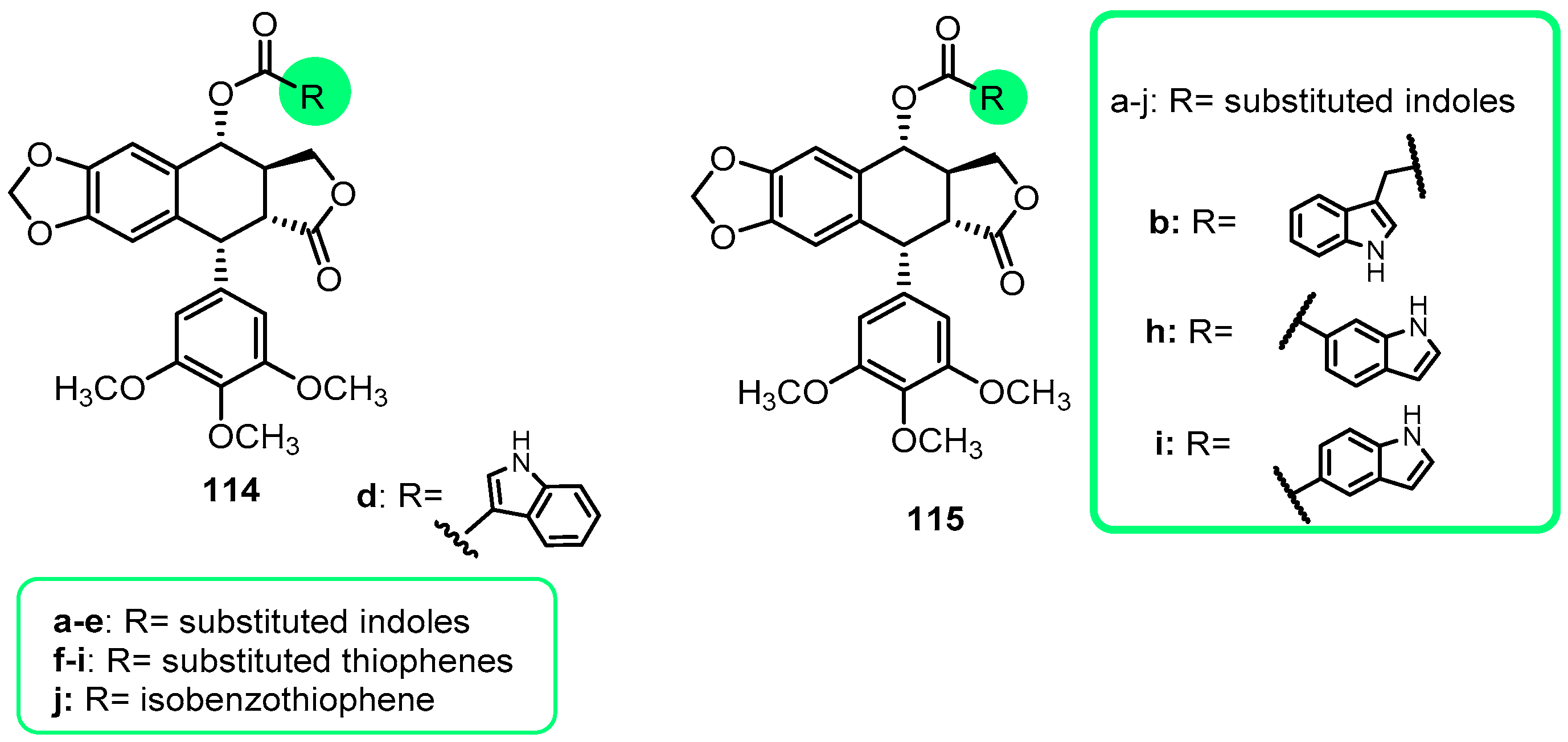{kind=link}
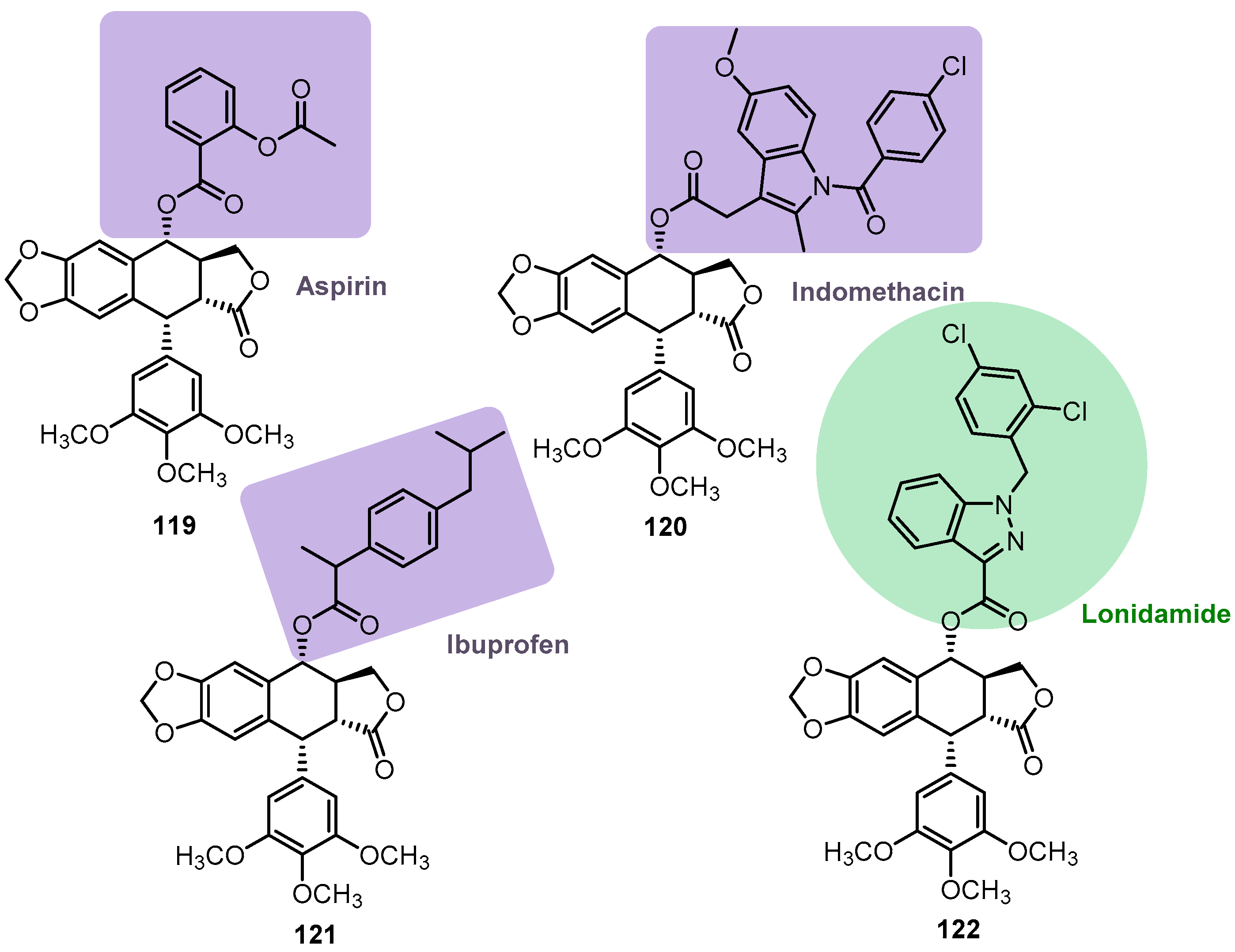{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
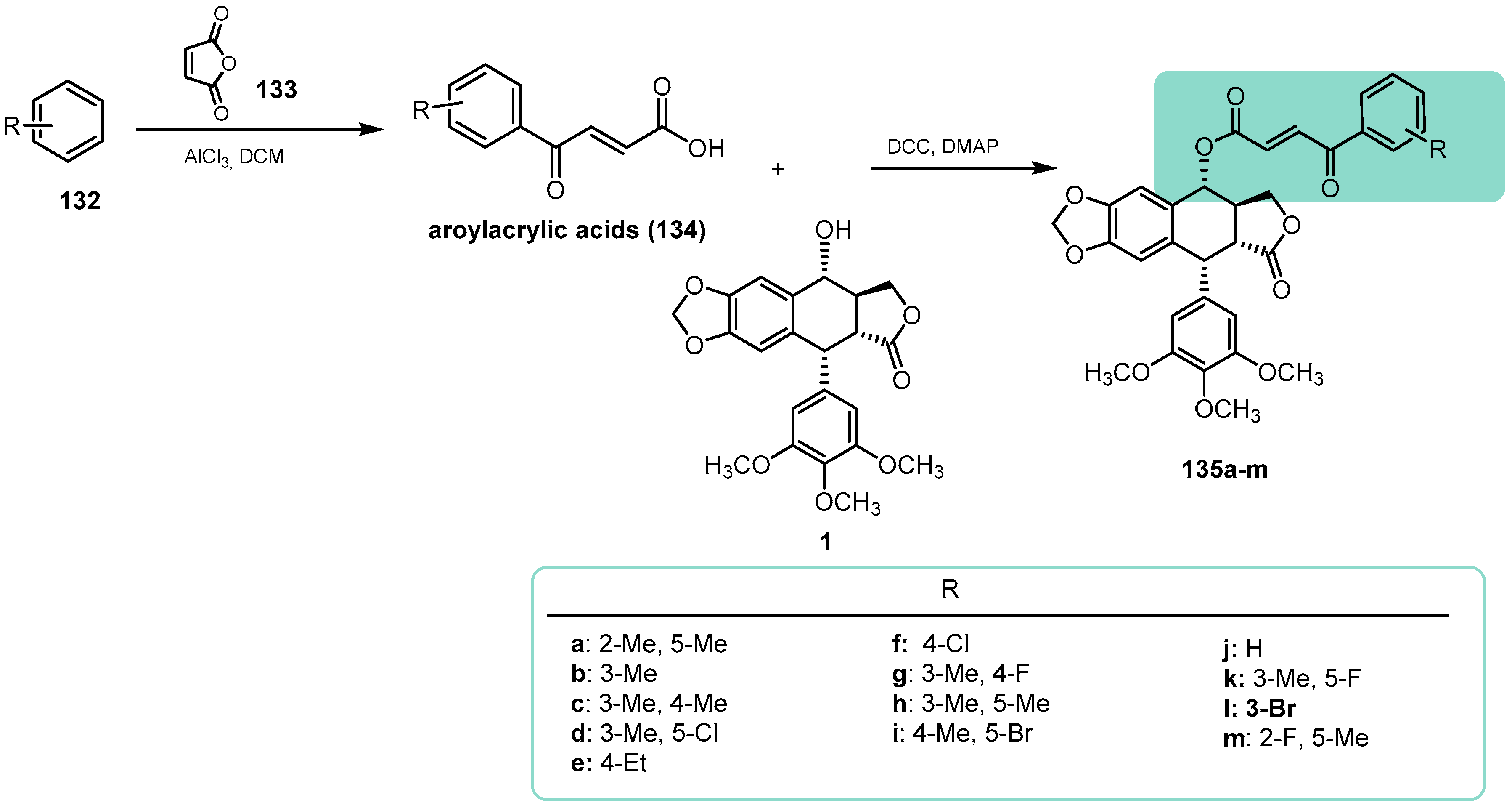{kind=link}
{kind=link}
{kind=link}
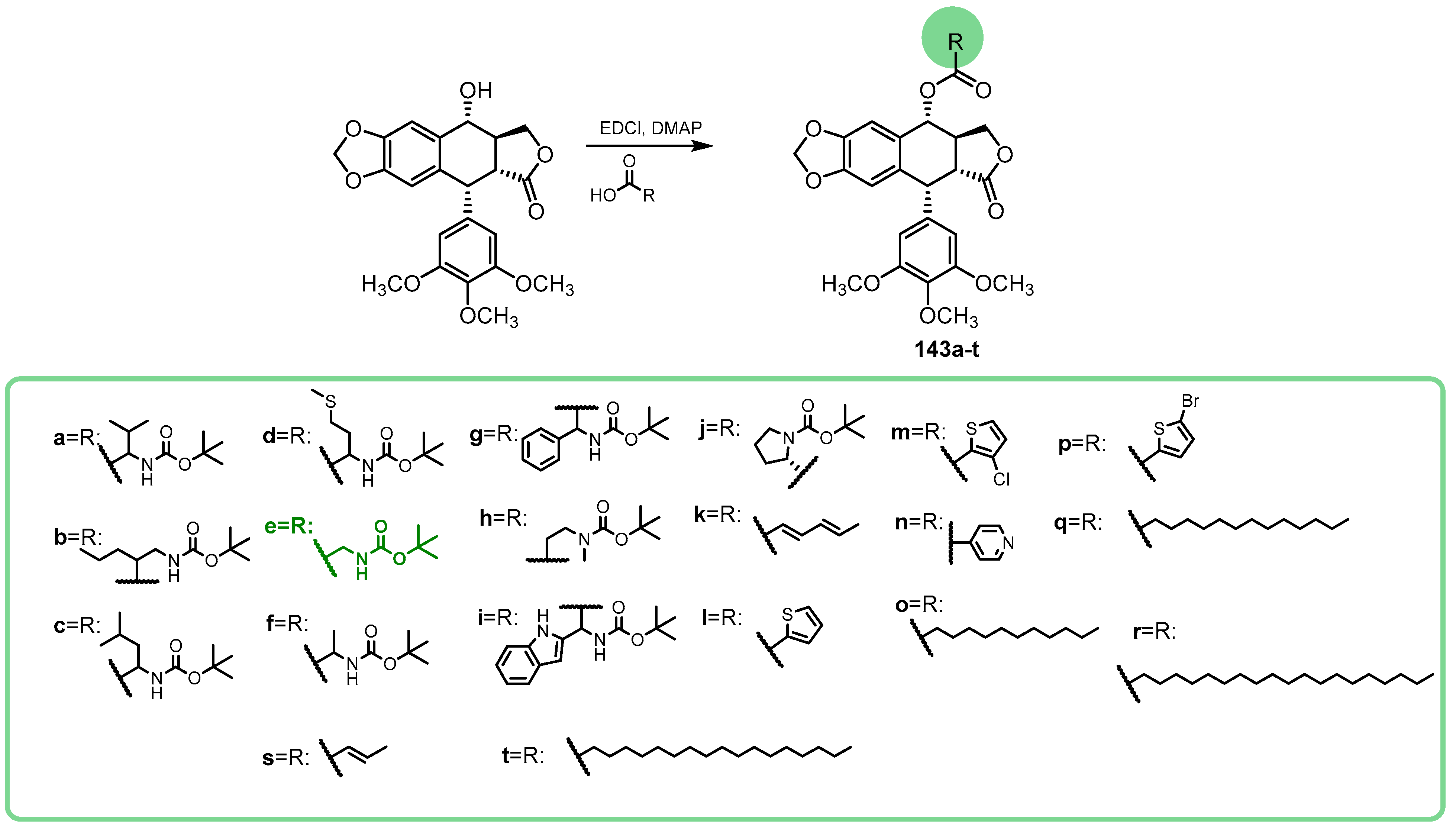{kind=link}
{kind=link}
{kind=link}
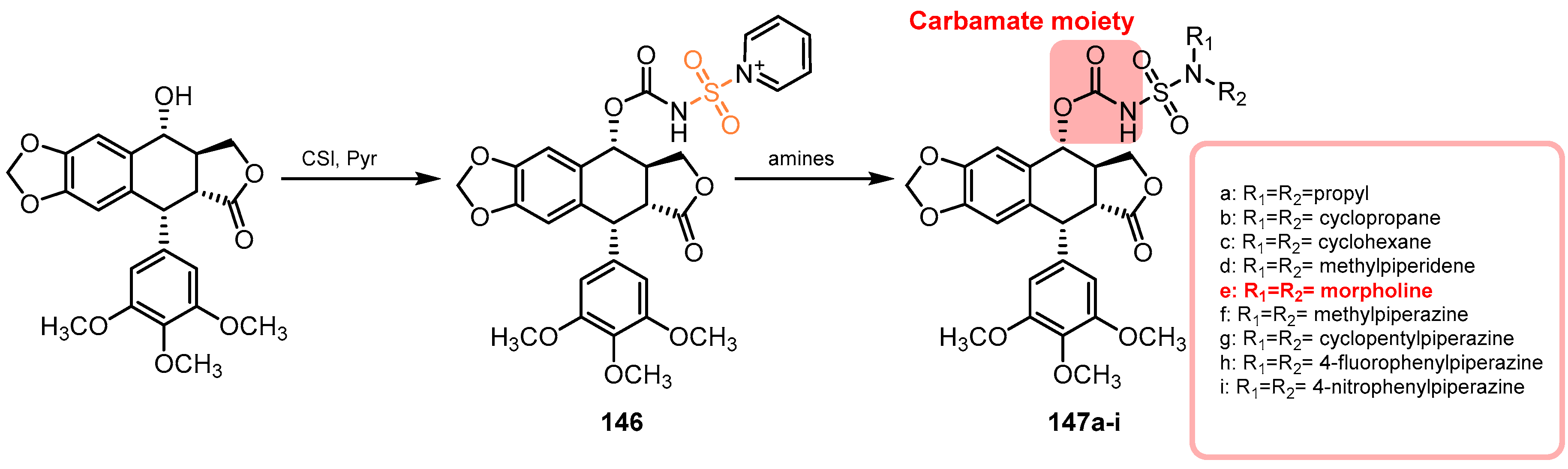{kind=link}
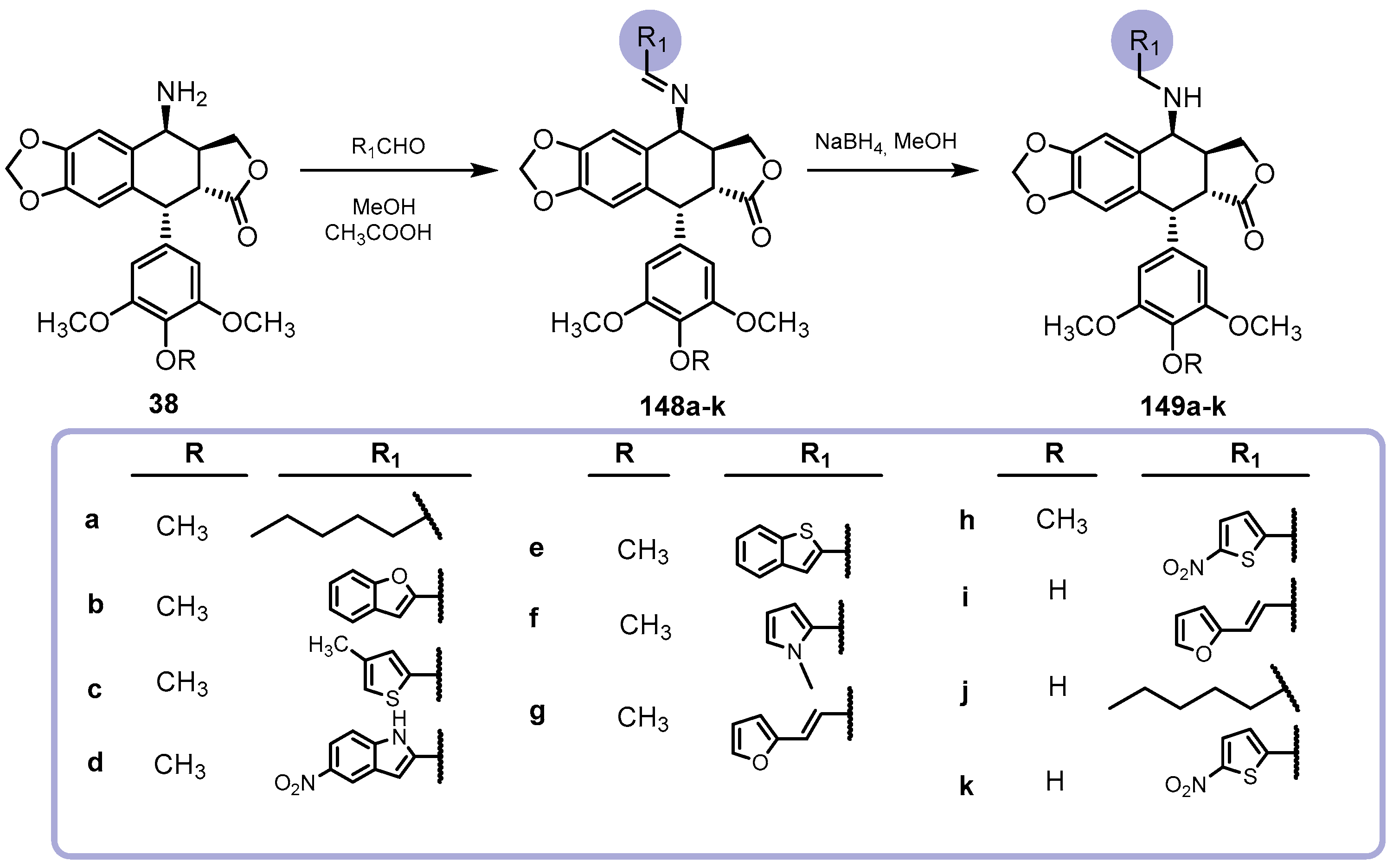{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. C-Ring Modifications of the Podophyllotoxin Skeleton
2.1. Hybrids of Podophyllotoxin with Different Natural Products
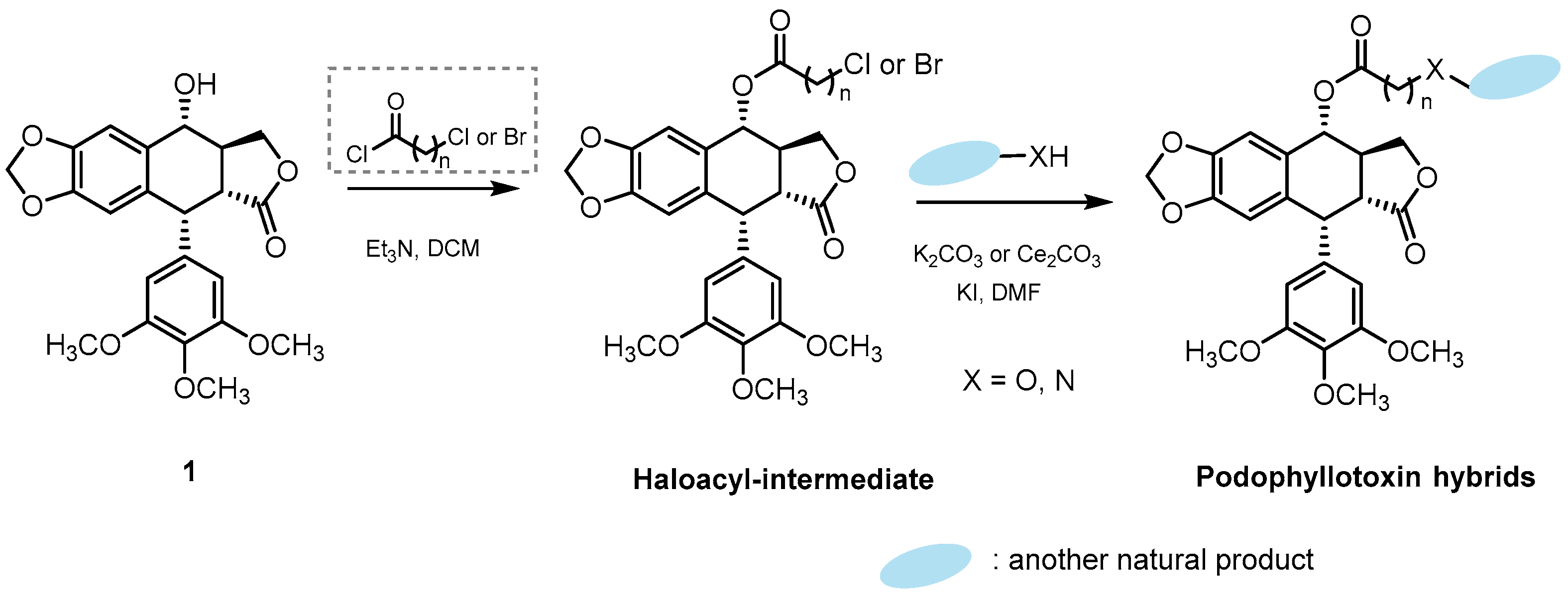
2.1.1. Podophyllotoxin Hybrids Synthesized through a Haloacyl Intermediate
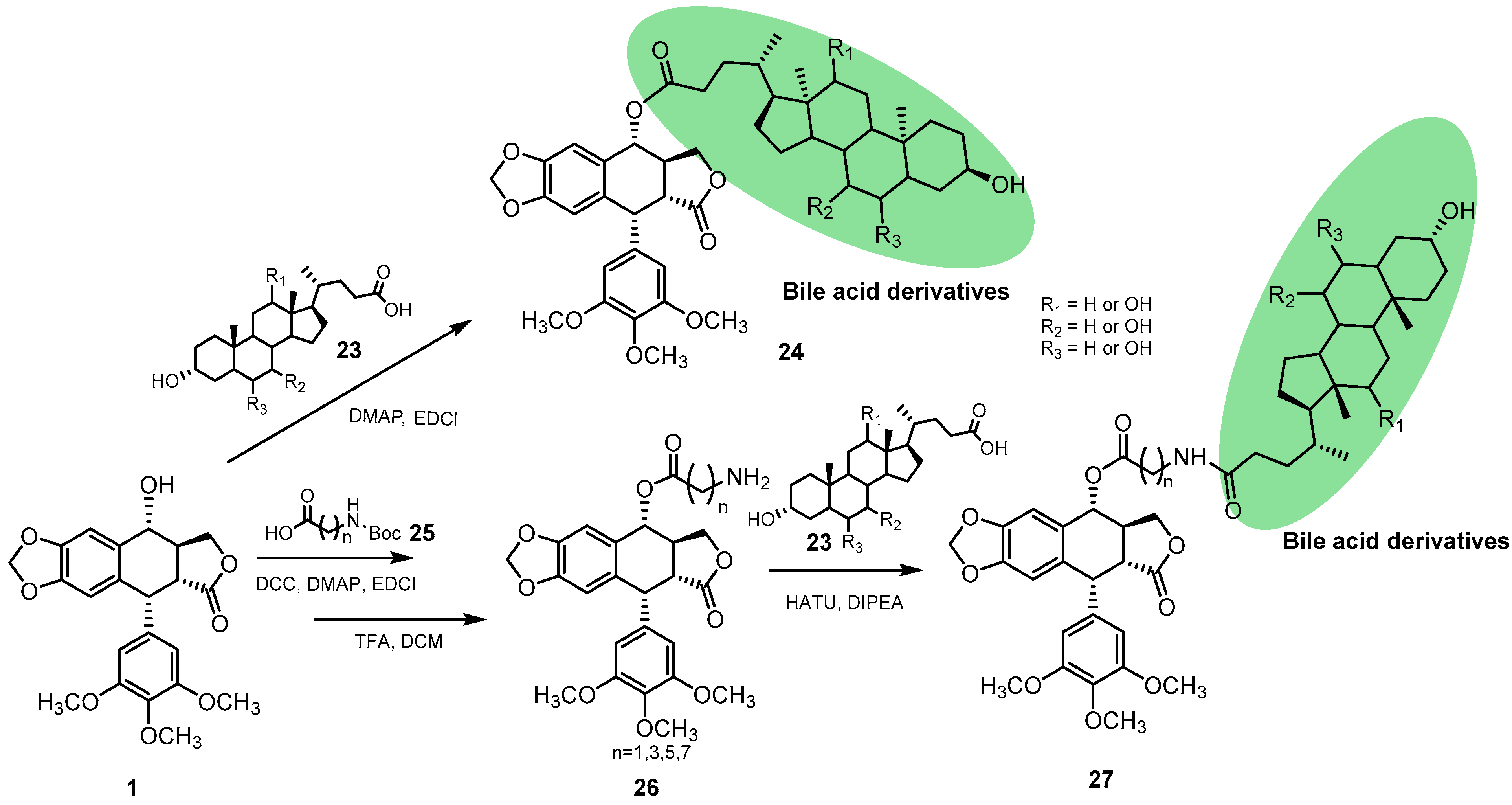
2.1.2. Podophyllotoxin Hybrids Synthesized via Direct Coupling with Natural-Product Derivatives Bearing a Carboxylic Group
2.1.3. Podophyllotoxin Hybrids with Other Natural Products Attached through a Triazole Ring
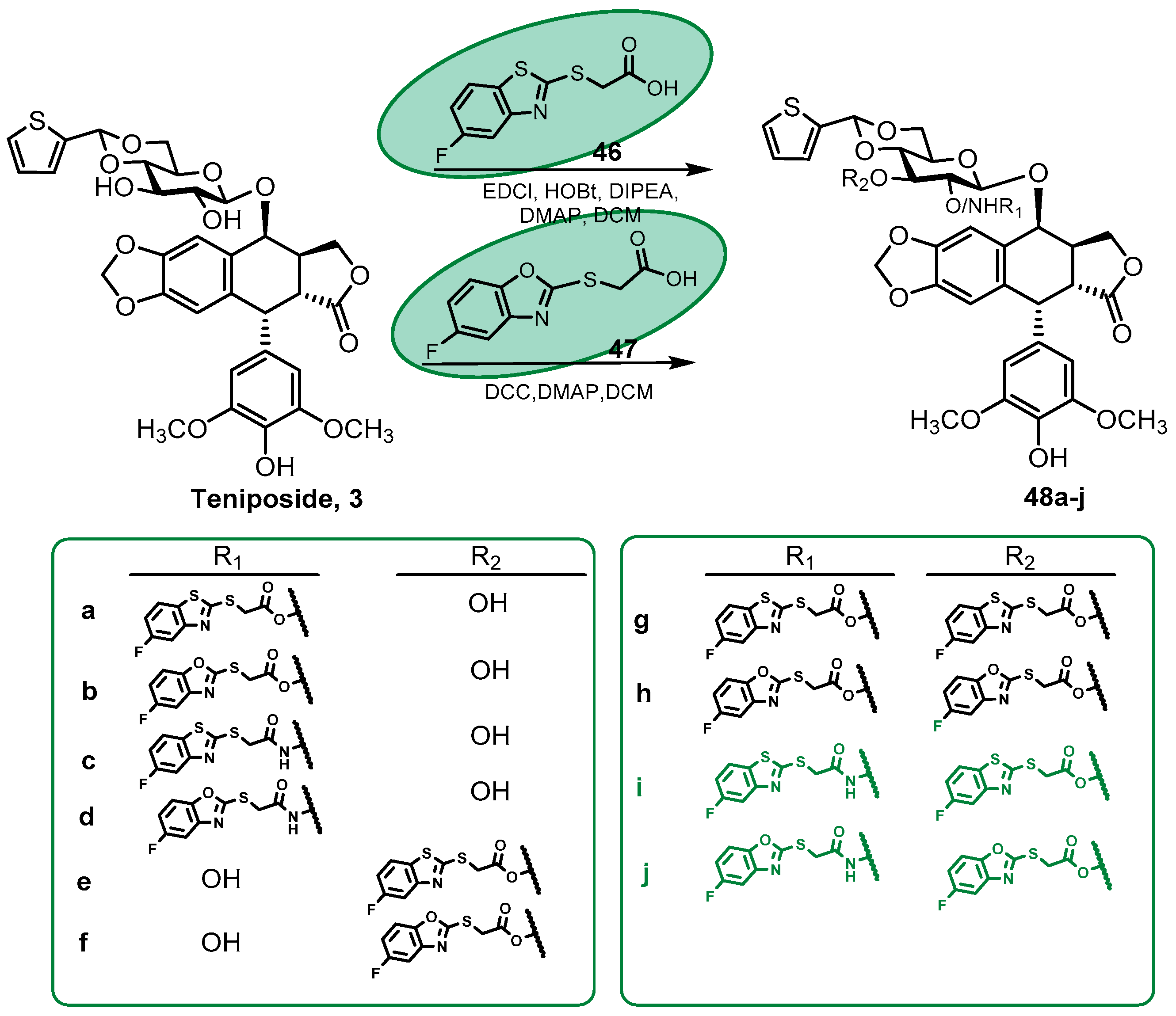
2.1.4. Podophyllotoxin Glycoconjugates
2.2. Podophyllotoxin Derivatives with Heterocyclic Systems
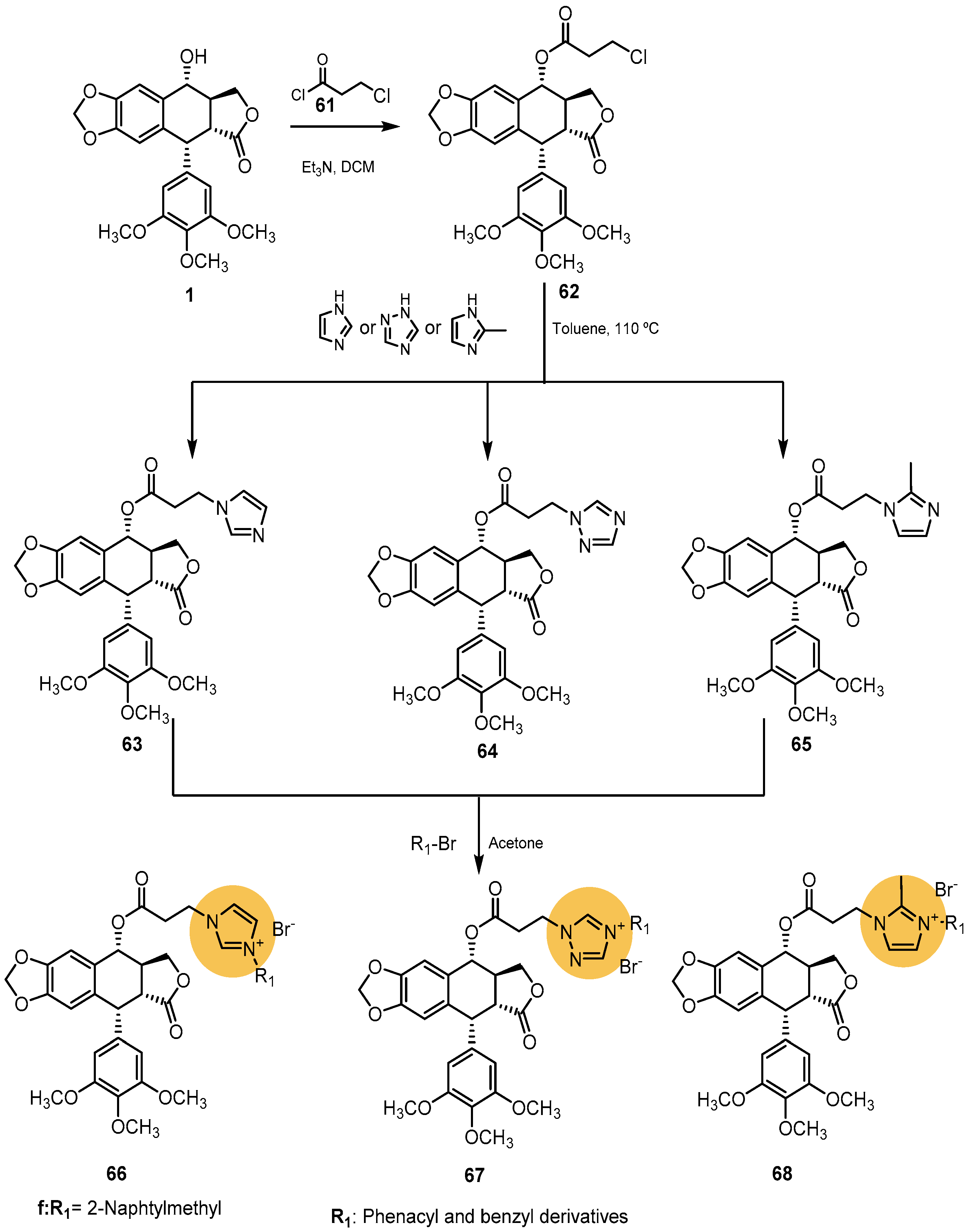
2.2.1. Five-Membered Heterocycles with Two Heteroatoms
2.2.2. Five-Membered Heterocycles with Three Heteroatoms
2.2.3. Six-Membered Heterocycles
2.2.4. Benzoheterocycle Derivatives
2.3. Hybrids of Podophyllotoxin with Other Moieties
3. D-Ring Modifications of the Podophyllotoxin Skeleton
4. E-Ring Modifications of the Podophyllotoxin Skeleton
5. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics. Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- WHO. Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 29 September 2023).
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Panda, M.; Biswal, B.K. Cell signaling and cancer: A mechanistic insight into drug resistance. Mol. Biol. Rep. 2019, 46, 5645–5659. [Google Scholar] [CrossRef] [PubMed]
- Paterson, I.; Anderson, E.A. The renaissance of natural products as drug candidates. Science 2005, 310, 451–453. [Google Scholar] [CrossRef] [PubMed]
- Clardy, J.; Walsh, C. Lessons from natural molecules. Nature 2004, 432, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Gordaliza, M. Natural products as leads to anticancer drugs. Clin. Transl. Oncol. 2007, 9, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, T.; Reker, D.; Schneider, P.; Schneider, G. Counting on natural products for drug design. Nat. Chem. 2016, 8, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Cragg, G.M.; Pezzuto, J.M. Natural Products as a Vital Source for the Discovery of Cancer Chemotherapeutic and Chemopreventive Agents. Med. Princ. Pract. 2016, 25, 41–59. [Google Scholar] [CrossRef]
- Zhao, W.; Cong, Y.; Li, H.M.; Li, S.; Shen, Y.; Qi, Q.; Zhang, Y.; Li, Y.Z.; Tang, Y.J. Challenges and potential for improving the druggability of podophyllotoxin-derived drugs in cancer chemotherapy. Nat. Prod. Rep. 2021, 38, 470–488. [Google Scholar] [CrossRef]
- Shah, Z.; Gohar, U.F.; Jamshed, I.; Mushtaq, A.; Mukhtar, H.; Zia-UI-Haq, M.; Ionut Toma, S.; Manea, R.; Moga, M.; Popovici, B. Podophyllotoxin: History, Recent Advances and Future Prospects. Biomolecules 2021, 11, 603. [Google Scholar] [CrossRef] [PubMed]
- Bohlin, L.; Rosén, B. Podophyllotoxin derivatives: Drug discovery and development. Drug Discov. Today 1996, 1, 343–351. [Google Scholar] [CrossRef]
- Gordaliza, M.; Castro, M.; Miguel del Corral, J.; San Feliciano, A. Antitumor Properties of Podophyllotoxin and Related Compounds. Curr. Pharm. Des. 2000, 6, 1811–1839. [Google Scholar] [CrossRef] [PubMed]
- Gordaliza, M.; García, P.A.; Miguel del Corral, J.M.; Castro, M.A.; Gómez-Zurita, M.A. Podophyllotoxin: Distribution, sources, applications and new cytotoxic derivatives. Toxicon 2004, 44, 441–459. [Google Scholar] [CrossRef] [PubMed]
- Guerram, M.; Jiang, Z.Z.; Zhang, L.Y. Podophyllotoxin, a medicinal agent of plant origin: Past, present and future. Chin. J. Nat. Med. 2012, 10, 161–169. [Google Scholar] [CrossRef]
- Baldwin, E.L.; Osheroff, N. Etoposide, topoisomerase II and cancer. Curr. Med. Chem. Anti-Cancer Agents 2005, 5, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Van Maanen, J.M.S.; Retèl, J.; De Vries, J.; Pinedo, H.M. Mechanism of Action of Antitumor Drug Etoposide: A Review. JNCI J. Natl. Cancer Inst. 1988, 80, 1526–1533. [Google Scholar] [CrossRef]
- Pitts, S.L.; Jablonksy, M.J.; Duca, M.; Dauzonne, D.; Monneret, C.; Arimondo, P.B.; Anklin, C.; Graves, D.E.; Osheroff, N. Contributions of the D-Ring to the Activity of Etoposide Against Human Topoisomerase IIα: Potential Interactions with DNA in the Ternary Enzyme-Drug-DNA Complex. Biochemistry 2011, 50, 5058. [Google Scholar] [CrossRef]
- Bromberg, K.D.; Burgin, A.B.; Osheroff, N. A Two-drug Model for Etoposide Action against Human Topoisomerase IIα. J. Biol. Chem. 2003, 278, 7406–7412. [Google Scholar] [CrossRef]
- Yu, X.; Che, Z.; Xu, H. Recent Advances in the Chemistry and Biology of Podophyllotoxins. Chem. A Eur. J. 2017, 23, 4467–4526. [Google Scholar] [CrossRef]
- Fan, H.; Zhu, Z.; Xian, H.; Wang, H.; Chen, B. Insight Into the Molecular Mechanism of Podophyllotoxin Derivatives as Anticancer Drugs. Front. Cell Dev. Biol. 2021, 9, 709075. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Gao, M.; Sun, Z.; Diao, Q.; Wang, P.; Gao, F. Recent advances of podophyllotoxin/epipodophyllotoxin hybrids in anticancer activity, mode of action, and structure-activity relationship: An update (2010–2020). Eur. J. Med. Chem. 2020, 208, 112830. [Google Scholar] [CrossRef]
- Zhang, X.; Rakesh, K.P.; Shantharam, C.S.; Manukumar, H.M.; Asiri, A.M.; Marwani, H.M.; Qin, H.L. Podophyllotoxin derivatives as an excellent anticancer aspirant for future chemotherapy: A key current imminent needs. Bioorg. Med. Chem. 2018, 26, 340–355. [Google Scholar] [CrossRef] [PubMed]
- Castro, M.Á.; Miguel del Corral, J.M.; García, P.A.; Victoria Rojo, M.; Bento, A.C.; Mollinedo, F.; Francesch, A.M.; San Feliciano, A. Lignopurines: A new family of hybrids between cyclolignans and purines. Synthesis and biological evaluation. Eur. J. Med. Chem. 2012, 58, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Castro, M.Á.; Miguel del Corral, J.M.; García, P.A.; Rojo, M.V.; De La Iglesia-Vicente, J.; Mollinedo, F.; Cuevas, C.; San Feliciano, A. Synthesis and biological evaluation of new podophyllic aldehyde derivatives with cytotoxic and apoptosis-inducing activities. J. Med. Chem. 2010, 53, 983–993. [Google Scholar] [CrossRef] [PubMed]
- Castro, M.A.; Miguel del Corral, J.M.; Gordaliza, M.; García, P.A.; Gómez-Zurita, M.A.; García-Grávalos, M.D.; De La Iglesia-Vicente, J.; Gajate, C.; An, F.; Mollinedo, F.; et al. Synthesis and Biological Evaluation of New Selective Cytotoxic Cyclolignans Derived from Podophyllotoxin. J. Med. Chem. 2004, 47, 1214–1222. [Google Scholar] [CrossRef]
- Sampath Kumar, H.M.; Herrmann, L.; Tsogoeva, S.B. Structural hybridization as a facile approach to new drug candidates. Bioorg. Med. Chem. Lett. 2020, 30, 127514. [Google Scholar] [CrossRef]
- Bansal, Y.; Silakari, O. Multifunctional compounds: Smart molecules for multifactorial diseases. Eur. J. Med. Chem. 2014, 76, 31–42. [Google Scholar] [CrossRef]
- Bérubé, G. An overview of molecular hybrids in drug discovery. Expert Opin. Drug Discov. 2016, 11, 281–305. [Google Scholar] [CrossRef]
- Fortin, S.; Bérubé, G. Advances in the development of hybrid anticancer drugs. Expert Opin. Drug Discov. 2013, 8, 1029–1047. [Google Scholar] [CrossRef]
- Choudhary, S.; Singh, P.K.; Verma, H.; Singh, H.; Silakari, O. Success stories of natural product-based hybrid molecules for multi-factorial diseases. Eur. J. Med. Chem. 2018, 151, 62–97. [Google Scholar] [CrossRef]
- Moss, G.P.; Mary, Q.; Road, M.E. Nomenclature of lignans and neolignans. Pure Appl. Chem 2000, 72, 1493–1523. [Google Scholar] [CrossRef]
- Kumar, A.; Kumar, V.; Alegria, A.E.; Malhotra, S. V Synthetic and Application Perspectives of Azapodophyllotoxins: Alternative Scaffolds of Podophyllotoxin. Curr. Med. Chem. 2011, 18, 3853–3870. [Google Scholar] [CrossRef]
- Yang, C.; Xie, Q.; Zeng, X.; Tao, N.; Xu, Y.; Chen, Y.; Wang, J. Novel hybrids of podophyllotoxin and formononetin inhibit the growth, migration and invasion of lung cancer cells. Bioorg. Chem. 2019, 85, 445–454. [Google Scholar] [CrossRef]
- Tay, K.-C.; Tan, L.T.-H.; Chan, C.K.; Hong, S.L.; Chan, K.-G.; Yap, W.H.; Pusparajah, P.; Lee, L.-H.; Goh, B.-H. Formononetin: A Review of Its Anticancer Potentials and Mechanisms. Front. Pharmacol. 2019, 10, 820. [Google Scholar] [CrossRef]
- Jiang, D.; Rasul, A.; Batool, R.; Sarfraz, I.; Hussain, G.; Tahir, M.M.; Qin, T.; Selamoglu, Z.; Ali, M.; Li, J.; et al. Potential Anticancer Properties and Mechanisms of Action of Formononetin. BioMed Res. Int. 2019, 2019, 5854315. [Google Scholar] [CrossRef]
- Ong, S.K.L.; Shanmugam, M.K.; Fan, L.; Fraser, S.E.; Arfuso, F.; Ahn, K.S.; Sethi, G.; Bishayee, A. Focus on Formononetin: Anticancer Potential and Molecular Targets. Cancers 2019, 11, 611. [Google Scholar] [CrossRef]
- Estrela, J.M.; Ortega, A.; Mena, S.; Rodriguez, M.L.; Asensi, M. Pterostilbene: Biomedical applications. Crit. Rev. Clin. Lab. Sci. 2013, 50, 65–78. [Google Scholar] [CrossRef]
- Kim, H.; Seo, K.H.; Yokoyama, W. Chemistry of Pterostilbene and Its Metabolic Effects. J. Agric. Food Chem. 2020, 68, 12836–12841. [Google Scholar] [CrossRef]
- Obrador, E.; Salvador-Palmer, R.; Jihad-Jebbar, A.; López-Blanch, R.; Dellinger, T.H.; Dellinger, R.W.; Estrela, J.M. Pterostilbene in Cancer Therapy. Antioxidants 2021, 10, 492. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, J.; Liu, L.; Zheng, C.; Wang, Y. Podophyllotoxin-pterostilbene fused conjugates as potential multifunctional antineoplastic agents against human uveal melanoma cells. RSC Adv. 2017, 7, 10601–10608. [Google Scholar] [CrossRef]
- Bai, G.; Zhao, D.; Ran, X.; Zhang, L.; Zhao, D. Novel Hybrids of Podophyllotoxin and Coumarin Inhibit the Growth and Migration of Human Oral Squamous Carcinoma Cells. Front. Chem. 2021, 8, 626075. [Google Scholar] [CrossRef]
- Gkionis, L.; Kavetsou, E.; Kalospyros, A.; Manousakis, D.; Garzon Sanz, M.; Butterworth, S.; Detsi, A.; Tirella, A. Investigation of the cytotoxicity of bioinspired coumarin analogues towards human breast cancer cells. Mol. Divers. 2021, 25, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Long, L.; Chen, Y.; Xu, Y.; Zhang, L. Design, synthesis and antineoplastic activity of novel hybrids of podophyllotoxin and indirubin against human leukaemia cancer cells as multifunctional anti-MDR agents. Bioorg. Med. Chem. Lett. 2018, 28, 1817–1824. [Google Scholar] [CrossRef] [PubMed]
- Eisenbrand, G.; Hippe, F.; Jakobs, S.; Muehlbeyer, S. Molecular mechanisms of indirubin and its derivatives: Novel anticancer molecules with their origin in traditional Chinese phytomedicine. J. Cancer Res. Clin. Oncol. 2004, 130, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, Z.; Wei, C.; Wang, J.; Xu, Y.; Bai, G.; Yao, Q.; Zhang, L.; Chen, Y. Anticancer potential of indirubins in medicinal chemistry: Biological activity, structural modification, and structure-activity relationship. Eur. J. Med. Chem. 2021, 223, 113652. [Google Scholar] [CrossRef] [PubMed]
- Blažević, T.; Heiss, E.H.; Atanasov, A.G.; Breuss, J.M.; Dirsch, V.M.; Uhrin, P. Indirubin and Indirubin Derivatives for Counteracting Proliferative Diseases. Evid.-Based Complement. Altern. Med. 2015, 2015, 654098. [Google Scholar] [CrossRef]
- Siddikuzzaman; Guruvayoorappan, C.; Berlin Grace, V.M. All trans retinoic acid and cancer. Immunopharmacol. Immunotoxicol. 2011, 33, 241–249. [Google Scholar] [CrossRef]
- Ni, X.; Hu, G.; Cai, X. The success and the challenge of all-trans retinoic acid in the treatment of cancer. Crit. Rev. Food Sci. Nutr. 2019, 59, S71–S80. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, J.; Liu, L.; Zheng, C.; Wang, Y. Synthesis and Antiproliferative Activity of Novel All-Trans-Retinoic Acid-Podophyllotoxin Conjugate towards Human Gastric Cancer Cells. Molecules 2017, 22, 628. [Google Scholar] [CrossRef]
- Sathish, M.; Kavitha, B.; Nayak, V.L.; Tangella, Y.; Ajitha, A.; Nekkanti, S.; Alari, A.; Shankaraiah, N. Synthesis of podophyllotoxin linked b-carboline congeners as potential anticancer agents and DNA topoisomerase II inhibitors. Eur. J. Med. Chem. 2018, 144, 557–571. [Google Scholar] [CrossRef]
- Moura, D.J.; Richter, M.F.; Boeira, J.M.; Pêgas Henriques, J.A.; Saffi, J. Antioxidant properties of β-carboline alkaloids are related to their antimutagenic and antigenotoxic activities. Mutagenesis 2007, 22, 293–302. [Google Scholar] [CrossRef]
- Osorio, E.J.; Robledo, S.M.; Bastida, J. Alkaloids with antiprotozoal activity. Alkaloids Chem. Biol. 2008, 66, 113–190. [Google Scholar]
- Alsayari, A.; Muhsinah, A.B.; Hassan, M.Z.; Ahsan, M.J.; Alshehri, J.A.; Begum, N. Aurone: A biologically attractive scaffold as anticancer agent. Eur. J. Med. Chem. 2019, 166, 417–431. [Google Scholar] [CrossRef] [PubMed]
- Mazziotti, I.; Petrarolo, G.; La Motta, C. Aurones: A Golden Resource for Active Compounds. Molecules 2021, 27, 2. [Google Scholar] [CrossRef] [PubMed]
- Paidakula, S.; Nerella, S.; Vadde, R.; Kamal, A. Design and synthesis of 4β-Acetamidobenzofuranone-podophyllotoxin hybrids and their anti-cancer evaluation. Bioorg. Med. Chem. Lett. 2019, 29, 2153–2156. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Feng, J.; Li, J.; Yu, Q.; Ji, J.; Wu, J.; Dai, W.; Guo, C. The gut microbiome-bile acid axis in hepatocarcinogenesis. Biomed. Pharmacother. 2021, 133, 111036. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Apte, U. Bile Acid Metabolism and Signaling in Cholestasis, Inflammation, and Cancer. Adv. Pharmacol. 2015, 74, 263–302. [Google Scholar] [PubMed]
- Park, J.; Choi, J.U.; Kim, K.; Byun, Y. Bile acid transporter mediated endocytosis of oral bile acid conjugated nanocomplex. Biomaterials 2017, 147, 145–154. [Google Scholar] [CrossRef]
- Gong, Y.; Zhang, X.; Zhang, Y.; Chu, F.; Li, G.; Zhang, H.; Xu, B.; Zhang, H.; Li, B.; Wang, P.; et al. Bile acids, carriers of hepatoma-targeted drugs? Pharmazie 2016, 71, 139–145. [Google Scholar]
- Cai, D.S.; Lou, S.Y.; Huo, S.; Yang, Y.Q.; Gao, F.; Pi, W.M.; Chen, K.D.; Wang, C.; Yang, X.Y.; Jiao, J.Y.; et al. Synthesis and biological activity evaluation of podophyllotoxin-linked bile acid derivatives as potential anti-liver cancer agents. Bioorg. Chem. 2022, 128, 106066. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Xu, B.; Yang, Y.; Zhang, X.; Fang, K.; Ma, T.; Wang, H.; Xue, N.; Chen, M.; Guo, W.; et al. Synthesis and biological evaluation of podophyllotoxin derivatives as selective antitumor agents. Eur. J. Med. Chem. 2018, 155, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Liu, P.; Li, P.; Huang, Z.; Yu, F.; Lei, T.; Chen, Y.; Cheng, Y.; Mu, Q.; Huang, H. Ligustrazine monomer against cerebral ischemia/reperfusion injury. Neural Regen. Res. 2015, 10, 832. [Google Scholar] [PubMed]
- Zou, J.; Gao, P.; Hao, X.; Xu, H.; Zhan, P.; Liu, X. Recent progress in the structural modification and phamacological activities of ligustrazine derivatives. Eur. J. Med. Chem. 2018, 147, 150–162. [Google Scholar] [CrossRef]
- Shao, H.; He, X.; Zhang, L.; Du, S.; Yi, X.; Cui, X.; Liu, X.; Huang, S.; Tong, R. Efficacy of Ligustrazine injection as adjuntive therapy in treating acute cerebral infarction: A systematic review and meta-analysis. Front. Pharmacol. 2021, 12, 761722. [Google Scholar] [CrossRef]
- Zi, C.; Yang, L.; Hu, Y.; Zhang, P.; Tang, H.; Zhang, B.; Shen, J.; Kong, Q.; Wang, Y.; Wang, X.; et al. Synthesis, antitumor activity, and molecular docking of (−)-epigallocatechin-3-gallate-4β-triazolopodophyllotoxin conjugates. J. Asian Nat. Prod. Res. 2021, 23, 772–780. [Google Scholar] [CrossRef]
- Zi, C.; Wang, Z.; Shi, J.; Shi, B.; Zhang, N.; Wu, Y.; Xie, Y.; Zhou, L.; Xiao, C.; Wang, X.; et al. Synthesis, cytotoxicity, and molecular docking of methylated (–)-epigallocatechin-3-gallate- 4 β -triazolopodophyllotoxin derivatives as novel antitumor agents. J. Chem. Res. 2021, 1, 4–10. [Google Scholar] [CrossRef]
- Bimonte, S.; Cascella, M. The Potential Roles of Epigallocatechin-3-Gallate in the Treatment of Ovarian Cancer: Current State of Knowledge. Drug Des. Dev. Ther. 2020, 14, 4245–4250. [Google Scholar] [CrossRef]
- Negri, A.; Naponelli, V.; Rizzi, F.; Bettuzzi, S. Molecular Targets of Epigallocatechin—Gallate (EGCG): A Special Focus on Signal Transduction and Cancer. Nutrients 2018, 10, 1936. [Google Scholar] [CrossRef]
- Pizzolato, J.F.; Saltz, L.B. The camptothecins. Lancet 2003, 361, 2235–2242. [Google Scholar] [CrossRef]
- Hsiang, Y.H.; Hertzberg, R.; Hecht, S.; Liu, L.F. Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. J. Biol. Chem. 1985, 260, 14873–14878. [Google Scholar] [CrossRef] [PubMed]
- Zi, C.; Yang, L.; Dong, F.; Kong, Q.; Ding, Z. Synthesis and antitumor activity of camptothecin-4β-triazolopodophyllotoxin. Nat. Prod. Res. 2019, 34, 2301–2309. [Google Scholar] [CrossRef] [PubMed]
- Duan, M.; Mahal, A.; Mohammed, B.; Zhu, Y.; Tao, H.; Mai, S.; Al-Haideri, M.; Zhu, Q. Synthesis and antitumor activity of new tetrahydrocurcumin derivatives via click reaction. Nat. Prod. Res. 2022, 36, 5268–5276. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.C.; Tsai, M.L.; Lai, C.S.; Wang, Y.J.; Ho, C.T.; Pan, M.H. Chemopreventative effects of tetrahydrocurcumin on human diseases. Food Funct. 2014, 5, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.S.; Ho, C.T.; Pan, M.H. The cancer chemopreventive and therapeutic potential of tetrahydrocurcumin. Biomolecules 2020, 10, 831. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.C.; Patchva, S.; Aggarwal, B.B. Therapeutic roles of curcumin: Lessons learned from clinical trials. Am. Assoc. Pharm. Sci. J. 2013, 15, 195–218. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Dong, F.; Yang, D.; Li, Y.; Ding, Z.; Zhou, J.; Jiang, Z.; Hu, J. Synthesis and anticancer activity of dimeric podophyllotoxin derivatives. Drug Des. Dev. Ther. 2018, 12, 3393–3406. [Google Scholar]
- Zempleni, J.; Wijeratne, S.S.K.; Hassan, Y.I. Biotin. Biofactors 2009, 35, 36–46. [Google Scholar] [CrossRef]
- León-Del-Río, A. Biotin in metabolism, gene expression, and human disease. J. Inherit. Metab. Dis. 2019, 42, 647–654. [Google Scholar] [CrossRef]
- Russell-Jones, G.; McTavish, K.; McEwan, J.; Rice, J.; Nowotnik, D. Vitamin-mediated targeting as a potential mechanism to increase drug uptake by tumours. J. Inorg. Biochem. 2004, 98, 1625–1633. [Google Scholar] [CrossRef]
- Chen, S.; Zhao, X.; Chen, J.; Chen, J.; Kuznetsova, L.; Wong, S.S.; Ojima, I. Mechanism-Based Tumor-Targeting Drug Delivery System. Validation of Efficient Vitamin Receptor-Mediated Endocytosis and Drug Release. Bioconjug. Chem. 2010, 21, 979–987. [Google Scholar] [CrossRef]
- Jiang, Z.; Yuan, S.; Hu, J. Design, Synthesis, and Biological Evaluation of Novel Biotinylated Podophyllotoxin Derivatives as Potential Antitumor Agents. Front. Chem. 2019, 7, 434. [Google Scholar]
- Gentry, A.C.; Pitts, S.L.; Jablonsky, M.J.; Bailly, C.; Graves, D.E.; Osheroff, N. Interactions between the Etoposide Derivative F14512 and Human Type II Topoisomerases: Implications for the C4 Spermine Moiety in Promoting Enzyme-mediated DNA Cleavage. Biochemistry 2011, 50, 3240–3249. [Google Scholar] [CrossRef] [PubMed]
- Pendleton, M.; Lindsey, R.H.; Felix, C.A.; Grimwade, D.; Osheroff, N. Topoisomerase II and leukemia. Ann. N. Y. Acad. Sci. 2014, 1310, 98. [Google Scholar] [CrossRef]
- Oviatt, A.A.; Kuriappan, J.A.; Minniti, E.; Vann, K.R.; Minarini, A.; De Vivo, M.; Osheroff, N.; Discovery, D.; Morego, V. Polyamine-containing etoposide derivatives as poisons of human type II topoisomerases: Differential effects on topoisomerase IIa and IIb. Bioorg. Med. Chem. Lett. 2018, 28, 2961–2968. [Google Scholar] [CrossRef] [PubMed]
- Tierny, D.; Serres, F.; Segaoula, Z.; Bemelmans, I.; Bouchaert, E.; Pétain, A.; Brel, V.; Couffin, S.; Marchal, T.; Nguyen, L.; et al. Phase i Clinical Pharmacology Study of F14512, a New Polyamine-Vectorized Anticancer Drug, in Naturally Occurring Canine Lymphoma. Clin. Cancer Res. 2015, 21, 5314–5323. [Google Scholar] [CrossRef]
- Boyé, P.; Floch, F.; Serres, F.; Segaoula, Z.; Hordeaux, J.; Pascal, Q.; Coste, V.; Courapied, S.; Bouchaert, E.; Rybicka, A.; et al. Randomized, double-blind trial of F14512, a polyaminevectorized anticancer drug, compared with etoposide phosphate, in dogs with naturally occurring lymphoma. Oncotarget 2020, 11, 671–686. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Yang, J.; Seeberger, P.H.; Yin, J. Glycoconjugates for glucose transporter-mediated cancer-specific targeting and treatment. Carbohydr. Res. 2020, 498, 108195. [Google Scholar] [CrossRef]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Calvaresi, E.C.; Hergenrother, P.J. Glucose conjugation for the specific targeting and treatment of cancer. Chem. Sci. 2013, 4, 2319–2333. [Google Scholar] [CrossRef]
- Arafa, H.M.M. Possible contribution of β-glucosidase and caspases in the cytotoxicity of glufosfamide in colon cancer cells. Eur. J. Pharmacol. 2009, 616, 58–63. [Google Scholar] [CrossRef]
- Cheng, J.; Zhao, W.; Yao, H.; Shen, Y.; Zhang, Y.; Li, Y.; Wongprasert, K.; Tang, Y. Discovery of 4,6-O-Thenylidene-β-D-glucopyranoside-(2″-acetamido,3″-acetyl-di-S-5-fluorobenzothizole/5-fluorobenzoxazole)-4′-demethylepipodophyllotoxin as Potential Less Toxic Antitumor Candidate Drugs by Reducing DNA Damage and Less Inhibition of. J. Med. Chem. 2020, 63, 2877–2893. [Google Scholar] [CrossRef]
- Zi, C.T.; Yang, L.; Kong, Q.H.; Li, H.M.; Yang, X.Z.; Ding, Z.T.; Jiang, Z.H.; Hu, J.M.; Zhou, J. Glucoside Derivatives of Podophyllotoxin: Synthesis, Physicochemical Properties, and Cytotoxicity. Drug Des. Dev. Ther. 2019, 13, 3683–3692. [Google Scholar] [CrossRef] [PubMed]
- Zi, C.; Yang, L.; Zhang, B.; Li, Y.; Ding, Z.; Jiang, Z.; Hu, J.; Zhou, J. Synthesis and Cytotoxicities of Novel Podophyllotoxin Xyloside Derivatives. Nat. Prod. Commun. 2019, 14, 1934578X19860668. [Google Scholar] [CrossRef]
- San Feliciano, A.; Miguel Del Corral, J.M.; Gordaliza, M.; Castro, M.A. 13C NMR data for several cyclolignans. Magn. Reson. Chem. 1993, 31, 868–875. [Google Scholar] [CrossRef]
- Zi, C.; Yang, L.; Gao, W.; Li, Y.; Zhou, J.; Ding, Z. Click Glycosylation for the Synthesis of 1,2,3- Triazole-Linked Picropodophyllotoxin Glycoconjugates and Their Anticancer Activity. Med. Chem. Drug Discov. 2017, 2, 5038–5044. [Google Scholar] [CrossRef]
- Nerella, S.; Kankala, S.; Gavaji, B. Synthesis of podophyllotoxin-glycosyl triazoles via click protocol mediated by silver (I)-N -heterocyclic carbenes and their anticancer evaluation as topoisomerase-II inhibitors. Nat. Prod. Res. 2021, 35, 9–16. [Google Scholar] [CrossRef]
- Ali, I.; Lone, M.; Al-Othman, Z.; Al-Warthan, A.; Sanagi, M. Heterocyclic Scaffolds: Centrality in Anticancer Drug Development. Curr. Drug Targets 2015, 16, 711–734. [Google Scholar] [CrossRef]
- Bozorov, K.; Zhao, J.; Aisa, H.A. 1,2,3-Triazole-containing hybrids as leads in medicinal chemistry: A recent overview. Bioorg. Med. Chem. 2019, 27, 3511–3531. [Google Scholar] [CrossRef]
- Beltran-Hortelano, I.; Alcolea, V.; Font, M.; Pérez-Silanes, S. The role of imidazole and benzimidazole heterocycles in Chagas disease: A review. Eur. J. Med. Chem. 2020, 206, 112692. [Google Scholar] [CrossRef]
- Verma, A.; Joshi, S.; Singh, D. Imidazole: Having versatile biological activities. J. Chem. 2013, 2013, 329412. [Google Scholar] [CrossRef]
- Shalini, K.; Sharma, P.K.; Kumar, N. Imidazole and its biological activities: A review. Der Chem. Sin. 2010, 1, 36–47. [Google Scholar]
- Cao, B.O.; Yang, S.; Li, W.; Chen, H.; Chen, Y.; Liu, Y.; Liu, B.I.N. GMZ-1 is a podophyllotoxin derivative that suppresses growth and induces apoptosis in adriamycin-resistant K562 / A02 cells through modulation of MDR1 expression. Mol. Med. Rep. 2018, 17, 474–478. [Google Scholar] [CrossRef]
- Yin, M.; Fang, Y.; Sun, X.; Xue, M.; Zhang, C.; Zhu, Z.; Meng, Y.; Kong, L.; Myint, Y.Y.; Li, Y.; et al. Synthesis and anticancer activity of podophyllotoxin derivatives with nitrogen-containing heterocycles. Front. Chem. 2023, 11, 1191498. [Google Scholar] [CrossRef] [PubMed]
- Ayati, A.; Emami, S.; Asadipour, A.; Shafiee, A.; Foroumadi, A. Recent applications of 1,3-thiazole core structure in the identification of new lead compounds and drug discovery. Eur. J. Med. Chem. 2015, 97, 699–718. [Google Scholar] [CrossRef] [PubMed]
- Ayati, A.; Emami, S.; Moghimi, S.; Foroumadi, A. Thiazole in the targeted anticancer drug discovery. Future Med. Chem. 2019, 11, 1929–1952. [Google Scholar] [CrossRef] [PubMed]
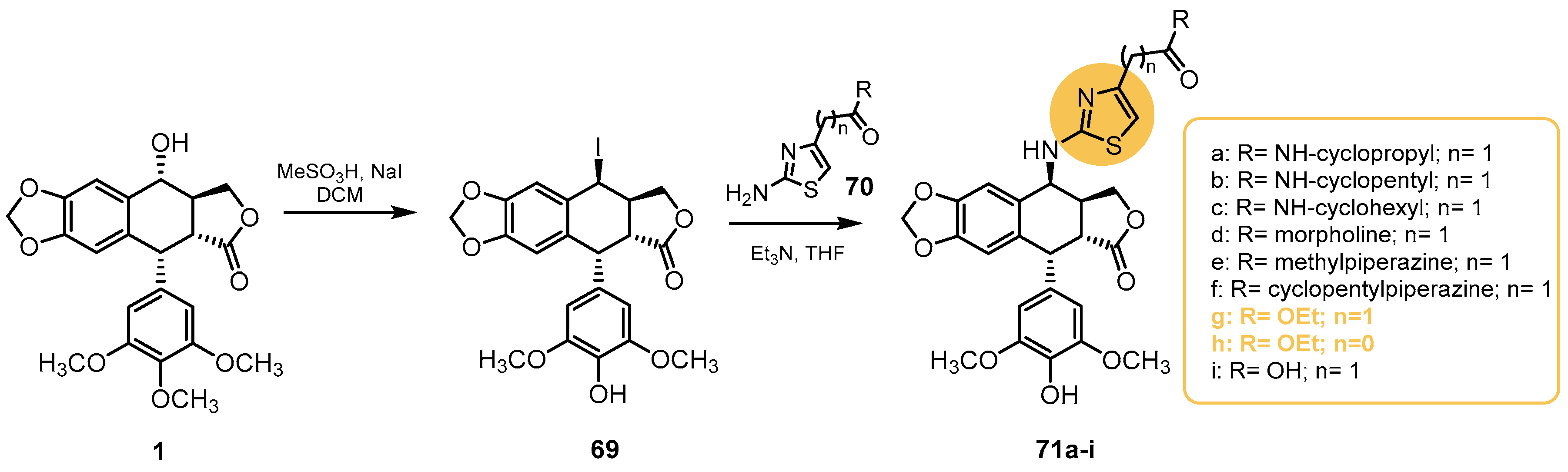
- Sang, C.; Tian, H.; Chen, Y.; Liu, J.; Chen, S.; Hui, L. Synthesis and biological evaluation of 4b-(thiazol-2-yl)amino-4′-O demethyl-4-deoxypodophyllotoxins as topoisomerase-II inhibitors. Bioorg. Med. Chem. Lett. 2018, 28, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Vágó, I.; Kalaus, G.; Greiner, I.; Kajtár-Peredy, M.; Brlik, J.; Szabó, L.; Szántay, C. Synthesis of vinca alkaloids and related compounds. Heterocycles 2001, 55, 873–880. [Google Scholar]
- Pairas, G.N.; Perperopoulou, F.; Tsoungas, P.G.; Varvounis, G. The Isoxazole Ring and Its N-Oxide: A Privileged Core Structure in Neuropsychiatric Therapeutics. ChemMedChem 2017, 12, 408–419. [Google Scholar] [CrossRef]
- Ji Ram, V.; Sethi, A.; Nath, M.; Pratap, R. Five-Membered Heterocycles. In The Chemistry of Heterocycles; Elsevier Inc.: Amsterdam, The Netherlands, 2019; pp. 149–478. ISBN 9780081010334. [Google Scholar]
- Yang, R.; Zhang, Y.; Xu, H. Synthesis of novel isoxazoline-containing podophyllotoxin/2′(2′6′)-(di) halogenopodophyllotoxin derivatives and their insecticidal/acaricidal activities. Bioorg. Med. Chem. Lett. 2018, 28, 1410–1416. [Google Scholar] [CrossRef]
- Fan, L.; Guo, Y.; Zhi, X.; Yu, X.; Xu, H. Stereoselective synthesis of 2α-chloropicropodophyllotoxins and insecticidal activity of their esters against oriental armyworm, mythimna separata walker. J. Agric. Food Chem. 2014, 62, 3726–3733. [Google Scholar] [CrossRef]
- Che, Z.; Yu, X.; Fan, L.; Xu, H. Insight into dihalogenation of E-ring of podophyllotoxins, and their acyloxyation derivatives at the C4 position as insecticidal agents. Bioorg. Med. Chem. Lett. 2013, 23, 5592–5598. [Google Scholar] [CrossRef]
- Zefirov, N.A.; Nurieva, E.V.; Mamaeva, A.V.; Vasilenko, D.A.; Sadovnikov, K.S.; Averina, E.B.; Kolchanova, A.Y.; Milaeva, E.R.; Zefirova, O.N. New podophyllotoxin and epipodophyllotoxin derivatives with substituted isoxazole fragments. Russ. Chem. Bull. 2023, 72, 1029–1035. [Google Scholar] [CrossRef]
- Huang, D.; Huang, M.; Liu, W.; Liu, A.; Liu, X.; Chen, X.; Pei, H.; Sun, J.; Yin, D.; Wang, X. Design, synthesis and biological evaluation of 1H-pyrazole-5-carboxamide derivatives as potential fungicidal and insecticidal agents. Chem. Pap. 2017, 71, 2053–2061. [Google Scholar] [CrossRef]
- Baraldi, P.G.; Simoni, D.; Moroder, F.; Manfredini, S.; Mucchi, L.; Dalla Vecchia, F.; Orsolini, P. Synthesis of 2-(5′-substituted isoxazol-3′-yl)-4-oxo-3-thiazolidinylalkanoic acids. J. Heterocycl. Chem. 1982, 19, 557–560. [Google Scholar] [CrossRef]
- Jackson, P.L.; Hanson, C.D.; Farrell, A.K.; Butcher, R.J.; Stables, J.P.; Eddington, N.D.; Scott, K.R. Enaminones An explanation of anticonvulsant activity and toxicity per Linus Pauling’s clathrate hypothesis. Eur. J. Med. Chem. 2012, 51, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Zhang, L.H.; Dolbier, W.R. A convenient new method for the bromination of deactivated aromatic compounds. Synlett 1999, 1999, 1245–1246. [Google Scholar] [CrossRef]
- Datta, R.L.; Chatterjbe, N.R. Halogenation. XVIII. Direct ionization by means of iodine and nitric acid. J. Am. Chem. Soc. 1919, 41, 292–295. [Google Scholar] [CrossRef]
- Trukhacheva, L.A.; Levina, V.I.; Grigor’ev, N.B.; Arzamastsev, A.P.; Dalinger, I.L.; Vatsadze, I.A.; Popova, G.P.; Shevelev, S.A.; Granik, V.G. Kinetics of hydrolysis of five-membered C-nitroheterocycles: Pyrazole, imidazole, 1,2,4-triazole, and isoxazole derivatives. Russ. Chem. Bull. 2005, 54, 2813–2819. [Google Scholar] [CrossRef]
- Dheer, D.; Singh, V.; Shankar, R. Medicinal attributes of 1,2,3-triazoles: Current developments. Bioorg. Chem. 2017, 71, 30–54. [Google Scholar] [CrossRef] [PubMed]
- Bonandi, E.; Christodoulou, M.S.; Fumagalli, G.; Perdicchia, D.; Rastelli, G.; Passarella, D. The 1,2,3-triazole ring as a bioisostere in medicinal chemistry. Drug Discov. Today 2017, 22, 1572–1581. [Google Scholar] [CrossRef]
- Jiang, X.; Hao, X.; Jing, L.; Wu, G.; Kang, D.; Liu, X.; Zhan, P. Recent applications of click chemistry in drug discovery. Expert Opin. Drug Discov. 2019, 14, 779–789. [Google Scholar] [CrossRef]
- Kaur, J.; Saxena, M.; Rishi, N. An Overview of Recent Advances in Biomedical Applications of Click Chemistry. Bioconjug. Chem. 2021, 32, 1455–1471. [Google Scholar] [CrossRef]
- Kolb, H.C.; Sharpless, K.B. The growing impact of click chemistry on drug discovery. Drug Discov. Today 2003, 8, 1128–1137. [Google Scholar] [CrossRef]
- Hou, W.; Zhang, G.; Luo, Z.; Su, L.; Xu, H. Click chemistry-based synthesis and cytotoxic activity evaluation of 4α-triazole acetate podophyllotoxin derivatives. Chem. Biol. Drug Des. 2019, 93, 473–483. [Google Scholar] [CrossRef]
- Vishnuvardhan, M.V.P.S.; Chandrasekhar, K.; Nayak, V.L.; Sayeed, I.B.; Alarifi, A.; Kamal, A. Click chemistry-assisted synthesis of triazolo linked podophyllotoxin conjugates as tubulin. Med. Chem. Commun. 2017, 8, 1817–1823. [Google Scholar] [CrossRef]
- Reddy, V.G.; Bonam, S.R.; Reddy, T.S.; Akunuri, R.; Naidu, V.G.M.; Nayak, V.L.; Bhargava, S.K.; Kumar, H.M.S.; Srihari, P.; Kamal, A. 4b-amidotriazole linked podophyllotoxin congeners: DNA topoisomerase-IIa inhibition and potential anticancer agents for prostate cancer. Eur. J. Med. Chem. 2018, 20, 595–611. [Google Scholar] [CrossRef]
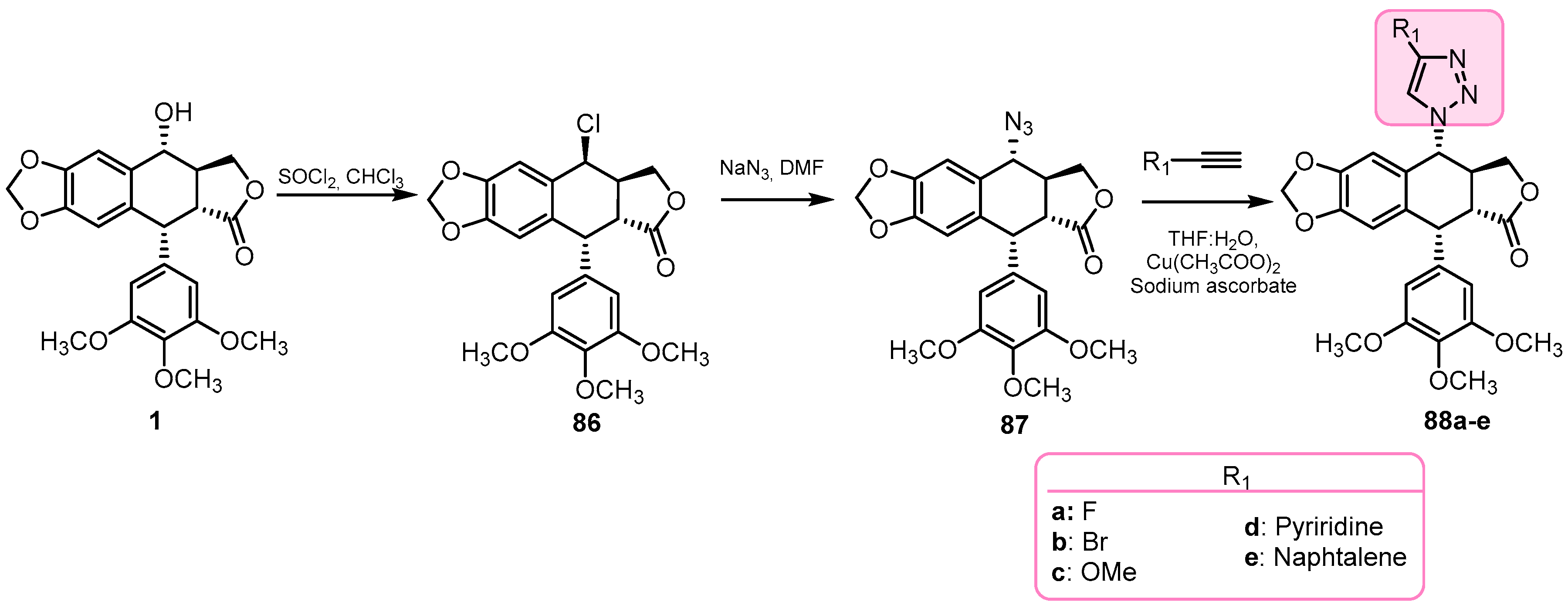
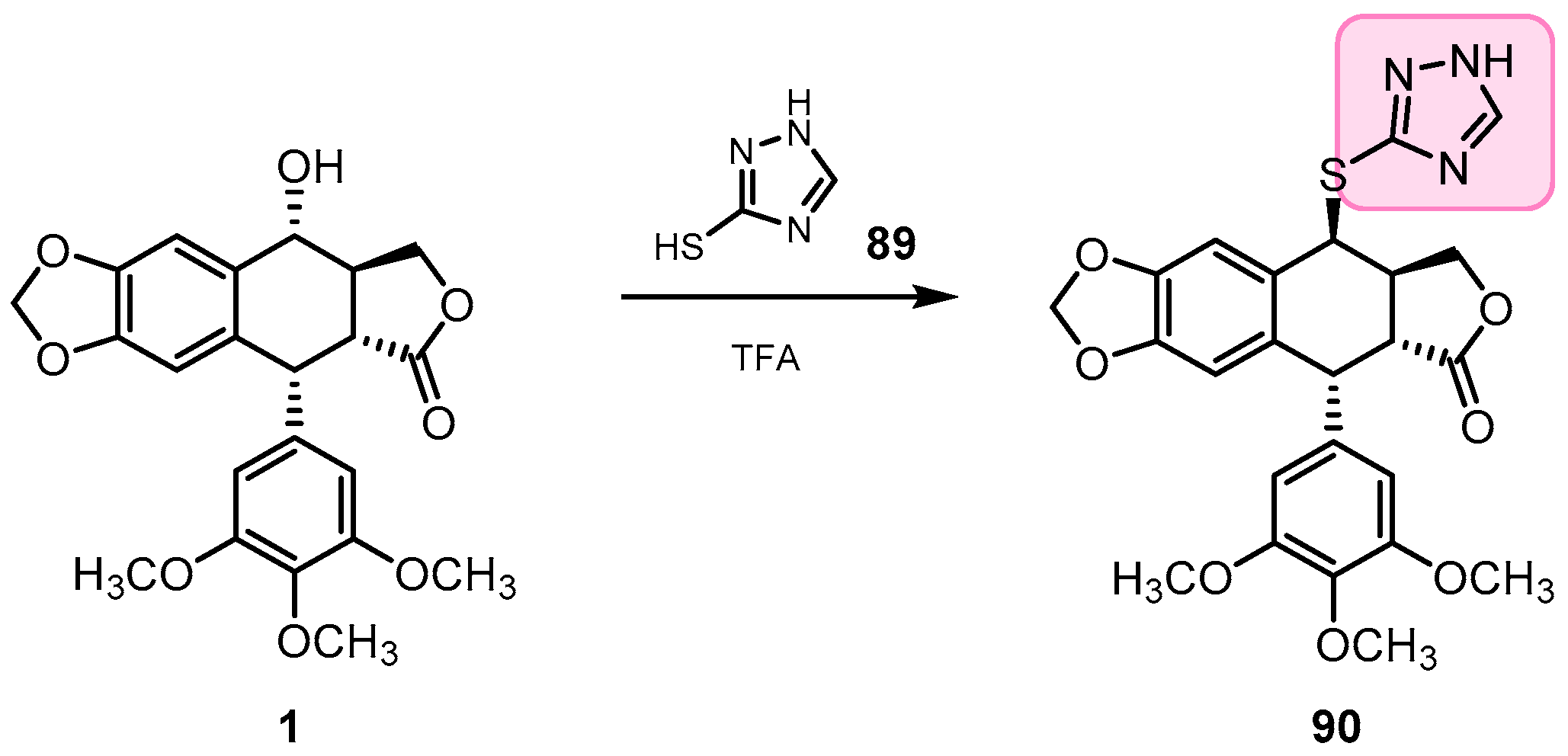
- Li, J.L.; Zhao, W.; Zhou, C.; Zhang, Y.X.; Li, H.M.; Tang, Y.L.; Liang, X.H.; Chen, T.; Tang, Y.J. Comparison of carbon-sulfur and carbon-amine bond in therapeutic drug: 4β-S-aromatic heterocyclic podophyllum derivatives display antitumor activity. Nat. Sci. Rep. 2015, 5, 14814. [Google Scholar] [CrossRef]
- Zhao, W.; Zhou, C.; Guan, Z.; Yin, P.; Chen, F.; Tang, Y. Structural insights into the inhibition of tubulin by the antitumor agent 4b-(1,2,4-triazol-3-ythio)-4-deoxypodophyllotoxin. ACS Chem. Biol. 2017, 12, 745–752. [Google Scholar] [CrossRef]
- Chawla, G.; Naaz, B.; Siddiqui, A. Exploring 1,3,4-Oxadiazole Scaffold for Anti-inflammatory and Analgesic Activities: A Review of Literature from 2005–2016. Mini-Rev. Med. Chem. 2018, 18, 216–233. [Google Scholar] [CrossRef]
- Bajaj, S.; Asati, V.; Singh, J.; Roy, P.P. 1,3,4-Oxadiazoles: An emerging scaffold to target growth factors, enzymes and kinases as anticancer agents. Eur. J. Med. Chem. 2015, 97, 124–141. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, A.; Pathak, D.; Shah, K. 1,3,4-oxadiazole and its derivatives: A review on recent progress in anticancer activities. Chem. Biol. Drug Des. 2021, 97, 572–591. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Wu, L.; Xin, W.Q.; Chen, X.; Hu, K. Synthesis and biological evaluation of novel 4β-(1,3,4-oxadiazole-2-amino)-podophyllotoxin derivatives. Bioorg. Med. Chem. Lett. 2012, 22, 4778–4782. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Liu, Y.; Li, L.; Zhao, Y.; Li, Z.; Wu, C.; Chen, L.; Hu, K. OAMDP, a novel podophyllotoxin derivative, induces apoptosis, cell cycle arrest and autophagy in hepatoma HepG2 cells. Cell Biollogy Int. 2018, 42, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Sun, W.; Wang, X.; Zhang, Y.; Qiu, H.; Qi, J.; Pang, Y.; Lu, G.; Wang, X.; Yu, F.; et al. Design, Synthesis, Biological evaluation and 3D−QSAR Analysis of podophyllotoxin dioxazole combination as tubulin targeting anticancer agents. Chem. Biol. Drug Des. 2017, 90, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Xu, X.; Ma, Y.; Luo, Y.; Wang, Z.; Yang, M. Discovering Podophyllotoxin Derivatives as Potential Anti-Tubulin Agents: Design, Synthesis and Biological Evaluation. Biol. Chem. Chem. Biol. 2020, 5, 10526–10536. [Google Scholar] [CrossRef]
- Ling, Y.; Hao, Z.Y.; Liang, D.; Zhang, C.L.; Liu, Y.F.; Wang, Y. The expanding role of pyridine and dihydropyridine scaffolds in drug design. Drug Des. Dev. Ther. 2021, 15, 4289–4338. [Google Scholar] [CrossRef]
- Tahir, T.; Ashfaq, M.; Saleem, M.; Rafiq, M.; Shahzad, M.I.; Kotwica-Mojzych, K.; Mojzych, M. Pyridine Scaffolds, Phenols and Derivatives of Azo Moiety: Current Therapeutic Perspectives. Molecules 2021, 26, 4872. [Google Scholar] [CrossRef]
- Wang, H. Podophyllum derivatives containing fluorine atom in the 3-position of 2-aminopyridine improved the antitumor activity by inducing P53-dependent apoptosis. Med. Chem. Res. 2017, 26, 1279–1290. [Google Scholar] [CrossRef]
- Wang, H.; Feng, J.; Zhou, T.; Wei, L.; Zhou, J. International Journal of Biochemistry and Cell Biology P-3F, a microtubule polymerization inhibitor enhances P53 stability through the change in localization of RPS27a. Int. J. Biochem. Cell Biol. 2017, 92, 53–62. [Google Scholar] [CrossRef]
- Li, Y.; Wang, T.; Sun, Y.; Huang, T.; Li, C.; Fu, Y.; Li, Y.; Li, C. p53-Mediated PI3K/AKT/mTOR Pathway Played a Role in Ptox Dpt -Induced EMT Inhibition in Liver Cancer Cell Lines. Oxidative Med. Cell. Longev. 2019, 2019, 2531493. [Google Scholar]
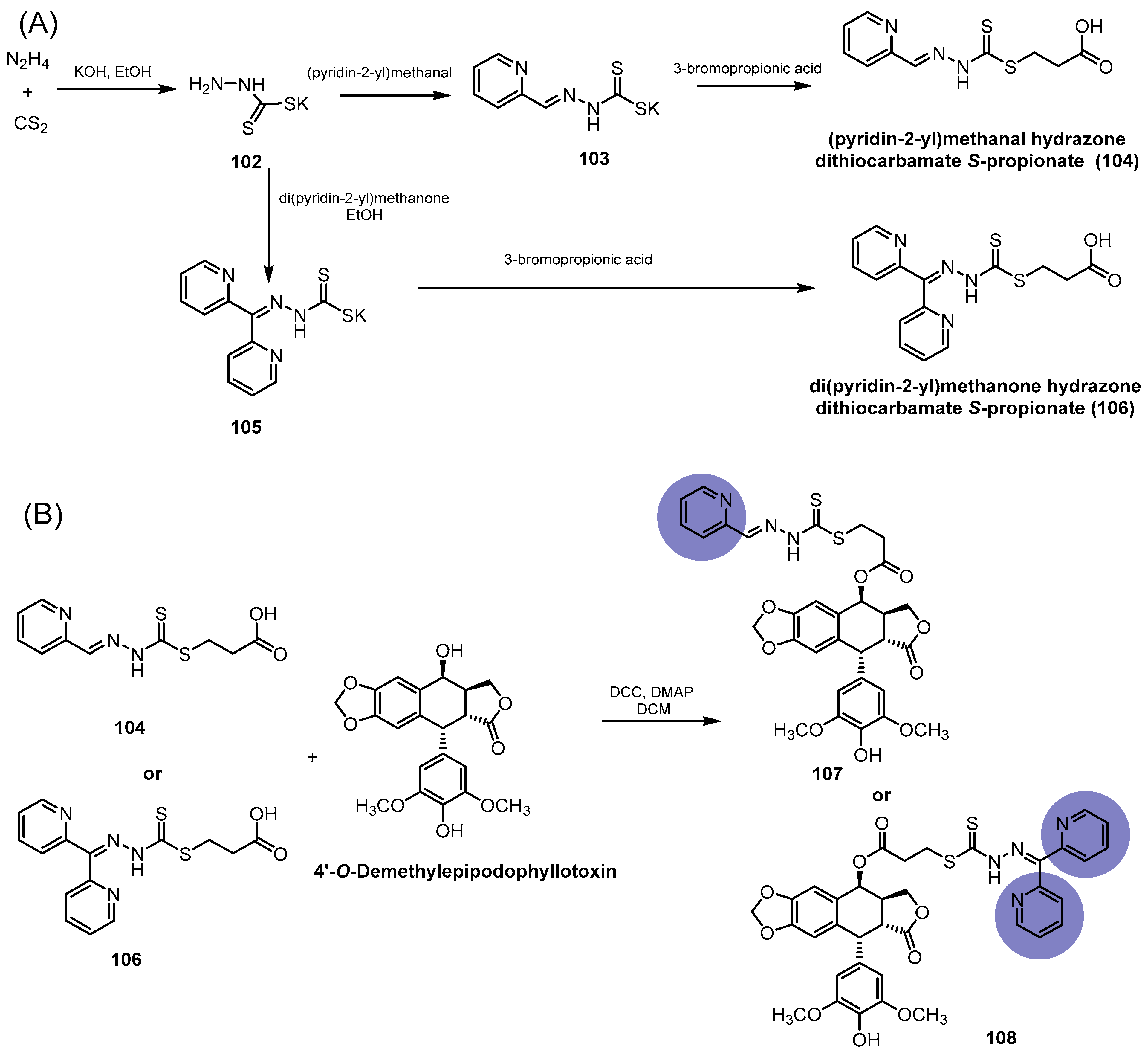
- Wang, T.; Fu, Y.; Huang, T.; Liu, Y.; Wu, M.; Yuan, Y.; Li, S.; Li, C. Copper Ion Attenuated the Antiproliferative Activity of Di-2-pyridylhydrazone Dithiocarbamate Derivative; However, There Was a Lack of Correlation between ROS Generation and Antiproliferative Activity. Molecules 2016, 21, 1088. [Google Scholar] [CrossRef]
- Li, Y.; Huang, T.; Fu, Y.; Wang, T.; Zhao, T.; Guo, S.; Sun, Y.; Yang, Y.; Id, C.L. Antitumor activity of a novel dual functional podophyllotoxin derivative involved PI3K/AKT/mTOR pathway. PLoS ONE 2019, 14, e0215886. [Google Scholar] [CrossRef]
- Jain, A.; Chaudhary, J.; Khaira, H.; Chopra, B.; Dhingra, A. Piperazine: A Promising Scaffold with Analgesic and Anti-inflammatory Potential. Drug Res. 2021, 71, 62–72. [Google Scholar] [CrossRef]
- Zhang, R.H.; Guo, H.Y.; Deng, H.; Li, J.; Quan, Z.S. Piperazine skeleton in the structural modification of natural products: A review. J. Enzyme Inhib. Med. Chem. 2021, 36, 1165–1197. [Google Scholar] [CrossRef]
- Singh, K.; Siddiqui, H.H.; Shakya, P.; Bagga, P.; Kumar, A.; Khalid, M.; Arif, M.; Alok, S. Piperazine-A Biologically Active Scaffold. Int. J. Pharceutical Sci. Res. 2015, 6, 4145–4158. [Google Scholar]
- Markandewar, R.A.; Baseer, M.A. Exploring Pharmacological Significance of Piperazine Scaffold. World J. Pharm. Res. 2016, 5, 1409–1420. [Google Scholar]
- Sun, W.; Ji, Y.; Wan, Y.; Han, H.; Lin, H.; Qi, J.; Wang, X.; Yang, Y. Design and synthesis of piperazine acetate podophyllotoxin ester derivatives targeting tubulin depolymerization as new anticancer agents. Bioorg. Med. Chem. Lett. 2017, 27, 4066–4074. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, T.; Jin, X.; Lin, K.; Dai, X.; Gao, T.; Huang, G.; Fan, M.; Ma, L.; Liu, Z.; et al. Synthesis and biological evaluation of cytotoxic activity of novel podophyllotoxin derivatives incorporating piperazinyl-cinnamic amide moieties. Bioorg. Chem. 2022, 123, 105761. [Google Scholar] [CrossRef] [PubMed]
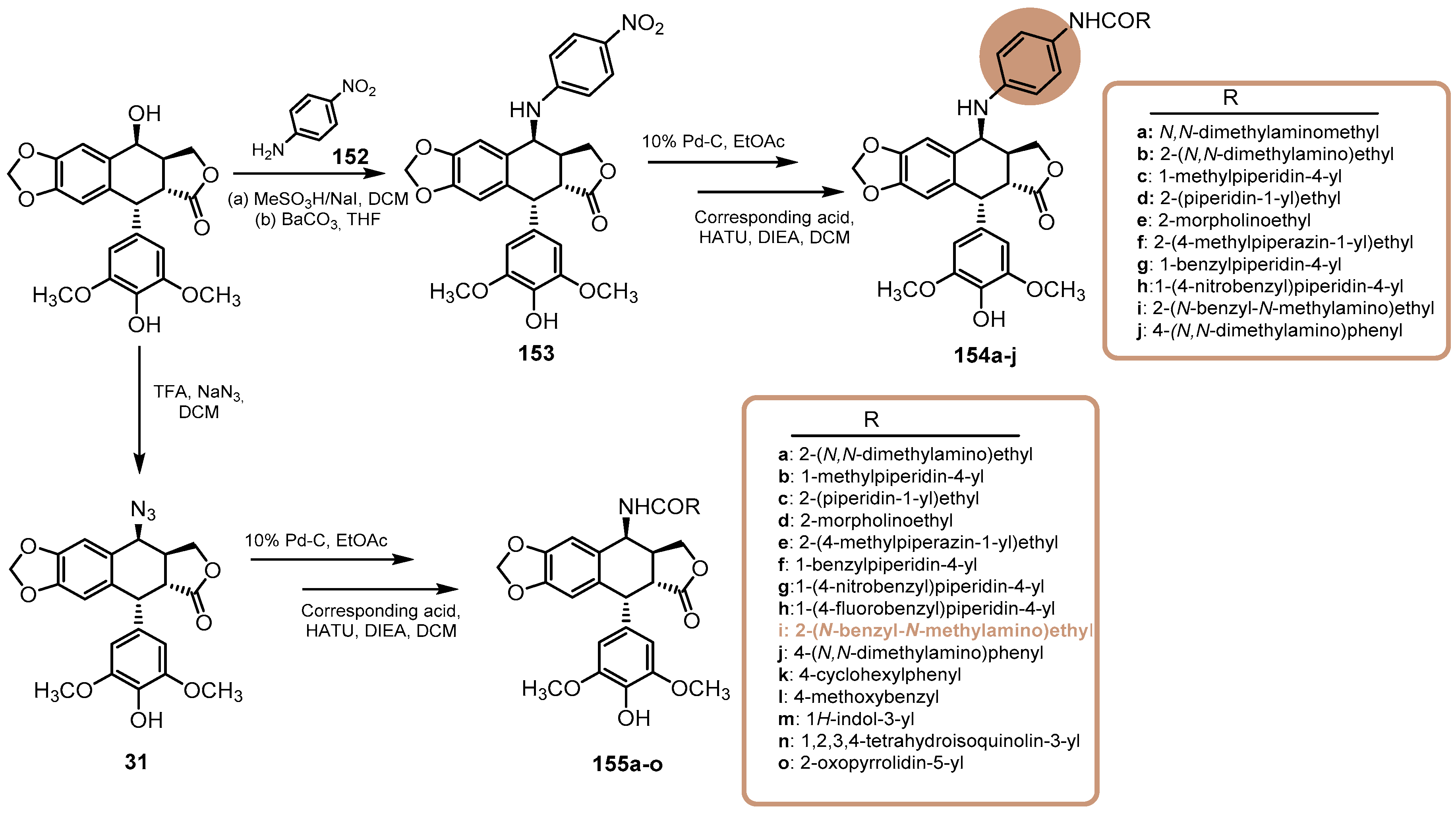
- Wu, Z.R.; Deng, W.; He, D. Synthesis and biological evaluation of prodrugs for nitroreductase based 4-β-amino-4′-Demethylepipodophyllotoxin as potential anticancer agents. Med. Chem. Res. 2022, 31, 1099–1108. [Google Scholar] [CrossRef]
- Dadashpour, S.; Emami, S. Indole in the target-based design of anticancer agents: A versatile scaffold with diverse mechanisms. Eur. J. Med. Chem. 2018, 150, 9–29. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Lin, H.; He, D.; Ren, Y.; Sun, W.; Liang, L. Novel Podophyllotoxin Derivatives as Potential Tubulin Inhibitors: Design, Synthesis, and Antiproliferative Activity Evaluation. Chem. Biodivers. 2018, 15, e1800289. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zeng, X.; Ren, X.; Tao, N.; Yang, C.; Xu, Y.; Chen, Y.; Wang, J. Design, synthesis, and biological evaluation of indole carboxylic acid esters of podophyllotoxin as antiproliferative agents. Med. Chem. Res. 2019, 28, 81–94. [Google Scholar] [CrossRef]
- Zhao, W.; He, L.; Xiang, T.; Tang, Y. Discover 4b-NH-(6-aminoindole)-4-desoxy-podophyllotoxin with nanomolar-potency antitumor activity by improving the tubulin binding affinity on the basis of a potential binding site nearby colchicine domain. Eur. J. Med. Chem. 2019, 170, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhao, W.; Tang, Y. Discover the leading compound of 4b-S-(5-fluorobenzoxazole)-4deoxy-4′-demethylepipodophyllotoxin with millimolar potency toxicity by modifiyng the molecule structure of 4′-demethylepipodophyllotoxin. Eur. J. Med. Chem. 2018, 158, 951–964. [Google Scholar] [CrossRef]
- Zang, L.; Zheng, J.; Rong, Y.; Chengli, Y.; Long, L.; Xu, Y.; Chen, Y.; Wang, J.; Yao, Q. Synthesis, antitumor evaluation and molecular docking study of a novel podophyllotoxin-Ionidamine hybrid. Med. Chem. Res. 2018, 27, 2231–2238. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, L.; Zheng, C.; Wang, Y.; Nie, X.; Shi, D. Synthesis and biological evaluation of novel podophyllotoxin-NSAIDs conjugates as multifunctional anti-MDR agents against resistant human hepatocellular carcinoma Bel-7402/5-FU cells. Eur. J. Med. Chem. 2017, 131, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Sun, G.; Sun, X.; Li, F.; Zhao, L.; Zhong, R.; Peng, Y. The potential of lonidamine in combination with chemotherapy and physical therapy in cancer treatment. Cancers 2020, 12, 3332. [Google Scholar] [CrossRef]
- Bhutia, Y.D.; Babu, E.; Ganapathy, V. Re-programming tumour cell metabolism to treat cancer: No lone target for lonidamine. Biochem. J. 2016, 473, 1503–1506. [Google Scholar] [CrossRef]
- Nath, K.; Guo, L.; Nancolas, B.; Nelson, D.S.; Shestov, A.A.; Lee, S.-C.; Roman, J.; Zhou, R.; Leeper, D.B.; Halestrap, A.P.; et al. Mechanism of antineoplastic activity of lonidamide. Biochim. Biophys. Acta 2016, 1866, 151–162. [Google Scholar]
- Zhang, L.; Rong, Y.; Zheng, J.; Yang, C.; Chen, Y.; Wang, J.; Wei, G. Desing, synthesis and biological evaluation of novel nitric oxide-donating podophyllotoxin derivatives as potential antiproliferative agents against multi-drug resistant leukemia cells. RSC Adv. 2018, 8, 34266–34274. [Google Scholar] [CrossRef]
- Yang, Y.; Qi, S.; Shi, R.; Yao, J.; Wang, L.; Yuan, H. Induction of apoptotic DNA fragmentation mediated by mitochondrial pathway with caspase-3-dependent BID cleavage in human gastric cancer cells by a new nitroxyl spin-labeled derivative of podophyllotoxin. Biomed. Pharmacother. 2017, 90, 131–138. [Google Scholar] [CrossRef]
- Tian, X.; Wang, Y.; Yang, M.; Chen, Y. Synthesis and antitumor activity of spin labeled derivatives of podophyllotoxin. Life Sci. 1996, 60, 511–517. [Google Scholar] [CrossRef]
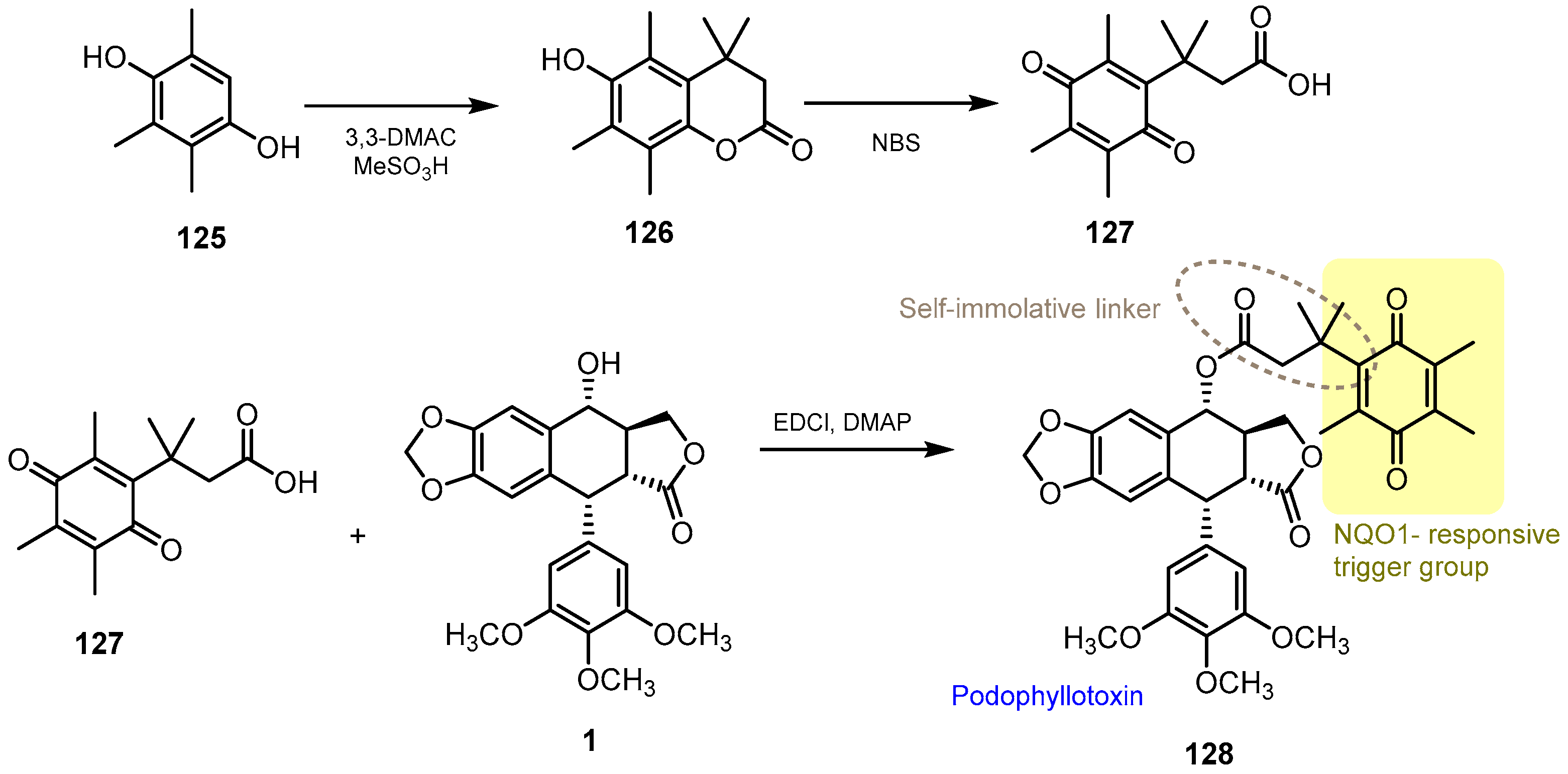
- Qu, Y.; Zhang, C.; Ma, X.; Gao, Y.; Liu, J.; Wu, L. Synthesis and biological evaluation of NQO1-activated prodrugs of podophyllotoxin as antitumor agents. Bioorg. Med. Chem. 2020, 28, 115821. [Google Scholar] [CrossRef]
- Belinsky, M.; Jaiswal, A.K. NAD(P)H:Quinone oxidoreductase I (DT-diaphorase) expression in normal and tumor tissues. Cancer Metastasis Rev. 1993, 12, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Siegel, D.; Gustafson, D.L.; Dehn, D.L.; Han, J.Y.; Boonchoong, P.; Berliner, L.J.; Ross, D. NAD(P)H:quinone oxidoreductase 1: Role as a superoxide scavenger. Mol. Pharmacol. 2004, 65, 1238–1247. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Sun, F.; Meng, J.; Cao, X.; Zhao, S.; Wang, C.; Li, L.; Pei, J. Design, semi-synthesis and bioactivity evaluation of novel podophyllotoxin derivatives as potent anti-tumor agents. Bioorg. Chem. 2022, 126, 105906. [Google Scholar] [CrossRef] [PubMed]
- Salehi, B.; Quispe, C.; Chamkhi, I.; El Omari, N.; Balahbib, A.; Sharifi-Rad, J.; Bouyahya, A.; Akram, M.; Iqbal, M.; Docea, A.O.; et al. Pharmacological Properties of Chalcones: A Review of Preclinical Including Molecular Mechanisms and Clinical Evidence. Front. Pharmacol. 2021, 11, 592654. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Lu, X.; Zhang, H.-J.; Li, N.; Xiao, Y.; Zhu, H.-L.; Ye, Y.-H. Synthesis, structure, and biological assay of cinnamic amides as potential EGFR kinase inhibitors. Med. Chem. Res. 2013, 22, 986–994. [Google Scholar] [CrossRef]
- Plazuk, D.; Wieczorek, A.; Błauz, A.; Rychlik, B. Synthesis and biological activities of ferrocenyl derivatives of paclitaxel. Medchemcomm 2012, 3, 498–501. [Google Scholar] [CrossRef]
- Wieczorek, A.; Blauz, A.; Makal, A.; Rychlik, B.; Plazuk, D. Synthesis and evaluation of biological properties of ferrocenyl—Podophyllotoxin conjugates. Dalton Trans. 2017, 47, 10847–10858. [Google Scholar] [CrossRef] [PubMed]
- Wieczorek, A.; Błauż, A.; Żal, A.; Arabshahi, H.J.; Reynisson, J.; Hartinger, C.G.; Rychlik, B.; Plażuk, D. Ferrocenyl Paclitaxel and Docetaxel Derivatives: Impact of an Organometallic Moiety on the Mode of Action of Taxanes. Chem. A Eur. J. 2016, 22, 11413–11421. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Chen, J.; Ju, P.; Ma, L.; Chen, L.; Ma, W.; Zheng, T. Synthesis and Biological Evaluation of 4 β -N-Acetylamino Substituted Podophyllotoxin Derivatives as Novel Anticancer Agents. Agents Front. Chem. 2019, 7, 253. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Li, D.; Wei, M.; Du, R.; Yan, Z. The ester derivatives obtained by C-ring modification of podophyllotoxin induce apoptosis and inhibited proliferation in PC-3M cells via down-regulation of PI3K/Akt signaling pathway. Bioorg. Med. Chem. Lett. 2021, 46, 128174. [Google Scholar] [CrossRef]
- Bader, A.; Bkhaitan, M.M.; Abdalla, A.N.; Abdallah, Q.M.A.; Ali, H.I.; Sabbah, D.A.; Albadawi, G.; Abushaikha, G.M. Design and Synthesis of 4-O-Podophyllotoxin Sulfamate Derivatives as Potential Cytotoxic Agents. Evid.-Based Complement. Altern. Med. 2021, 2021, 6672807. [Google Scholar] [CrossRef]
- Yang, Z.; Zhou, Z.; Luo, X.; Luo, X.; Luo, H.; Luo, L.; Yang, W. Desing and Synthesis of Novel Podophyllotoxins Hybrids and the Effects of Different functional Groups on Cytotoxicity. Molecules 2022, 27, 220. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Brindisi, M. Organic Carbamates in Drug Design and Medicinal Chemistry. J. Med. Chem. 2015, 58, 2895–2940. [Google Scholar] [CrossRef]
- Matošević, A.; Bosak, A. Carbamate group as structural motif in drugs: A review of carbamate derivatives used as therapeutic agents. Arh. Hig. Rada Toksikol. 2020, 71, 285–299. [Google Scholar] [CrossRef]
- Xu, X.; Guan, X.; Feng, S.; Ma, Y.; Chen, S.; Hui, L. Chemistry Letters One-pot synthesis and biological evaluation of N-(aminosulfonyl)-4-podophyllotoxin carbamates as potential anticancer agents. Bioorg. Med. Chem. Lett. 2017, 27, 2890–2894. [Google Scholar] [CrossRef]
- Tian, D.; Chen, H.; Luo, G.; Liang, C. Synthesis and cytotoxicity of heterocyclic amine derivatives of podophyllotoxin. Chem. Nat. Compd. 2020, 56, 858–862. [Google Scholar] [CrossRef]
- Hu, C.; Zhu, X.; Wang, G.; Wu, X.; Han, H.; Lu, G.; Qi, J.; Pang, Y.; Yang, R.; Wang, X.; et al. Design, synthesis and anti-cancer evaluation of novel podophyllotoxin derivatives as potent tubulin-targeting agents. Med. Chem. Res. 2018, 27, 351–365. [Google Scholar] [CrossRef]
- Xi, W.; Sun, H.; Bastow, K.F.; Xiao, Z.; Lee, K.H. Identification of Novel 4′-O-Demethyl-epipodophyllotoxin Derivatives as Antitumor Agents Targeting Topoisomerase II. Molecules 2022, 27, 5029. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sun, H.; Xiao, Z.; Zhang, G.; Zhang, D.; Bao, X.; Li, F.; Wu, S. DNA damage and apoptosis induced by a potent orally podophyllotoxin derivative in breast cancer. Cell Commun. Signal. 2018, 16, 52. [Google Scholar] [CrossRef]
- Castro, M.A.; García, P.A.; Hernández, Á.P.; Díez, D. An overview on heterocyclic podophyllotoxin derivatives. Targets Heterocycl. Syst. 2015, 19, 28–61. [Google Scholar]
- Hernández, Á.P.; Díez, P.; García, P.A.; Miguel Del Corral, J.M.; Pérez-Andrés, M.; Díez, D.; San Feliciano, A.; Fuentes, M.; Castro, M.Á. New Hybrids Derived from Podophyllic Aldehyde and Diterpenylhydroquinones with Selectivity toward Osteosarcoma Cells. ACS Med. Chem. Lett. 2018, 9, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Hernández, Á.P.; Díez, P.; García, P.A.; Pérez-Andrés, M.; Ortega, P.; Jambrina, P.G.; Díez, D.; Castro, M.Á.; Fuentes, M. A Novel Cytotoxic Conjugate Derived from the Natural Product Podophyllotoxin as a Direct-Target Protein Dual Inhibitor. Molecules 2020, 25, 4258. [Google Scholar] [CrossRef] [PubMed]
- Munday, R. Autoxidation of naphthohydroquinones: Effects on metals, chelating agents, and superoxide dismutase. Free Radic. Biol. Med. 1997, 22, 689–695. [Google Scholar] [CrossRef]
- Pedroza, D.A.; De Leon, F.; Valera-Ramírez, A.; Lema, C.; Aguilera, R.J.; Mito, S. The cytotoxic effec of 2-acylated-1,4-naphthohydroquinones on leukemia/lymphoma cells. Bioorg. Med. Chem. 2014, 22, 842–847. [Google Scholar] [CrossRef]
- García, P.A.; Hernández, Á.P.; San Feliciano, A.; Castro, M.A. Bioactive prenyl- and terpenyl-quinones/Hydroquinones of marine origin. Mar. Drugs 2018, 16, 292. [Google Scholar] [CrossRef]
- Hernández, Á.P.; Chamorro, P.; Rodríguez, M.L.; Miguel del Corral, J.M.; García, P.A.; Francesch, A.; San Feliciano, A.; Castro, M.Á. New antineoplastic naphthohydroquinones attached to labdane and rearranged diterpene skeletons. Molecules 2021, 26, 474. [Google Scholar] [CrossRef]
- Roa-Linares, V.C.; Miranda-Brand, Y.; Tangarife-Castaño, V.; Ochoa, R.; García, P.A.; Castro, M.Á.; Betancur-Galvis, L.; San Feliciano, A. Anti-herpetic, anti-dengue and antineoplastic activities of simple and heterocycle-fused derivatives of terpenyl-1,4-naphthoquinone and 1,4-anthraquinone. Molecules 2019, 24, 1279. [Google Scholar] [CrossRef]
- Hernández, Á.-P.; Díez, P.; García, P.A.; Pérez-Andrés, M.; Veselinova, A.; Jambrina, P.G.; San Feliciano, A.; Díez, D.; Fuentes, M.; Castro, M.Á. Improving Properties of Podophyllic Aldehyde-Derived Cyclolignans: Design, Synthesis and Evaluation of Novel Lignohydroquinones, Dual-Selective Hybrids against Colorectal Cancer Cells. Pharmaceutics 2023, 15, 886. [Google Scholar] [CrossRef] [PubMed]
- Nerella, S.; Kankala, S.; Paidakula, S.; Gavaji, B. Synthesis of D-ring modified acid hydrazide derivatives of podophyllotoxin and their anticancer studies as Tubulin inhibiting agents. Bioorg. Chem. 2020, 94, 103384. [Google Scholar] [CrossRef] [PubMed]
- Popiołek, Ł. Updated Information on Antimicrobial Activity of Hydrazide–Hydrazones. Int. J. Mol. Sci. 2021, 22, 9389. [Google Scholar] [CrossRef] [PubMed]
- Mali, S.N.; Thorat, B.R.; Gupta, D.R.; Pandey, A. Mini-Review of the Importance of Hydrazides and Their Derivatives—Synthesis and Biological Activity. Eng. Proc. 2021, 11, 21. [Google Scholar]
- Ganaie, B.; Banday, J.A.; Baht, B.A.; Ara, T. Synthesis and In Vitro Anticancer Activity of Triazolyl Analogs of Podophyllotoxin, a Natural Occurring Lignin. Russ. J. Org. Chem. 2021, 57, 2039–2047. [Google Scholar] [CrossRef]
- Miguel del Corral, J.M.; Gordaliza, M.; Castro, M.A.; López-Vázquez, M.L.; García-Grávalos, M.D.; Broughton, H.B.; San Feliciano, A. Bioactive isoxazoline and oxime derivatives from 7-ketolignans. Tetrahedron 1997, 53, 6555–6564. [Google Scholar] [CrossRef]
- Xiang, R.; Guan, X.; Hui, L.; Jin, Y.; Chen, S. Investigation of the anti-angiogenesis effects induced by deoxypodophyllotoxin-5-FU conjugate C069 against HUVE cells. Bioorg. Med. Chem. Lett. 2017, 27, 713–717. [Google Scholar] [CrossRef]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-Fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef]
- Guan, X.W.; Xu, X.H.; Feng, S.L.; Tang, Z.B.; Chen, S.W.; Hui, L. Synthesis of hybrid 4-deoxypodophyllotoxin-5-fluorouracil compounds that inhibit cellular migration and induce cell cycle arrest. Bioorg. Med. Chem. Lett. 2016, 26, 1561–1566. [Google Scholar] [CrossRef]
- Kwon, J.; Lee, N.; Kang, A.; Ahn, I.; Choi, I.; Song, J.; Hwang, S.; Um, H.; Choi, J.; Kim, J.; et al. JNC-1043, a novel podophyllotoxin derivative, exerts anticancer drug and radiosensitizer effects in colorectal cancer cells. Molecules 2022, 27, 7008. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.J.; Fan, H.Y.; Yu, X.H.; Tang, Y.L.; Jiang, J.; Liang, X.H. Advances of podophyllotoxin and its derivatives: Patterns and mechanisms. Biochem. Pharmacol. 2022, 200, 115039. [Google Scholar] [CrossRef] [PubMed]
- Charlton, J.L. Antiviral activity of lignans. J. Nat. Prod. 1998, 61, 1447–1451. [Google Scholar] [CrossRef] [PubMed]
- Sudo, K.; Konno, K.; Shigeta, S.; Yokota, T. Inhibitory effects of podophyllotoxin derivatives on herpes simplex virus replication. Antivir. Chem. Chemother. 1998, 9, 263–267. [Google Scholar] [CrossRef]
- Brand, Y.M.; Roa-Linares, V.; Santiago-Dugarte, C.; del Olmo, E.; López-Pérez, J.L.; Betancur-Galvis, L.; Gallego-Gómez, J.C.; San Feliciano, A. A new host-targeted antiviral cyclolignan (SAU-22.107) for Dengue Virus infection in cell cultures. Potential action mechanisms based on cell imaging. Virus Res. 2023, 323, 198995. [Google Scholar] [CrossRef]
- Hensel, A.; Bauer, R.; Heinrich, M.; Spiegler, V.; Kayser, O.; Hempel, G.; Kraft, K. Challenges at the Time of COVID-19: Opportunities and Innovations in Antivirals from Nature. Planta Med. 2020, 86, 659–664. [Google Scholar] [CrossRef]
- Miao, Y.; Fan, L.; Jian-Yong, L. Potential treatments for COVID-19 related cytokine storm-Beyond corticosteroids. Front. Immunol. 2020, 11, 1445. [Google Scholar] [CrossRef]
- Zhang, J.; Zhao, H.; Feng, Y.; Xu, X.; Yang, Y.; Zhang, P.; Lu, Z.; Zhang, T. Topoisomerase 2 inhibitor etoposide promotes interleukin-10 production in LPS-induced macrophages via upregulating transcription factor Maf and activating PI3K/Akt pathway. Int. Immunopharmacol. 2021, 101, 108264. [Google Scholar] [CrossRef]
- Johnson, T.S.; Terrell, C.E.; Millen, S.H.; Katz, J.D.; Hildeman, D.A.; Jordan, M.B. Etoposide Selectively Ablates Activated T Cells To Control the Immunoregulatory Disorder Hemophagocytic Lymphohistiocytosis. J. Immunol. 2014, 192, 84–91. [Google Scholar] [CrossRef]
- Resendez, A.; Tailor, D.; Graves, E.; Malhotra, S.V. Radiosensitization of Head and Neck Squamous Cell Carcinoma (HNSCC) by a Podophyllotoxin. ACS Med. Chem. Lett. 2019, 10, 1314–1321. [Google Scholar] [CrossRef]

































































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miranda-Vera, C.; Hernández, Á.P.; García-García, P.; Díez, D.; García, P.A.; Castro, M.Á. Podophyllotoxin: Recent Advances in the Development of Hybridization Strategies to Enhance Its Antitumoral Profile. Pharmaceutics 2023, 15, 2728. https://doi.org/10.3390/pharmaceutics15122728
Miranda-Vera C, Hernández ÁP, García-García P, Díez D, García PA, Castro MÁ. Podophyllotoxin: Recent Advances in the Development of Hybridization Strategies to Enhance Its Antitumoral Profile. Pharmaceutics. 2023; 15(12):2728. https://doi.org/10.3390/pharmaceutics15122728
Chicago/Turabian StyleMiranda-Vera, Carolina, Ángela Patricia Hernández, Pilar García-García, David Díez, Pablo Anselmo García, and María Ángeles Castro. 2023. "Podophyllotoxin: Recent Advances in the Development of Hybridization Strategies to Enhance Its Antitumoral Profile" Pharmaceutics 15, no. 12: 2728. https://doi.org/10.3390/pharmaceutics15122728
APA StyleMiranda-Vera, C., Hernández, Á. P., García-García, P., Díez, D., García, P. A., & Castro, M. Á. (2023). Podophyllotoxin: Recent Advances in the Development of Hybridization Strategies to Enhance Its Antitumoral Profile. Pharmaceutics, 15(12), 2728. https://doi.org/10.3390/pharmaceutics15122728